Molecular docking for virtual screening of natural product databases
Dik-Lung
Ma
*a,
Daniel Shiu-Hin
Chan
a and
Chung-Hang
Leung
*b
aDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong. E-mail: edmondma@hkbu.edu.hk
bCentre for Cancer and Inflammation Research, School of Chinese Medicine, Hong Kong Baptist University, Kowloon Tong, Hong Kong. E-mail: duncanl@hkbu.edu.hk
First published on 20th June 2011
Abstract
Molecular docking enables the extraordinary structural diversity of natural products to be harnessed in an efficient manner. In this mini-review, we highlight recent examples of the use of molecular docking in virtual screening for the identification of bioactive molecules from natural product databases.
 Dik-Lung Ma | Dik-Lung Ma completed his PhD in 2004 at the University of Hong Kong under the supervision of Prof. C.-M. Che. He spent the years 2005–09 at the University of Hong Kong, the Hong Kong Polytechnic University, and the Scripps Research Institute with Prof. C.-M. Che, Prof. K.-Y. Wong, and Prof. R. Abagyan. His research mainly focuses on luminescent sensing for biomolecules and metal ions, computer-aided drug discovery, and inorganic medicines. In 2010 he was appointed Assistant Professor at the Hong Kong Baptist University. |
 Daniel Shiu-Hin Chan | Daniel Shiu-Hin Chan completed his BSc degree in Chemistry at the University of New South Wales, Sydney, Australia. He is currently appointed as a Research Assistant at the Department of Chemistry at the Hong Kong Baptist University. His research interests include development of luminescent oligonucleotide-based assays for biomolecules and metal ions. |
 Chung-Hang Leung | Chung-Hang Leung completed his PhD in 2002 at City University of Hong Kong under the supervision of Prof. W.-F. Fong. After completing a five-year post-doctoral fellowship at the Department of Pharmacology, Yale University with Prof. Y.-C. Cheng, he was appointed Research Assistant Professor at the University of Hong Kong and then at the Hong Kong Baptist University. His primary research interests are in anticancer and anti-inflammatory drug discovery, molecular pharmacology and immunology. |
Introduction
The 1990's heralded a paradigm shift in the way pharmaceutical research was conducted around the world. With new tools and techniques supplied by molecular biology, millions of research dollars were invested into the development of highly efficient high-throughput screening (HTS) in vitro and in vivo methodologies that were capable of screening hundreds of thousands of compounds each year.1–4 This growth was accompanied by exciting advances in the field of combinatorial chemistry.5–7 Whereas a typical bench chemist could be expected to possibly make a few hundred novel compounds each year, the new strategies afforded by combinatorial chemistry easily drove that number into the thousands. The pharmaceutical industry eagerly adopted these new technologies, which promised to usher in a new golden age of drug discovery. However, medicinal chemists were soon confronted with painfully disappointing results and the realization dawned that the chemical space was too vast to be systemically explored experimentally.8 Today, despite the increasing financial investment into research and development, the number of new drugs approved for the market has fallen to an all-time low. Bereft of promising leads, large pharmaceutical companies have increasingly shifted towards mergers and acquisitions in order to fill their pipelines.9,10 Thus, researchers are continually seeking to develop novel methodologies for the efficient discovery of new chemical scaffolds for pharmaceutical investigations. This review focuses on the emerging use of computer-aided high-throughput molecular docking methodologies to identify bioactive NPs against important therapeutic targets, and aims to summarize the recent successes in this area.Role of natural products in drug research
Nature has provided us with a fascinating cornucopia of complex molecular scaffolds unparalleled in function and diversity. These natural products (NPs) have been refined over evolutionary time scales for optimal interactions with biomolecules. As the production of any new compound by a living organism entails increased costs, the natural selection hypothesis predicts that each natural product retained by an organism must be of value to the producer. Before the advent of modern synthetic medicinal chemistry, natural products were the only source of medicinal compounds available to mankind. Pharmacological investigations into traditional and herbal remedies have yielded many significant discoveries for modern medicine.11–16 For example, the herb Artemisia annua has been used in traditional Chinese medicine for thousands of years for treating malaria and other skin diseases, but it was only several decades ago that the active ingredient, artemisinin, was isolated.17–19 Huperzine A, isolated from the plant moss Huperzia serrta, is an acetylcholinesterase inhibitor investigated for Alzheimer's disease, though the plant itself has been used for centuries by Chinese herbalists for treating swelling, fever and blood disoders.20–22 Another highly important drug of natural origin is the anti-cancer paclitaxel (taxol), isolated from the tree bark of Taxus brevifolia, and used for the treatment of breast, ovarian, lung, head and neck cancer.23 The total number of natural compounds produced by plants has been estimated to be over 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.24 However, this is believed to represent only a small fraction of the total number of existing NPs, considering the huge number of undocumented species in the vast unchartered regions of the biosphere. Marine organisms, in particular, are a tremendous source of secondary metabolites possessing unique structural diversities yet to be tapped by the medicinal chemist.25–27
000.24 However, this is believed to represent only a small fraction of the total number of existing NPs, considering the huge number of undocumented species in the vast unchartered regions of the biosphere. Marine organisms, in particular, are a tremendous source of secondary metabolites possessing unique structural diversities yet to be tapped by the medicinal chemist.25–27
Until recently, NPs were still regarded as a fundamental cornerstone of pharmaceutical research.28–32 However, amidst the rapid developments and promises of HTS and combinatorial chemistry, the realm of natural products fell somewhat by the wayside. Many major pharmaceutical companies have halted or significantly cut natural product research. A survey of new chemical entities by Newman and Cragg between the years 1981–2006 shows that NPs and drugs derived directly from natural compounds occupied only 5% and 23% of this class, respectively, compared to 30% for synthetic compounds.33 It has also been noted that the number of NP-based drugs in clinical trials has fallen 30% between 2001 and 2008.34
Tellingly, the shift away from NP research in pharmaceutical corporations was engendered not by any intrinsic faults with the class of natural compounds as a whole, but by the incompatibility of NP with the highly automated drug discovery methodologies of HTS. The reasons are many-fold. Firstly, NPs are often regarded as highly structurally complex, containing any number of chiral centres, fused ring systems, and reactive functional groups like the hydroxyl. Pursuing the synthesis of a complicated NP or NP derivative is hardly a trivial task, requiring the construction of intricate ring systems together with a laborious strategy of protection and deprotection steps that are not readily amenable to combinatorial methods. Secondly, the extraction and purification of NPs from plant, marine and microbial sources can be very difficult, and the precious compounds are often obtained in only minute quantities. Issues with supply can become a major bottleneck in NP drug discovery especially if the NP itself is to be utilized as a benchmark or as a synthetic intermediate for lead optimization and further in vivo testing. Furthermore, structural elucidation still has to be performed on any assay hit obtained from a natural source, whether from bioassay-guided fractionation or the isolation of a single active compound, and its structure would have to be compared to the synthetically-generated NP. Thirdly, the HTS screening of crude NP extracts can sometimes be problematic due to highly polar or lipophilic components in the sample matrix, leading to false positives or false negatives in the assay result. The screening of crude extract libraries also entails a stringent identification and dereplication program to prevent the expenditure of resources on known or otherwise uninteresting compounds. A further complication arises due to the fact that individual compounds in an extract are likely to be present at vastly different concentrations, such that levels of trace components may be too low for detection, while inactive, highly abundant compounds may be registered due to non-specific inhibition. Additionally, the intrinsic optical properties of uncharacterized NPs in the extract may interfere with fluoresence-based assay methodologies. Lastly, the synergistic or antagonistic effects of two or more components may vanish upon isolation of the primary constituent. By comparison, the screening of large libraries of pure, characterized, synthetic compounds present in known quantities is regarded as a much easier endeavour.
However, the failure of combinatorial chemistry to yield a significant increase in the number of drug candidates despite the powerful testing capabilities of HTS prompted a rethink of the nature and quality of the chemical libraries themselves. The major drawback to combinatorial compounds lies in the implicit requirement for efficient reaction and purification methodologies that whilst able to produce large numbers of compounds at high yield and low cost, severely limits the structural diversity of the chemical library. According to a study performed by Feher and co-workers, combinatorial compounds tend to contain fewer chiral centers compared to natural compounds.35 They also contain more rotable single bonds and aromatic rings, an artifact of the construction process that involves joining many building blocks together in simple reactions. On the other hand, NPs tend to contain more rigid scaffolds comprising fused, unsaturated ring systems, potentially exploiting the entropic advantages conferred by rigidity in order to achieve the high biochemical affinities and specificities characteristic of NPs.35
Today, HTS is rarely performed using purely combinatorial, unfocused libraries. Instead, efforts have focused on “privileged” synthetic databases that seek to occupy a more biologically relevant space within the vast universe of possible chemical structures. Such libraries include databases of existing drugs (drug repurposing),36–38 as well as those based upon the principles of “diversity-orientated synthesis” (DIOS)39–41 and “biology-orientated synthesis” (BIOS).42–44 A comprehensive review of all these methodologies is outside the scope of this work, and the reader is directed to the excellent reviews that have been published on these topics.36–44 However, despite the best of these efforts, HTS screening of such carefully constructed libraries can still realistically only sample but a small portion of the entire chemical space at a time. This is where computer-aided drug discovery can make an important contribution.
Virtual screening in drug discovery
Virtual screening has recently emerged as a powerful technique complementing traditional HTS technologies. Virtual screening can be broadly defined as the use of computational analysis of a database of chemical structures to identify possible drug candidates for a specific pharmaceutical target, often a particular enzyme or receptor. The major advantage derived from virtual screening is the tremendous reduction in time and resources required to screen a chemical library of known compounds in a drug discovery project. By identifying non-binding compounds in silico, the number of compounds to be tested in vitro can be dramatically reduced, sometimes by orders of magnitudes. Due to this “weeding out” or elimination of inactive chemical structures, the hit rates in the in vitro assays are often much higher compared to conventional HTS without preliminary virtual screening. Furthermore, the number of compounds in even the largest chemical library is but a small fraction of the total possible number of compounds that could be synthesized in principle; estimates range up to 1020–24 for the total number of molecules accessible using known synthetic methods.45 Computer-aided screening is thus a valuable tool to help medicinal chemists decide what to synthesize, an advantage over the somewhat random nature of combinatorial libraries used previously.The use of virtual screening methodologies in drug discovery and development has been widely reviewed.46–51 Today, computational chemistry and chemoinformatics play a key role in early phase drug research, by identifying the most promising candidates for experimental investigations. Two major strategies have been employed for virtual screening: pharmacophore modeling and molecular docking (Fig. 1). Within the realm of pharmacophore modeling, the two techniques of structure-based phamacophore modeling and ligand-based pharmacophore modeling can be distinguished. In the former, the knowledge of the three-dimensional (3D) structure of the biomolecular target is analyzed to identify features of the binding site that are important for ligand binding affinity and selectivity. The 3D structure may be solved by X-ray crystallography, NMR studies or constructed by homology with related proteins, and over 73000 of such structures are freely available at the Protein Data Bank (PDB).52 An X-ray structure of the biomolecular target that is co-crystallized with a ligand is especially advantageous since specific features of the ligand–target interaction can be readily detected. Some computational softwares such as LIGANDSCOUT are able to automatically construct a pharmacophore model by identifying and performing calculations on the relevant interactions between the small molecule and the receptor. These ligand–targets are classified into features such as hydrogen bonding, charge transfer, and lipophilic interactions to construct at 3D pharmacophore model. In ligand-based pharmacophore modeling, a 3D structure of the biomolecular target is not required. Rather, a collection of known ligands is assembled in order to derive representative electrostatic and steric features that are then used to construct the pharmacophore model. After the construction of the 3D pharmacophore model, a chemical database is screened against the model in order to extract ligands that possess the complementary functional groups in the correct spatial arrangement. Since the affinity calculation is based only on the geometric fit of ligand atoms and groups to the chemical features of the model, the strain on computational resources is significantly lower compared to molecular docking. This makes pharmacophore modeling suitable as a pre-filter for more demanding virtual screening methodologies.53 However, a disadvantage is that the calculated affinity values are often not accurate, however, they are still broadly useful for eliminating non-binders. Ligand-based pharmacophore modeling also requires prior knowledge of a set of active compounds, which makes it use unsuitable for novel biomolecular targets. An additional drawback of the pharmacophoric method is that in the absence of structural or mechanistic information about the target, it is not always possible to anticipate which particular structural features of the ligands is important for the receptor–ligand interaction. Lastly, due to the inherent nature of pharmacophore modeling, screening usually reveals hits that are structurally similar to the known ligands, rather than leads with novel modes of binding. The use of pharmacophore modeling in NP drug research is not the focus of this review, and the reader is directed to excellent reviews published recently by the groups of Langer54 and Wolber.55
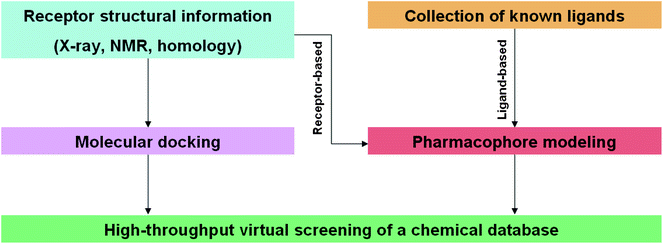 | ||
| Fig. 1 Schematic flowchart outlining the two major strategies employed in in silico virtual screening. | ||
Compared to the relatively established technique of pharmacophore modeling, 3D molecular docking is considered more complex and computer-intensive. However, exponential advances in computing processing power and capabilities have increased the popularity of molecular docking methods (also called structure-based or receptor-based virtual screening) in drug discovery and development. The use of molecular docking avoids some of the aforementioned drawbacks involved with pharmacophore modeling. Molecular docking requires knowledge of the 3D structure of the biomolecular target with or without a bound ligand, at atomic resolution. Use of a co-crystal structure of a biomolecular target with a ligand entails several advantages. Firstly, the search area for the in silico docking can be restricted to the binding site only (alternatively, the binding site may be predicted from homology with similar proteins). An unnecessarily large search area can increase the rate of false positives due to the binding of virtual molecules outside the actual binding site. Furthermore, this can also lead to wastage in computer resources. Secondly, prior knowledge of critical ligand–target interactions can help identify false positives in the virtual screening. Third, more accurate docking calculations can be performed since the target is in its active or induced conformation. A computational model of the target is constructed from the 3D structure, and ligands from the chemical database are sequentially docked against the receptor. Most docking programs incorporate ligand flexibility into the docking calculations so that the binding geometry of the ligand can be corrected predicted. However, the target is usually assumed to be mostly rigid, as the explicit inclusion of receptor flexibility in the docking calculations would be too computationally demanding. Some docking algorithms attempt to model receptor flexibility in an indirect way, for example by tolerating some degree of steric clashing with the ligand without the explicit repacking of the receptor side chains, or by using several alternative receptor binding site conformations for docking and then merging the results. Some modern docking algorithms are able to explicitly model receptor flexibility, but this is usually constrained to the ligand binding domain in order to conserve computing resources.56,57 A RMSD of <2 Å from the predicted binding pose to the X-ray crystal structure is considered satisfactory.
After the optimal binding poses have been predicted for each compound in the database, the next step is to rank or score the structures to determine their relative binding affinities. Scoring functions perform calculations involving statistical potentials or weighted interaction terms that have been previously calibrated with “training sets” of known binders and non-binders. However, it has been noted that scoring functions represent the major weakness in docking programs; while the docking algorithm is relatively accurate at predicting the preferred binding mode, the ability of the scoring functions to distinguish strong and weak binders still leads to a significant number of false positives in the virtual screen. Consensus scoring, the use of multiple scoring functions in concert, has been found to significantly improve hit rates compared to the use of a single scoring function.58–61
As discussed in the previous section, despite the astonishing structural diversity and fascinating molecular architecture exhibited NPs, their use in HTS has been somewhat neglected compared to purely synthetic libraries based on drug-likeness, DIOS or BIOS. The screening of isolated natural compounds and NP extracts has often been regarded as too “dirty” and too expensive, and involving additional time and labour-consuming efforts in isolation, fractionation, characterization, and dereplication. Computational methods open up new possibilities for screening natural compounds by bypassing many of these tedious steps. For example, non-binders can be predicted in silico, avoiding the wastage of scarce NPs. Additionally, practical difficulties associated with bioassay-guided fractionation and dereplication are made irrelevant. One minor drawback is that it is no longer possible to isolate totally new bioactive compounds, as from a NP extract.
It is common knowledge that the results of any HTS exercise is ultimately predicated upon the quality of the compound collection itself. Poorly designed libraries lacking sufficient diversity may result in few hits. Fortunately, NP libraries possess an intrinsic advantage in diversity of molecular scaffolds due to their exquisite relationships with biomolecules that have been streamlined over evolutionary time scales. Furthermore, many NP compound collections also contain NP-like structures that are based upon naturally occurring scaffolds. One should also note that NPs often contain more than one violation of Lipinski's “rule-of-fives”,62 especially with regards to molecular weight and hydrogen bond acceptors, and too-strict adherence to these rules in a screening campaign would certainly act to negate the inherent advantages of structural diversity conferred by the use natural compounds. A summary of some available NP databases is presented in Table 1.
| Database | Company | Number of compounds | Link |
|---|---|---|---|
| ZINC | Bioinfomatics and Chemical Informatics Research Center (BCIRC) | 89425 |
http://zinc.docking.org/vendor0/npd/index.html |
| MEGabolite | Analyticon Discovery | 4700 |
http://www.ac-discovery.com |
| NatDiverse | Analyticon Discovery | >20000 |
http://www.ac-discovery.com |
| IBS Database | InterBioScreen Ltd | >45000 |
http://www.ibscreen.com/natural.shtml |
| Super Natural Database | Charitè, Medical Faculty of the Humboldt-University | 45917 |
http://bioinformatics.charite.de/supernatural/ |
| CHEMnetBASE (Dictionary of Natural Products) | Chapman & Hall | >226000 |
http://www.chemnetbase.com/ |
| CHEMnetBASE (Dictionary of Marine Natural Products) | Chapman & Hall | >34000 |
http://www.chemnetbase.com/ |
| Natural Products Database (NPD) | Molecular Diversity Preservation International (MDPI) | 22048 |
http://www.mdpi.org/ |
| Natural Compound Library (NCL) | TimTec | 640 | http://www.timtec.net/ |
| Natural Derivative Library (NDL) | TimTec | 3000 |
http://www.timtec.net/ |
| SPECS Natural Products | SPECS, Inc. | 400 | http://www.specs.net/ |
| CNPD | NeoTrident Technology Ltd | 57000 |
http://www.neotrident.com/newweb/index.asp |
Recent successes of molecular docking of natural products for drug discovery
We discuss here some recent examples of the use of molecular docking for the discovery of bioactive NPs in the last decade. For ease of access, we have grouped docking targets into three classes: enzyme–substrate interactions, receptor–ligand interactions (including protein–protein interactions) and DNA interactions.Natural products targeting the enzyme–substrate interaction
In 2004, Toney and co-workers screened the National Cancer Institute (NCI) diversity set of 1853 compounds of both natural and synthetic origin against 3CLproproteinase of the severe acute respiratory syndrome coronavirus (SARS-CoV), the causative agent for a pandemic that swept Southeast Asian regions in 2003.63 The compounds were docked against the X-ray crystal structure of 3CLpro (PDB entry: 1P9S) using the AutoDock program. The highest scoring compound was found to be the natural product sabadinine, which showed a binding energy of −11.6 kcal mol−1 and a clustering of 9 out of 10 docked conformers within 0.5 Å. However, sabadinine did not affect murine coronavirus replication at 100 μM, as gauged by syncytium formation and cytopathic effects.Sangma and co-workers (2005) utilized a combined approach of molecular docking and neural networks to identify inhibitors of HIV-1reverse transcriptase (RT) and HIV-1protease (PR) from a Thai Medicinal Plants Database.64 2684 compounds were docked against the X-ray co-crystal structures of HIV-1 RT with nevirapine or calanolide A (PDB entry: 1VRT) and HIV-1 PR with XK-263 (PDB entry: 1HVR) using AutoDock, and then the results were applied to a neural network based on a self-organizing map (SOM) in order to reduce the size of the hit list. In the SOM approach, the reference structures are analyzed to find potential pharmacophoric groups, and for easier analysis, a map was generated that contained only the distances between three points of certain pharmacophoric groups. The same maps were generated from the docking hit list and then superimposed onto the reference map, and only the compounds having their features represented in the common regions were reselected. This successive SOM screening could be repeated as many times as necessary (by using different features) to reduce the size of the candidate pool. For example, AutoDock identified 562 (out of 2684 compounds) docking hits for HIV-1 PR, but this was reduced to 135 and then 13 after successive rounds of SOM screening. However, most of these compounds had already been reported to show anti-HIV activity; no biological validation was performed in this work. This study demonstrates that using a pharmacophoric SOM neural network after molecular docking is a computationally inexpensive method of reducing the size of the virtual screening hit list.
In 2006, the group of Moro identified ellagic acid, a naturally occurring derivative of tannic acid, as an inhibitor of casein kinase 2 (CK2), a putative oncogene in animal and cellular models.65 Using an in-house database of around 2000 NPs, they utilized a consensus docking and scoring approach that includes four docking algorithms (MOE-Dock, Glide, Fred and Gold) and five scoring functions (MOE-Score, GlideScore, GoldScore, ChemScore and Xscore) to dock and rank the NP database against the X-ray crystal structure of CK2 (PDB entry: 1JWH). Significantly, ellagic acid ranked in the top 5% of all five scoring functions. Furthermore, all four docking programs predicted the same lowest energy binding conformation for ellagic acid. This promising in silico result was validated by the biological verification experiments, which showed that ellagic acid represented the most potent CK2inhibitor reported at that time (Ki = 20 nM). Kinetic analysis revealed ellagic acid to be a competitive inhibitor with respect to ATP, which was consistent with the molecular docking results that showed binding of the small molecule to the ATP-binding domain. Importantly, ellagic acid was shown to be at least 72-fold more potent against CK2kinase (IC50 = 0.04 μM) compared to a panel of 11 other kinases (IC50 > 2.9 μM), a remarkable result considering the ostentatiously promiscuous structural features of ellagic acid (a planar compound containing four hydroxyl moieties) and the fact that this virtual screening hit was not optimized at all. This early work helped to demonstrate the power of molecular docking for identifying potent and selective NPs against therapeutic biomolecular targets.
In 2008, Fu et al. identified Jadomycin C as an inhibitor of Aurora B kinase by employing molecular docking of the Microbial Natural Products Database containing about 15000 natural compounds of microbial origin against the X-ray co-crystal structure of Aurora-B cocrystallized with the Hesperadin (PDB entry: 2BFY).66 Aurora kinases play key roles in cell division through the regulation of centrosome maturation and separation, microtubulue-kinetochore attachment, and chromosome alignment and segregation. Consequently, Aurora kinases have been found to be overexpressed in a variety of cancers, and inhibitors of such kinases, including Hesperadin, ZM447439 and VX-680, have been showed significant anticancer activity in vitro and in vivo. Using the docking program FlexX, a hit list of the 150 top scoring structures were obtained from the virtual screening campaign, of which 22 were procured for experimental testing. The most promising candidate, Jadomycin B, could inhibit the activity of Aurora-B in vitro with an IC50 value of 10.5 μM and a Ki value of 6.8 μM. Jadomcyin was also able to inhibit the proliferation of a variety of cancer cell lines with IC50 values in the range of 10–26 μM. However, because Jadomycin B induced apoptosis without blocking the cell cycle, the putative target mechanism of Aurora-B inhibition, the authors concluded that Jadomycin B could potentially inhibit other kinases more effectively resulting in apoptosis. Due to the highly conserved nature of the kinaseATP-binding domain, achieving selectivity amongst the 518 protein kinases in the human genome is often the paramount concern in kinase inhibitor discovery projects. It would have therefore been interesting for the authors to investigate the activity of Jadomycin B against panel of different kinases using both in silico and in vitro methods, in order to evaluate the ability of molecular docking to predict the selectivity profile of their NP screening hit.
In 2011, our research group reported the discovery of the NP-like 6,6′′-biapigenin as only the second inhibitor of NEDD8-activating enzyme (NAE) using molecular docking.67NAE is an analogue of the ubiquitin E1 enzyme that is involved with regulating the ubuiqitination and degradation of the subset of proteins regulated by E3 cullin-RING ligases, including cancer-related substrates such as p-IκBα and c-myc. The first inhibitor of NAE reported, MLN4924, displayed efficacy against both solid and hematological human cancer cell lines.68 We performed high-throughput molecular docking of the Analyticon MEGAbolite and NatDiverse databases of over 20000 NP and NP-like structures against the X-ray crystal structure of the quaternary APPBP1-UBA3-NEDD8-ATP complex (PDB: 1R4N) using the docking software Molsoft. The 10 highest-scoring compounds were purchased and tested in biological validation experiments, and the biflavonoid 6,6′′-biapigenin emerged as the top candidate, with micromolar potencies against NAE in enzyme and cell-based assays. While 6,6′′-biapigenin itself has not been found in nature, it has been synthesized from 6,6′′-biapigenin hexaaceate, in turn obtained from the naturally occurring succedaneaflavanone (6,6′′-binarigenin). Interestingly, molecular modeling analysis of the ligand–receptor interaction indicated a binding mode very different to that of ATP or the nucleotide mimic NAEinhibitorMLN4924, tentatively suggesting that 6,6′′-biapigenin can be considered a new class of NAEinhibitor (Fig. 2). Significantly, 6,6′′-biapigenin was only the second NAEinhibitor reported to date. While further kinetic or structural evidence would be required to definitively establish the mechanism of inhibition of NAE by 6,6′′-biapigenin, this study highlights some of the chief advantages of molecular docking over ligand-based pharmacophore screening. Firstly, for a relatively new biomolecular target such as NAE, ligand-based pharmacophore modeling is often impossible due to the fact that inhibitors of the target have not yet been discovered. Secondly, since the structures in the screening library are computed against the pharmacophoric model of known inhibitors, pharmacophore screening is inherently unable to identify bioactive compounds with a novel mode of binding, and such compounds would be the first to fall out of the virtual screening campaign due to their incongruous binding mode. Besides its use in virtual screening, molecular docking can also be used to explore the mode and mechanism of enzyme inhibition by natural products. For example, Monti et al. have used Molsoft to analyze the binding of the marine natural products including scalaradial69 and petrosaspongiolide M70 to phospholipase A2, a target for inflammation.
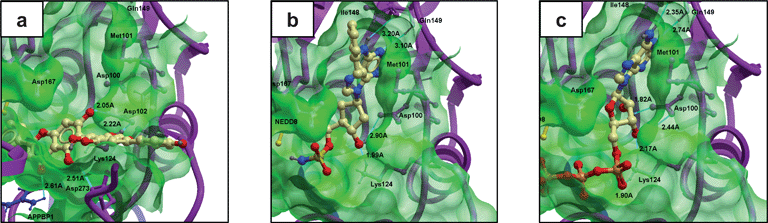 | ||
| Fig. 2 Molecular model of a) virtual screening hit 6,6′′-biapigenin, b) MLN4924 and c) ATP bound to the NAE heterodimer generated by virtual ligand docking. Molecular modeling analysis revealed a putatively different binding mode of 6,6′′-biapigenin compared to MLN4924 or ATP.67 | ||
Natural products targeting the receptor–ligand interaction
In 2003, Liu et al. screened the China Natural Products Database of 50000 compounds against a structural model of the potassiumion channel using Dock.71 The 3D model of eukaryotic ShakerK+channel was constructed based on homology with the crystal structure of prokaryotic Streptomyces Kcsa K+channel (PDB entry: 1BL8). The search area for the molecular docking was restricted to the extracellularporebinding site formed by the tetramer. Attention was focused on potential ligands that interacted with the residues surrounding the entry of the “ion-selective filter” of the channel so that the compounds could inhibit K+channel function. To reduce the size of the initial hit list, the top 200 compounds were reoptimized using the Sybyl molecular mechanics force field, yielding a final collection of 14 compounds of which 4 were procured for biological testing. All 4 compounds inhibited the K+channel in the electrophysiological assay by whole-cell voltage-clamp recording in dissociated hippocampal rat neurons, with 20–1000-fold higher potency than tetraethylammonium, a well-known K+channel blocker.
Brinton and co-workers (2005) docked an in-house database of 25000 plant-based NP and NP derivatives against the X-ray co-crystal structure of estrogen receptor-β (ERβ) with the isoflavone genistein (PDB entry: 1QKM) using the docking software Gold.72ERβ is postulated to play key roles in maintaining estrogen-inducible neuronal morphological plasticity, brain development, and cognition. On the other hand, ERα is more predominantly expressed in the breast and the endometrium. Therefore, ERagonists for the treatment of neurodegenerative diseases should display high selectivity for ERβ over ERα to avoid unwanted profilerative effects in other tissues or organs. The top 500 scoring molecules from the molecular docking were filtered by visual inspection and 100 compounds were manually selected for further analysis by the docking program Affinity, to refine the binding modes predicted by Gold. The final result was that 31 compounds were selected that possessed the critical hydrogen bond with His475 of ERβ as well as necessary hydrophilic and hydrophobic characteristics matching that of endogenous 17β-estradiol or genistein. Of the 12 tested compounds, 5 molecules displayed significant selectivity for ERβ over ERα (3 of these exhibited over 100-fold selectivity). However, the neurobiological effects of these compounds will require further in vivo investigations as the agonistic or antagonistic activities of weak estrogenic binders are known to be dependent on a number of factors, such as the concentration of the drug, the concentrations of endogenous estrogens, as well as the level of transcriptional coactivators or corepressers present in the particular tissue.73
Peroxisome proliferator-activated receptor-gamma (PPAR-γ) plays a critical role in lipid and glucose homeostasis, and inhibitors of PPAR-γ, such as the synthetic thiazolidinediones, are potential therapeutic agents for the treatment of type II diabetes.74 Notable side effects associated with such compounds, including fluid retention, weight gain, and cardiac hypertropy have recently stimulated Hibbs and co-workers (2008) to discover new NP scaffolds as PPAR-γagonists.75 The authors docked an in-house natural product database of 200 compounds against two structurally distinct X-ray co-crystal structures of PPAR-γ using rigid-receptor docking with Glide. One PPAR-γ structure was co-crystallized with the thiazolidinedione rosiglitazone (PDB entry: 1FM6), while the other incorporated the tyrosine-based analogue farglitazar (PDB entry: 1FM9), one of the earliest anti-diabetic lead compounds. Termed “multiple rigid-receptor docking”, the authors proposed that the use of two structurally distinct induced-fit conformations of the protein would admit a wider range of ligands, particularly if the hits achieved ligand–receptor interactions comparable to one or both of the known inhibitors. The 20 best-scoring compounds for each of the two conformations were pooled together, and duplicates (those compounds scoring highly for both conformations) were removed, to achieve a hit list of 29 unique candidates, that were observed to fall into three structural classes: the flavonoids, the gingeroids, and the ginkolides. However, visual inspection of the binding poses revealed that only the flavonoids were able to engage key protein residues in the transcriptional activation funtion 2 domain (AF-2 helix) characteristic of PPAR-γagonists, and all of the flavonoids achieved much better docking scores with the farglitazar-induced receptor rather than the rosiglitazone-induced receptor. Biological validation using a cellular transcriptional factor assay confirmed this analysis as only 6 out of the 29 compounds showed biological activity, and all 6 were from the flavonoid family, with activities approximately 3.6–21-fold times less potent than rosiglitazone. Considering the considerable flexibility exhibited by the PPAR-γ ligand binding domain, the authors used an induced-fit docking (IFD) protocol to further analyze the binding mode of the flavonoids. Receptor flexibility was modeled using the protein structure prediction and refinement package Prime in combination with Glide. Significantly, induced-fit docking of the flavonoids into the inferior model (the rosiglitazone-induced receptor) resulted in significant structural rearrangements in the protein side chains of the ligand-binding domain, reproducing the superior, lower-energy binding pose observed in the farglitazar model. This study showed that induced-fit docking can be used to reveal a more plausible binding mode than by rigid-docking alone, allowing for even significant rearrangements of the protein scaffold in order to better accommodate the docked ligand. However, the protein side-chain prediction and geometry minimization calculations are more computationally demanding compared to those in rigid-receptor docking. Furthermore, as with all molecular models, the induced-fit binding pose would still have to be verified with experimental techniques such as NMR or X-ray crystallography.
In 2008, the group of Ngai and Archer reported the discovery of high-affinity agonists of an olfactory G protein-coupled receptor (GPCR) using both receptor-based and ligand-based virtual screening.76 The GPCR responds preferentially to long chain amino acids such as lysine and arginine, as well as to other amino acids with lower affinity. This relatively non-specific binding profile is characteristic of odorant receptors, which allows the olfactory system to recognize a diversity of chemical structures exceeding the actual number of olfactory receptors in the human genome. The authors wished to identify novel high-affinity agonists of the V2R vomeronasal receptor-like goldfish receptor 5.24 for use as molecular probes to study receptor structure and olfactory function. Interestingly, while a database comprising over 1.6 million commercially available compounds was used for the molecular docking and pharmacophore-screening, the most active hit compounds were all naturally occurring, linear amino acid derivatives, and these were found to be more potent agonists than the natural amino acids. For example, diaminopimelic acid is a component of Gram-negative bacterial cell walls and is used as an intermediate bacterial biosynthetic pathways for lysine and peptidoglycans. Furthermore, the hit rate for the common virtual hits that scored highly in both the molecular docking and pharmacophore screening was extremely high (16/36 = 44%), with activity defined as >40% maximum receptor activity at 20 μM of the compound. Four of these compounds were active as olfactory receptoragonistsin vivo as measured by electrophysiological recordings of goldfish olfactory epithelium. Furthermore, molecular modeling was performed to analyse specific features of the amino acid ligands that may confer agonist or antagonist activities, based on the structural analysis of the receptor's ligand binding domain.
Compared to enzyme–substrate or ligand–receptor interactions, targeting the protein–protein interface is usually regarded as more difficult due to the relatively large and featureless nature of most protein–protein binding pockets, that lack clearly-defined binding crevices or mechanism-based contacts.77 In 2010, our research group wondered if the enormous structural diversity of NPs could be harnessed to discover novel small molecule inhibitors of tumor necrosis factor-α (TNF-α).78 TNF-α is a multifunctional cytokine that acts as a central biological mediator for critical immune responses, including inflammation, infection, and apoptotic cell death. Dysregulation of TNF-α has been implicated in cases of tumorigenesis, diabetes, and especially in autoinflammatory diseases such as rheumatoid arthritis, psoriatic arthritis and Crohn's disease.79 Using the software Molsoft, over 20000 NP and NP-like structures from the Analyticon MEGAbolite and NatDiverse databases were docked against the X-ray co-crystal structure of TNF-α dimer with SPD304 (PDB code: 2AZ5), the strongest TNF-α small molecule inhibitor reported to date. The top 16 compounds from the virtual screening were obtained and tested in vitro, and two NP-like compounds, a pyrazole-linked quinuclidine and an indolo[2,3-a]quinolizidine, emerged as the top candidates. Despite the mostly hydrophobic, formless nature of the TNF-α binding pocket, both compounds occupied a similar region in the binding site compared to SPD304 (Fig. 3). Furthermore, both compounds were observed to be large enough to contact both subunits of the TNF-α dimer simultaneously, thus occupying and blocking the binding site for the third TNF-α subunit and preventing the formation of the biologically active TNF-α trimer complex. Molecular modeling analysis indicated the absence of extensive hydrogen bonding or salt bridge formation which was consistent with our knowledge of the TNF-α binding pocket and with the reported binding mode SPD304. The indoloquinolizidine (IC50 = ca. 10 μM) was found to be more potent against TNF-α in the receptor binding assay compared to SPD304 (IC50 = 22 μM by a comparable ELISA), while the quinuclidine (IC50 = ca. 5 μM) had comparable activity to SPD304 (IC50 = ca. 3 μM) against cellular TNF-α induced NF-κBluciferase activity. These compounds also represented only the third and fourth examples of TNF-α inhibition by a small molecule inhibitor at that time. This study demonstrates that despite the seemingly intractable nature of the protein–protein interface, molecular modeling can be an effective tool for harnessing the structural diversity of NPs towards identifying novel inhibitors of the protein–protein interaction.
![Low-energy binding conformations of a) screening hit quinuclidine, b) screening hit indolo[2,3-a]quinolizidine, c) SPD304 bound to TNF-α dimer and d) superimposition of quinuclidine (orange), indolo[2,3-a]quinolizidine (yellow) and SPD304 (blue) generated by virtual ligand docking.78](/image/article/2011/SC/c1sc00152c/c1sc00152c-f3.gif) | ||
| Fig. 3 Low-energy binding conformations of a) screening hit quinuclidine, b) screening hit indolo[2,3-a]quinolizidine, c) SPD304 bound to TNF-α dimer and d) superimposition of quinuclidine (orange), indolo[2,3-a]quinolizidine (yellow) and SPD304 (blue) generated by virtual ligand docking.78 | ||
Natural products targeting DNA
DNA-targeting NPs are some of the most well-known anti-cancer drugs. For example, mitomycin is a potent DNA cross-linker from Streptomyces caespitosus or Streptomyces lavendulae that finds use as a chemotherapeutic against gastrointestinal, breast, and bladder cancers,80 while the actinomycins, also from Streptomyces, are chemotherapeutics and antibiotics that bind DNA at the transcription initiation complex and inhibiting RNA polymerase.81 Recently, non-canonical secondary structures of DNA such as the G-quadruplex have emerged as attractive targets for therapeutic intervention due to their putative association with oncogene promoter sequences such as c-myc, bcl-2, VEGF, KRAS, and c-kit.82–84 While the NP telomestatin is the most potent G-quadruplex ligand known to date,85 the majority of G-quadruplex binding ligands reported in the literature have been synthetic in origin. In 2010, our research group applied high-throughput structure-based virtual screening methods to identify natural product ligands of the c-mycG-quadruplex, as potential chemotherapeutic agents.86 In order to develop a high-throughput screening platform for G-quadruplex binding ligands, a model of the intramolecular c-mycG-quadruplex 1:2:1 loop isomer was constructed using the X-ray crystal structure of the closely related intramolecular human telomeric G-quadruplexDNA (PDB entry: 1KF1), since neither the NMR nor X-ray crystallographic information for the predominant c-myc 1:2:1 loop isomer was available. Using Molsoft, we docked the Analyticon MEGAbolite and NatDiverse databases of over 20000 NP and NP-like structures against our the model of the c-mycG-quadruplex, and the top 5 scoring compounds were tested in an in vitro assay. The naphthopyrone pigment fonsecin B, isolated from the fungus Aspergillus fonsecaeus, emerged as the top candidate. Fonsecin B was found to inhibit Taq-mediated extension through stabilization of the G-quadruplex secondary structure (IC50 = ca. 20 μM), with potency comparable to the well-known G-quadruplex binder TMPyP4. The molecular docking analysis revealed that fonsecin B was stacked against the end of the G-quadruplex at the 3′-terminus, with potential electrostatic interactions with a potassium ion in the central channel (Fig. 4). Importantly, this study represented the first large scale high-throughput virtual screening of a NP database against the c-mycG-quadruplex. To our knowledge, no other study has been published that utilizes high-throughput molecular docking to discover novel NP or NP-like nucleic acid-binding compounds. With a growing awareness of the important regulatory roles played by non-canonical DNA or RNA motifs in human biology, as well as the ready availability of such nucleic acid structures in the PDB, we believe that this is an area that definitely deserves further attention in the literature.
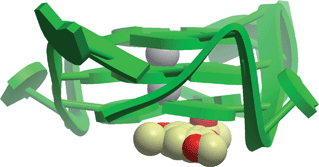 | ||
| Fig. 4 Hypothetical molecular model of virtual screening hit fonsecin B with the c-mycG-quadruplex.86 | ||
Conclusions
Computational technologies are a practical solution to the horrific experimental costs associated with high-throughput screening of large compound libraries. A variety of modeling techniques are available for today's medicinal chemists for the rapid and efficient discovery of lead molecules against biomolecular targets. Meanwhile, natural products are re-emerging as a valuable source of bioactive scaffolds that display remarkable chemical diversity in structure and function. The use of virtual screening technologies ameliorates many of the problems associated with the incompatability of natural products with high-throughput screening. The combination of virtual screening and natural products allows the medicinal chemist to harness the extraordinary potential of natural products in an efficient and inexpensive manner. Molecular docking, while regarded as more complex and computationally demanding compared to pharmacophore modeling, has the potential to accurately predict binding affinities of screening hits as well as potentially reveal lead structures with novel modes of binding. As scoring algorithms become more refined, together with the continuous improvement in computer processing power and capabilities, we believe that molecular docking has great promise in virtual lead discovery. In this review, we have highlighted some recent examples of the use of molecular docking for the identification of bioactive compounds from natural product databases. These works demonstrate that molecular modeling can be used to reveal natural products as highly potent and selective enzyme inhibitors, blockers of protein-protein interactions, as well as ligands targeting non-canonical DNA structures. Given that most of these studies were published within the last three years, we are confident that this powerful combination of molecular docking and natural compounds will continue to thrive as an active area of research in the coming years.This work is supported by the Hong Kong Baptist University (FRG2/09-10/070 and FRG2/10-11/008), Centre for Cancer and Inflammation Research, School of Chinese Medicine (CCIR-SCM, HKBU) and The Hong Kong Anti-Cancer Society (HKACS) Research Grant.
Notes and references
- J. J. Burbaum and N. H. Sigal, Curr. Opin. Chem. Biol., 1997, 1, 72–78 CrossRef CAS.
- R. P. Hertzberg and A. J. Pope, Curr. Opin. Chem. Biol., 2000, 4, 445–451 CrossRef CAS.
- K. H. Bleicher, H.-J. Böhm, K. Müllee and A. I. Alanine, Nat. Rev. Drug Discovery, 2003, 2, 369–378 CrossRef CAS.
- P. B. Fernandes, Curr. Opin. Chem. Biol., 1998, 2, 597–603 CrossRef CAS.
- J. W. Szostak, Chem. Rev., 1997, 97, 347–348 CrossRef CAS.
- H. Mario Geysen, F. Schoenen, D. Wagner and R. Wagner, Nat. Rev. Drug Discovery, 2003, 2, 222–230 CrossRef.
- E. M. Gordon, M. A. Gallop and D. V. Patel, Acc. Chem. Res., 1996, 29, 144–154 CrossRef CAS.
- S. S. Phatak, C. C. Stephan and C. N. Cavasotto, Expert Opin. Drug Discovery, 2009, 4, 947–959 Search PubMed.
- S. Shibayama, K. Tanikawa, R. Fujimoto and H. Kimura, Drug Discovery Today, 2008, 13, 86–93 Search PubMed.
- L. Schweizer and D. Z. Knyphausen-Aufsess, in Handbook of Bioentrepreneurship, ed. Z. J. Acs and D. B. Audretsch, SpringerNew York, 2008, pp. 129–144 Search PubMed.
- B. Patwardhan and R. A. Mashelkar, Drug Discovery Today, 2009, 14, 804–811 Search PubMed.
- D. Normile, Science, 2003, 299, 188–190 Search PubMed.
- W. Y. Jiang, Trends Pharmacol. Sci., 2005, 26, 558–563 CrossRef CAS.
- M. Heinrich, Phytochem. Lett., 2008, 1, 1–5 Search PubMed.
- M. Heinrich, Frontiers in Pharmacology, 2010, 1, 8 Search PubMed.
- L. Bohlin, U. Goeransson, C. Alsmark, C. Weden and A. Backlund, Phytochem. Rev., 2010, 9, 279–301 Search PubMed.
- N. J. White, Science, 2008, 320, 330–334 CrossRef CAS.
- D. L. Klayman, Science, 1985, 228, 1049–1055 CrossRef CAS.
- S. R. Meshnick, T. E. Taylor and S. Kamchonwongpaisan, Microbiol. Rev., 1996, 60, 301–305 CAS.
- D. H. Cheng, H. Ren and X. C. Tang, NeuroReport, 1996, 8, 97–101 Search PubMed.
- J.-S. Liu, Y.-L. Zhu, C.-M. Yu, Y.-Z. Zhou, Y.-Y. Han, F.-W. Wu and B.-F. Qi, Can. J. Chem., 1986, 64, 837–839 CAS.
- A. P. Kozikowski and W. Tueckmantel, Acc. Chem. Res., 1999, 32, 641–650 CrossRef CAS.
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325–2327 CrossRef.
- L. Zhang, A. L. Demain, A. L. Demain and L. Zhang, in Natural Products, Humana Press, 2005, pp. 3–29 Search PubMed.
- A. M. Burja, B. Banaigs, E. Abou-Mansour, J. G. Burgess and P. C. Wright, Tetrahedron, 2001, 57, 9347–9377 CrossRef CAS.
- J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2006, 23, 26–78 RSC.
- B. Haefner, Drug Discovery Today, 2003, 8, 536–544 CrossRef CAS.
- A. L. Harvey, Drug Discovery Today, 2008, 13, 894–901 CrossRef CAS.
- W. R. Strohl, Drug Discovery Today, 2000, 5, 39–41 CrossRef.
- M. S. Butler, J. Nat. Prod., 2004, 67, 2141–2153 CrossRef CAS.
- F. E. Koehn and G. T. C. Carter, Nat. Rev. Drug Discovery, 2005, 4, 206–220 CrossRef CAS.
- D. J. Newman, J. Med. Chem., 2008, 51, 2589–2599 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS.
- J. W.-H. Li and J. C. Vederas, Science, 2009, 325, 161–165 CrossRef.
- M. Feher and J. M. Schmidt, J. Chem. Inf. Comput. Sci., 2003, 43, 218–227 CrossRef CAS.
- M. S. Boguski, K. D. Mandl and V. P. Sukhatme, Science, 2009, 324, 1394–1395 Search PubMed.
- D. W. Carley, IDrugs, 2005, 8, 306–309 Search PubMed.
- D. W. Carley, IDrugs, 2005, 8, 310–313 Search PubMed.
- S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS.
- M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46–58 CrossRef.
- D. S. Tan, Nat. Chem. Biol., 2005, 1, 74–84 CrossRef CAS.
- K. H. Altmann, Chem. Biol., 2007, 14, 443–451 CrossRef CAS.
- A. Nören-Müller, I. Reis-Corrêa, H. Prinz, C. Rosenbaum, K. Saxena, H. J. Schwalbe, D. Vestweber, G. Cagna, S. Schunk, O. Schwarz, H. Schiewe and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10606–10611 CrossRef.
- I. Reis-Corrêa, A. Nören-Müller, H.-D. Ambrosi, S. Jakupovic, K. Saxena, H. Schwalbe, M. Kaiser and H. Waldmann, Chem. – Asian J., 2007, 2, 1109–1126 CrossRef.
- P. Ertl, J. Chem. Inf. Comput. Sci., 2003, 43, 374–380 CrossRef CAS.
- R. Abagyan and M. Totrov, Curr. Opin. Chem. Biol., 2001, 5, 375–382 Search PubMed.
- B. K. Shoichet, S. L. McGovern, B. Wei and J. J. Irwin, Curr. Opin. Chem. Biol., 2002, 6, 439–446 CrossRef CAS.
- C. McInnes, Curr. Opin. Chem. Biol., 2007, 11, 494–502 CrossRef CAS.
- D. B. Kitchen, H. Decornez, J. R. Furr and J. Bajorath, Nat. Rev. Drug Discovery, 2004, 3, 935–949 CrossRef CAS.
- S. Ghosh, A. Nie, J. An and Z. Huang, Curr. Opin. Chem. Biol., 2006, 10, 194–202 CrossRef CAS.
- C. N. Cavasotto and A. J. W. Orry, Curr. Top. Med. Chem., 2007, 7, 1006–1014 Search PubMed.
- F. C. Bernstein, T. F. Koetzle, G. J. B. Williams, J. E. F. Meyer, M. D. Brice, J. R. Rodgers, O. Kennard, T. Shimanouchi and M. Tasumi, J. Mol. Biol., 1977, 112, 535–542 CAS.
- M. Loewer and E. Proschak, Molecular Informatics, 2011, 30, 398–404 Search PubMed.
- F. Petersen, R. Amstutz, J. M. Rollinger, H. Stuppner and T. Langer, in Natural Compounds as Drugs Volume I, BirkhäuserBasel, 2008, pp. 211–249 Search PubMed.
- D. Schuster and G. Wolber, Curr. Pharm. Des., 2010, 16, 1666–1681 Search PubMed.
- C. N. Cavasotto and N. Singh, Curr. Comput.-Aided Drug Des., 2008, 4, 221–234 Search PubMed.
- F. Spyrakis, A. B. Chanal, X. Barril and F. J. Luque, Curr. Top. Med. Chem. (Sharjah, United Arab Emirates), 2011, 11, 192–210 Search PubMed.
- R. D. Clark, A. Strizhev, J. M. Leonard, J. F. Blake and J. B. Matthew, J. Mol. Graphics Modell., 2002, 20, 281–295 CrossRef CAS.
- J.-M. Yang, Y.-F. Chen, T.-W. Shen, B. S. Kristal and D. F. Hsu, J. Chem. Inf. Model., 2005, 45, 1134–1146 Search PubMed.
- P. S. Charifson, J. J. Corkery, M. A. Murcko and W. P. Walters, J. Med. Chem., 1999, 42, 5100–5109 CrossRef CAS.
- M. Feher, Drug Discovery Today, 2006, 11, 421–428 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef.
- J. H. Toney, S. Navas-Martin, S. R. Weiss and A. Koeller, J. Med. Chem., 2004, 47, 1079–1080 Search PubMed.
- C. Sangma, D. Chuakheaw, N. Jongkon, K. Saenbandit, P. Nunrium, P. Uthayopas and S. Hannongbua, Comb. Chem. High T. Scr., 2005, 8, 417–429 Search PubMed.
- G. Cozza, P. Bonvini, E. Zorzi, G. Poletto, M. A. Pagano, S. Sarno, A. Donella-Deana, G. Zagotto, A. Rosolen, L. A. Pinna, F. Meggio and S. Moro, J. Med. Chem., 2006, 49, 2363–2366 CrossRef CAS.
- D.-H. Fu, W. Jiang, J.-T. Zheng, G.-Y. Zhao, Y. Li, H. Yi, Z.-R. Li, J.-D. Jiang, K.-Q. Yang, Y. Wang and S.-Y. Si, Mol. Cancer Ther., 2008, 7, 2386–2393 CrossRef CAS.
- C.-H. Leung, D. S.-H. Chan, H. Yang, R. Abagyan, S. M.-Y. Lee and D.-L. Ma, Chem. Commun., 2011, 47, 2511–2513 RSC.
- T. A. Soucy, P. G. Smith, M. A. Milhollen, A. J. Berger, J. M. Gavin, S. Adhikari, J. E. Brownell, K. E. Burke, D. P. Cardin, S. Critchley, C. A. Cullis, A. Doucette, J. J. Garnsey, J. L. Gaulin, R. E. Gershman, A. R. Lublinsky, A. McDonald, H. Mizutani, U. Narayanan, E. J. Olhava, S. Peluso, M. Rezaei, M. D. Sintchak, T. Talreja, M. P. Thomas, T. Traore, S. Vyskocil, G. S. Weatherhead, J. Yu, J. Zhang, L. R. Dick, C. F. Claiborne, M. Rolfe, J. B. Bolen and S. P. Langston, Nature, 2009, 458, 732–736 Search PubMed.
- M. C. Monti, A. Casapullo, C. N. Cavasotto, A. Napolitano and R. Riccio, ChemBioChem, 2007, 8, 1585–1591 CrossRef CAS.
- M. C. Monti, A. Casapullo, C. N. Cavasotto, A. Tosco, P. F. Dal, A. Ziemys, L. Margarucci and R. Riccio, Chem.–Eur. J., 2009, 15, 1155–1163 Search PubMed.
- H. Liu, Y. Li, M. Song, X. Tan, F. Cheng, S. Zheng, J. Shen, X. Luo, R. Ji, J. Yue, G. Hu, H. Jiang and K. Chen, Chem. Biol., 2003, 10, 1103–1113 CrossRef CAS.
- L. Zhao and R. D. Brinton, J. Med. Chem., 2005, 48, 3463–3466 CrossRef CAS.
- J.-Å. Gustafsson, Curr. Opin. Chem. Biol., 1998, 2, 508–511 Search PubMed.
- M. Ricote, A. C. Li, T. M. Willson, C. J. Kelly and C. K. Glass, Nature, 1998, 391, 79–82 CrossRef.
- N. K. Salam, T. H. W. Huang, B. P. Kota, M. S. Kim, Y. Li and D. E. Hibbs, Chem. Biol. Drug Des., 2008, 71, 57–70 Search PubMed.
- N. Triballeau, E. Van Name, G. Laslier, D. Cai, G. Paillard, P. W. Sorensen, R. Hoffmann, H.-O. Bertrand, J. Ngai and F. C. Acher, Neuron, 2008, 60, 767–774 Search PubMed.
- J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001–1009 CrossRef CAS.
- D. S.-H. Chan, H.-M. Lee, F. Yang, C.-M. Che, C. C. L. Wong, R. Abagyan, C.-H. Leung and D.-L. Ma, Angew. Chem. Int. Ed., 49, pp. 2860–2864 Search PubMed.
- B. B. Aggarwal, Nat. Rev. Immunol., 2003, 3, 745–756 CrossRef CAS.
- S. T. Crooke and W. T. Bradner, Cancer Treat. Rev., 1976, 3, 121–139 Search PubMed.
- R. P. Perry and D. E. Kelley, J. Cell. Physiol., 1970, 76, 127–139 Search PubMed.
- H. J. Lipps and D. Rhodes, Trends Cell Biol., 2009, 19, 414–422 CrossRef.
- A. Arola and R. Vilar, Curr. Top. Med. Chem., 2008, 8, 1405–1415 CrossRef CAS.
- J.-L. Mergny and C. Helene, Nat. Med., 1998, 4, 1366–1367 CrossRef CAS.
- M.-Y. Kim, H. Vankayalapati, K. Shin-ya, K. Wierzba and L. H. Hurley, J. Am. Chem. Soc., 2002, 124, 2098–2099 CrossRef CAS.
- H.-M. Lee, D. S.-H. Chan, F. Yang, H.-Y. Lam, S.-C. Yan, C.-M. Che, D.-L. Ma and C.-H. Leung, Chem. Commun., 2010, 46, 4680–4682 RSC.
| This journal is © The Royal Society of Chemistry 2011 |