DNA polyfluorophores as highly diverse chemosensors of toxic gases†
Chi-Kin
Koo
,
Florent
Samain
,
Nan
Dai
and
Eric T.
Kool
*
Department of Chemistry, Stanford University, Stanford, California 94305-5080, United States. E-mail: kool@stanford.edu; Fax: +650 725 0259; Tel: +650 724 4741
First published on 14th July 2011
Abstract
We describe a new molecular design for sensing toxic gases in air that employs DNA-polyfluorophores as reporters. In these polyfluorophores (ODFs), the DNA backbone is used as a scaffold to arrange several fluorescent aromatic hydrocarbons/heterocycles as DNA base replacements in a photophysically interacting stack. A library of 256 different tetramer ODFs was constructed on PEG-polystyrene beads and tested for optical responses to a set of toxic gases, SO2, H2S, MeSH, NH3, NHMe2, HCl, Cl2, and BF3. A set of 15 responding sequences was resynthesized, characterized, and cross-screened under a microscope against all eight gases at 1000 ppm. Responses were measured by changes in fluorescence wavelength and intensity, which were quantified as Δ(R, G, B, L) data from bead images. The data show a large range of responses, including lighting up, quenching, and color changes; remarkably, some single sensors showed all three types of responses to the varied analytes. Statistical methods were used to identify small sets of only three chemosensors that could be used to distinguish all eight analytes. The results establish that ODFs on surfaces can bind and report on small, simple gas molecules with highly varied responses. The DNA-like structure of the sensor molecules confers a number of potential advantages including simple synthesis, high diversity, and rapid sensor discovery.
Introduction
The search for versatile and low-cost sensors for toxic gases has been a longstanding challenge for chemists and engineers. Toxic gas detection has important applications in environmental protection, industrial safety, and in security. Examples include gases emitted from automobiles and factories (CO, SO2, NOx) that are subjects of air-quality monitoring in urban areas. Similarly, the detection of H2S and low-molecular-weight thiols is important for fossil fuel refining industries and wastewater treatment plants.1 Further, industrial manufacturing and agriculture emit ammonia and low-molecular-weight amines. Although highly sensitive equipment exists for detecting these species, such instruments are typically expensive, and their size and power needs require them to be situated in laboratories rather than at the sites where the gases occur. Thus there is interest in approaches to gas sensing that offer lower cost, greater simplicity and improved portability.The individual detection of some of these toxic substances has been successfully achieved with semi-conductive metal-oxides2 or functionalized single-walled carbon nanotubes.3,4 Such transducer sensors rely on the redox properties of the gaseous analytes, which may not be specific enough to differentiate closely related compounds with similar chemical properties. Moreover, such sensors can be limited by high operating temperature5 and sensitivity to conditions.6 An alternative to redox-based sensing is optical sensing; a few recent reports have demonstrated optical detection of gases based on organic polymers,7 inorganic complexes8 and lanthanide nano-particles.9 In general, however, the use of optical sensing in detecting toxic gases is relatively less explored than other approaches.
Owing to the structural simplicity of small-molecule gases, it can be difficult to design specific artificial receptors for the detection of each analyte. Therefore, the use of multi-sensor arrays with pattern-based recognition has been proposed for the detection and differentiation of such closely related small molecules.10 Outside of the gas sensing application, the pattern-based sensing approach11 has been applied to practical applications in discriminating metal ions,12 small organic compounds,13 biomolecules14 and even species of bacteria and eukaryotic cell types.15
Pattern-based sensing has not been generally pursued with redox-based sensors, possibly because of limited dimensionality of the observed data. Optical methods, on the other hand, are well-suited for pattern-based recognition because optical signals (absorption or fluorescence) can be separated into different colors (wavelengths) which give many additional signal dimensions beyond intensity.16 One recent example involved the use of optical absorbance of chemically responsive dyes in a colorimetric cross-reactive chemical array that yielded responses to a number of toxic gases.17 Although absorbance-based signals are operationally simple from an equipment standpoint, fluorescence signaling offers potential benefits, such as greater dynamic range of response.
To take advantage of fluorescence in chemosensing, we have developed a fluorogenic DNA-like scaffold to build sensors for small organic molecules.18 Oligodeoxyfluorosides (ODFs) are short deoxyribose-phosphodiester-linked oligomers with DNA nucleobases replaced by a wide range of aromatic fluorophores. As is the case with natural DNA bases, the fluorophores are covalently bonded to the sugar-phosphate backbone and are arrayed consecutively in a geometry that promotes direct chromophore stacking. The photophysical properties are sequence-dependent, and single ODFs often exhibit properties different from the monomer components, due to complex photophysical interactions among the chromophores, including FRET, exciplex, excimer, H-dimer, and other mechanisms.19 This results in a marked diversity of electronic properties, where small changes in sequence can result in large differences in fluorescence.20
Recent experiments have tested the ability of ODFs to act as reporters in sensing metal ions in aqueous solution21 and of vapors of organic liquids.18 However, the possibility of sensing smaller and much simpler gaseous molecules remains unexplored, and raises a number of questions. For example, can small inorganic gas molecules bind to ODFs without substantial ability to stack with ODF chromophores? Moreover, larger organic species may also interact with surfaces containing ODFs by simple chemisorption, while small room-temperature gases are likely to be less well absorbed. In addition, larger organic species are more efficient at photophysical interaction (quenching, energy transfer etc.) than very small molecules are; thus can such small molecules induce any measurable changes in fluorescence? And importantly, can ODFs differentiate between such small and chemically simple species? Here we describe the first tests of these issues for the ODF design, and report the discovery of a small set of chemosensors that can differentiate among eight common toxic gases, including sulfur dioxide (SO2), hydrogen sulfide (H2S), methanethiol (MeSH), ammonia (NH3), dimethylamine (NHMe2), chlorine (Cl2), hydrogen chloride (HCl) and boron trifluoride (BF3).
Results
Oligodeoxyfluoroside design
Our primary goal for this study was to study the ability of ODFs on solid supports to report on exposure to toxic gases through the perturbation of electronic interactions between the stacked fluorophores. The four fluorescent monomers (Fig. 1) were selected for their well-established photophysical properties, planar structure, photostability, and wide range of emission across the visible spectrum. Zinc porphyrin was included for its red emission and for its potential to coordinate gases at the metal center. The monomers were C-deoxyribosides with alpha configuration and were expected to undergo stable aromatic–aromatic stacking, allowing strong electronic communication.22 The compounds were prepared as 5′-dimethoxytrityl, 3′-cyanoethylphosphoramidite derivatives, so as to be compatible with standard oligonucleotide solid-phase synthesis methods.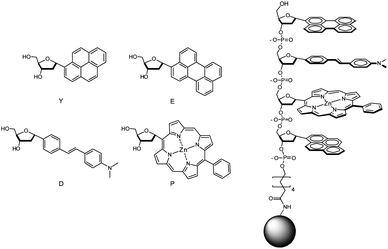 | ||
| Fig. 1 Structures of fluorescent monomers and sensors in this study. Monomer nucleosides are shown with their one-letter abbreviations. Sensors are tetramers (sequence S13 (5′-EDPY-3′) is shown as an example) covalently attached by an amide linkage to PEG-PS beads. | ||
Because it was not possible to predict which sequences and combinations of monomers would yield responses to the gases, we prepared a library of tetramer-length ODFs in all possible (256) combinations. The compounds were prepared on 130 μm PEG-polystyrene (PEG-PS) beads using split-and-mix methodology. This provided the convenient separation of each compound on a separate bead, so that responses could be easily screened and sequences identified.
Screening results
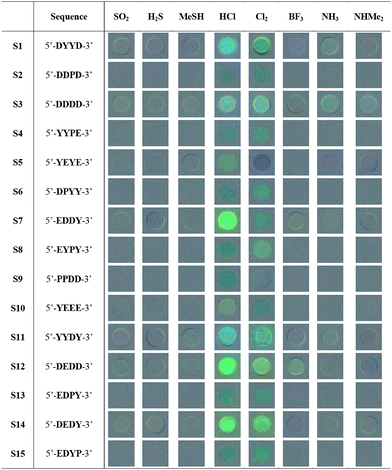
For this initial study, eight common toxic gases were tested in the screening: SO2, H2S, MeSH, NH3, NHMe2, HCl, Cl2, and BF3. The analytes were chosen to have diverse acid–base and redox properties, and chemically similar pairs of compounds were included (e.g., H2S and MeSH; NH3 and NHMe2) to test the selectivity of the sensors. We then used these analytes to screen the library for candidate sensors with the strongest responses. The beads from the library were exposed to each toxic gas (1% in air) in a closed quartz cuvette equipped with a septum. We initially chose this relatively high concentration so as to capture a wide range of responders; later studies were carried out at lower gas concentrations (see below). Air was used as diluent to account for any effects of atmospheric gases on the responses. Fluorescence was monitored under an epifluorescence microscope, and images were taken before (as blank with air alone) and after 2, 7 and 15 min of exposure at room temperature. To evaluate possible spectral changes in response to the analytes, images were processed with commercial image-processing software to construct graphical 50% gray-based difference maps of the beads (see Table 1 and the Supporting Information (ESI†)). For each analyte, at least two beads that showed the strongest responses were picked and sequence-decoded by EC-GC. Notably, a wide range of responses was commonly seen with each single analyte (see examples in ESI†), ranging from lighting-up to quenching and color shifts. Aside from the few strong responders, many ODFs in the library showed very little response, suggesting the importance of monomer composition and/or sequence in interaction with the gases, and underscoring the value of screening to eliminate inactive sequences and isolate useful candidate chemosensors.
|
Evaluation of sensing properties of ODFs
Based on the screening and decoding results, we selected 15 ODF sequences for re-synthesis by standard automated DNA synthesis methods. The ODFs were prepared on PEG-PS beads for sensing analysis (see Experimental Section) and in the same synthesis column, a portion of each compound was synthesized on controlled pore glass, cleaved from the support, purified by reversed phase HPLC, and characterized by mass spectrometry and UV/Vis absorption spectroscopy (see ESI†).In order to evaluate the sensing properties of each of re-synthesized sequence, a full cross-screening study of these 15 ODF sequences with the eight analytes (120 responses total) was performed. Results were averaged over at least 5 sensor beads for each analyte/sensor combination, to provide a measure of variability and error limits. Table 1 shows qualitative responses by the difference images of the beads containing the sensor sequences. These before/after blended images reveal that a substantial fraction of beads gives strong changes, including quenching, lighting up, and color shifts. In addition, many beads show small changes that are difficult to see by eye but which are evident in the quantitative data (see below). Inspection of the overall trends shows that the strongest responses were induced by exposure to HCl and Cl2, while the gases SO2 and MeSH yielded the smallest average fluorescence changes. The different sensors also varied considerably in their intensity and diversity of response; examples of the strongest responders included 5′-DDDD-3′ (S3), 5′-EDDY-3′ (S7), 5′-YYDY-3′ (S11), and 5′-DEDD-3′ (S12) (note that we use the 5′->3′ sequence convention of DNA).
To further evaluate the abilities of the ODF chemosensor design, we examined quantitative changes in emission, which confirm a marked diversity in responses (Fig. 2). The plots in the figure show the numerical changes upon analyte exposure, broken into red, green and blue wavelengths (ΔR, ΔG, ΔB) as well as overall luminosity (ΔL) on a standard ±256-unit RGB scale. In general, many of the sequences gave multiple emission enhancement responses towards most of the selected analytes, except Cl2 and HCl, which gave numerically stronger red-to-green or red-to-blue color changes and quenching responses. The color-change plots also revealed some interesting responses towards chemically related analytes. For instance, the responses to H2S and MeSH were generally similar, except that S1 and S14 displayed stronger enhancement responses toward H2S while S3 and S11 exhibited stronger responses to MeSH. In contrast, sensors S5 and S7 gave distinctly different responses for these two analytes. For NH3 and NHMe2, the situation was similar in that most (12 of 15) sequences gave the same response, except for S1, S7 and S11, which gave strongly divergent responses.
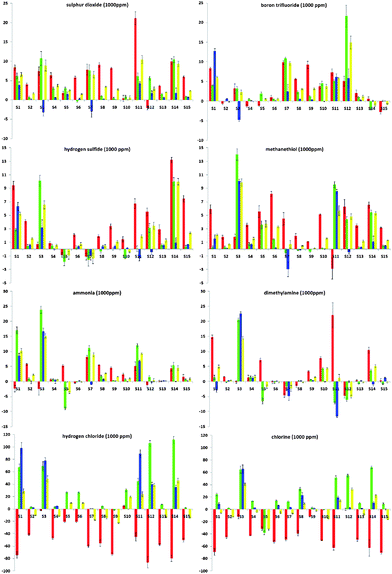 | ||
| Fig. 2 Quantitative color-change profiles in ΔR (red bar), ΔG (green), ΔB (blue) and ΔL (yellow) of all sensor sequences (S1 to S15) upon exposure to 1000 ppm of SO2, BF3, H2S, MeSH, NH3, HNMe2, HCl and Cl2 (x-axis: digital difference value, ±256 units; y-axis: S1 to S15). Values are averaged from at least five measurements. | ||
A closer look at the color-change plots with related sequences gives some evidence as to sequence dependence of the responses. For one striking example, S5 (5′-YEYE-3′) and S10 (5′-YEEE-3′) are of similar composition with one monomer difference. However, the difference in their responses toward Cl2 can even be recognized by the naked eye (Table 1). The corresponding color-change plots support this, showing a strong green quenching for S5 but not S10. Another remarkable example is given by S7 (5′-EDDY-3′) and S14 (5′-DEDY-3′), which are anagrams. This time, although the components are the same, the difference in the sequence of the fluorophores results in completely different response patterns toward BF3, H2S, MeSH and NHMe2 (Fig. 2). Indeed, the marked differences in the responses led S7 and S14 to be grouped into two distinct classes in the statistical analysis (see below) despite their identical composition.
Statistical analysis
To analyze the diversity of responses of the different chemosensors and to classify their patterns of response, we performed statistical analyses of the Δ(R, G, B, L) data. First, principal component analysis (PCA) was employed to evaluate the multidimensional analytical dispersion of the responses of the 15 sensor sequences. The analysis revealed a high dimensionality and diversity of responses, requiring 6 dimensions to capture 90% of the total variance. This suggests a high complexity of sensing mechanisms in the recognition events. A simple 2-D PCA scattering plot (Fig. 3A), whose F1 and F2 axes account for only 40.00% and 16.93% of the variance, shows a portion of the scattering but does not accurately reveal the full magnitude of scattering because of the limited ability to project the six dimensions onto the flat page.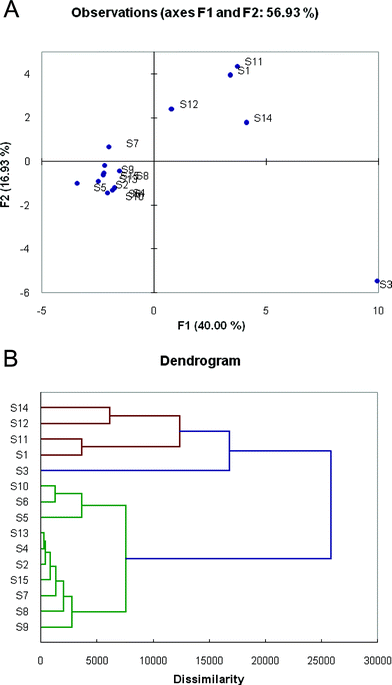 | ||
| Fig. 3 Variation in sensor responses to the eight toxic gases, as shown by Principal Component Analysis (PCA) and Agglomerative Hierarchical Clustering (AHC) analysis. (A) 2-D projection of the multidimensional principal components data for the sensors, showing scattering of the total responses. (B) Dendrogram showing similarities and relationships between response patterns of the 15 sensors. | ||
Because of the high dimensionality of the data, agglomerative hierarchical clustering (AHC) analysis proved to be more useful in analyzing the differences and similarities of the sensors. Fig. 3B shows a dendrogram of responses, illustrating classes of responses upon exposure to the different gases. There are three main groupings of sensors in the chart; sensor S3 is the most unique in its responses compared with all others, and forms its own group (marked in blue). A second group (red) is comprised of sensors S1, S11, S12, and S14, which showed significant similarities of response. The third group (green) comprises the remaining ten ODF sequences. Sensors from these three separate groups are most orthogonal in their responses, and if a pattern-based response were to be selected, a combination of sensors chosen from these separate groups would be most successful in differentiating the analytes.
We carried out PCA analysis on individual chemosensors from the three groups in order to evaluate how well they individually differentiate the analytes. Fig. S8 (ESI†) shows the quantitative color response plots for sensors S1, S3, S7, and S11, and data for S7 alone are shown Fig. 4. The scatterplots reveal that sensors S3 and S7 gave the best separation of all the analytes. Indeed, sensor S7 is able to differentiate between many of the gases alone.
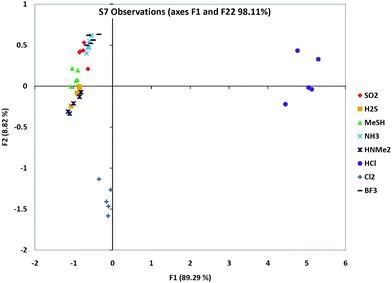 | ||
| Fig. 4 PCA scattering analysis of one sensor's (S7) responses to the eight analytes, showing which analytes were the furthest and closest in responses. Note that this is a 2-D representation of multidimensional data, and so does not show the full dispersion of the data. Plots for S1, S3 and S11 are given in ESI.† | ||
Optimal sequence set and pattern-based sensing
We then proceeded to identify a small set of sensors that could be employed in pattern-based discrimination capability among the eight analytes. In order to select the best sequence from each group in the AHC clustering, further PCA analyses were carried out on each ODF toward the eight gases (see ESI†) to assist in choice of sets with best scattering of the data. From the PCA plots of the sequences in Group A, it is clear that S1 and S11 showed the largest component values of this group (and thus well-separated responses). Similarly, S7 was selected from Group C for its diverse response towards these analytes. Including S3, the only choice from Group B, two sensor sets (S1, S3, S7 and S3, S7, S11) were selected. In addition, because of the general closeness in responses to SO2 and BF3, we also tested sets (S3, S7, S12) and (S3, S7, S14) because S12 and S14 gave the strongest differences for these two gases.Initial studies of these candidate 3-sensor sets showed that the strong responses to Cl2 and HCl obscured the scattering of the data for the other six gases (Fig. S10†). Since it was simple to use even a single sensor's response to discriminate between these two gases (see Fig. 3 above), we eliminated them from the subsequent PCA analysis. The results for four potential three-bead sets are shown in ESI,† and the scattering data for one selected set (S3, S7, S11) are plotted in Fig. 5. Results show that more than one three-bead set could successfully distinguish the six gases by their pattern response. Given that single beads in these sets could distinguish the remaining two gases (as shown above), the result is that all eight gases could be discriminated by a set of three ODF chemosensors.
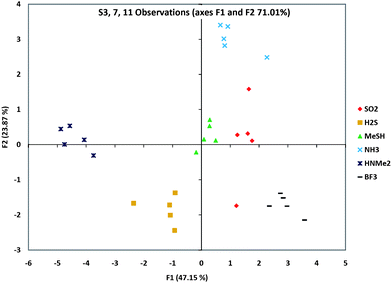 | ||
| Fig. 5 Gas discrimination as a pattern-based response. Shown is a PCA scattering analysis of a group of three sensors' (S3, S7 and S11) responses to six analytes, showing which analytes were the furthest and closest in responses. Data for five beads of each type are shown. Note that these are 2-D representations of multidimensional data. Scattering data for three other sets of three sensors are shown in the ESI.† | ||
Preliminary tests of sensitivity
Aside from selectivity of response, sensitivity in detecting low concentrations of analytes is one of the most important factors for virtually all sensor systems. For this reason we performed preliminary tests of sensitivity by lowering analyte concentration from ca. 1000 to 500, 50 and 20 ppm and observing ODF responses. Images taken before and after 15 min of exposure were then processed to determine color-change profiles in ΔR, ΔG, ΔB and ΔL. We observed that the overall response patterns were similar to those from the cross-screening experiments, but with decreasing response intensity as concentration was lowered (ESI, Fig. S12†). The experiments revealed that several of the analytes (SO2, NHMe2, NH3 and BF3) yielded clear selective responses at concentrations at least as low as 20 ppm. Others (H2S, MeSH, HCl and Cl2) gave reliable identification only at higher concentrations (100 ppm).Discussion
The current results establish that short DNA-like ODF sensor molecules can respond to small inorganic toxic gases with measurable and distinct changes in fluorescence. The diversity of response is surprisingly high, with single sensors showing markedly different responses to the different analytes. As a result of this diversity, the data show that a small set of only three chemosensors can be used to distinguish between eight gas analytes in a pattern-based response. Even closely related chemical species (e.g., MeSH and H2S; NH3 and NHMe2) could be differentiated.An appealing aspect of the current molecular approach is the simplicity of chemosensor structure and preparation. Only four monomer components were used, and one obtains different sensors merely by varying their order in the automated synthesis. Synthesis is rapid and straightforward on a commercial DNA synthesizer. Despite this simplicity, however, the chemical divergence of responses is large, which allows a small set of only three compounds to distinguish between a range of chemically varied species.
The high dimensionality of responses in this set of chemosensors can be attributed to the great variety of electronic interactions between different fluorophores. The experiments show that even closely related ODF sequences can give distinct responses to gases, and we even observed an example in which two sensors having identical composition but different order yielded different responses. The results suggest that complex electronic interactions occur between the fluorophores, and that binding of a gas moiety near one of the components can alter not just the monomer, but also its interactions, in a way that yields either a change in fluorescence intensity or in wavelength in the ODF as a whole.
It is of interest to consider the possible mechanisms by which the ODFs bind analytes and yield optical changes in response. Although the data in this initial study are limited, some early clues emerge. For example, acid–base properties could plausibly play a role; we note in this regard that most sequences gave similar responses towards the gases NH3 and NHMe2. Phosphate oxygens and an amine group on monomer D could reasonably act as H-bond acceptors for these species and possibly others. However, it is worth noting that S11 gave different responses to NH3 and NHMe2 (Table 1), stressing the likelihood that multiple mechanisms are active. Redox properties of analytes may play a role; for example, Cl2 is known to be disproportionated to HCl and HOCl with water, which could be adsorbed on the bead surface. HCl and Cl2 did yield similar responses for several of the sensors. However, these two gases induced very different responses with S5, S7 and S10, again supportive of multiple mechanisms. For the smaller and planar gases (e.g., H2S, Cl2, BF3) it also seems plausible that they could be trapped between the planar hydrocarbons, altering electronic communications. Yet another possible recognition pathway involves coordination of the Lewis-basic gases to the Zn(II) porphyrin moiety, which might disturb the aromatic-aromatic stacking and the electronic character of the porphyrin.23 We note, however, that only 6/15 sensor sequences contained this monomer. Finally, it should be pointed out that gases may also be trapped, concentrated or absorbed into the polymer substrate of the beads, concentrating their effects on the chemosensors attached there; further experiments with different types of supports will help clarify this in the future.
To our knowledge, this is the first report of the use of fluorescence pattern-based sensing of small gases. Most of the existing sensor arrays for inorganic toxic gases are based on potentiometric or colorimetric detection. Metal oxide semiconductors are well-known for their low detection limit but also for their high operating temperature.5b,24 For example, typical detection limits for NO2 and NH3 for commercial solid-state sensors range from 1–1000 ppm at a few hundred degrees Celsius.3,25 For these sensors, the gas selectivity relies solely on the redox properties of the gas analytes. In comparison, our ODF sensors offer much better selectivity at ppm levels even with structurally similar analytes.
For optical detection of vapors of organic liquids, there exists one previous report employing natural DNAs noncovalently doped with fluorescent dyes (one dye per sequence).26 That work measured only one dimension of response (fluorescence intensity), making multi-analyte differentiation difficult. In contrast, since the present study involves a different complex dye (sensing element) on each sensing molecule, multi-analyte differentiation can be easily achieved by multiple dimensions of fluorescence signal changes, including wavelength changes to the red or blue as well as intensity at each wavelength. Colorimetric (absorbance-based) sensor arrays by Suslick also have been used in the detection of toxic gases with ppm level sensitivity17 (and, impressively, sub-ppm levels with improved array methodology27). Without any optimization or signal amplification, the present ODF sensor system can currently reach sensitivity that is lower than the immediately dangerous to life or health (IDLH) concentrations of 7/8 of the gases tested (Table S2†). Further development is underway to evaluate whether ODFs can function at concentrations lower than those tested in these initial experiments. It will also be of interest to test the sensing responses in mixtures of toxic gases.
The current ODF sensing approach offers a number of promising aspects. ODFs give a much higher diversity than arrays that are composed of mixtures of a few pigments, in the present case offering up to 256 different structures from only four components. Yet higher diversity can be achieved, if desired, by using a larger set of monomers or longer oligomers.28 Second is the diversity of responses in single sensor compounds, which enables the discrimination of larger numbers of analytes with smaller numbers of sensors. A third advantage is the combinatorial nature of sensor sequence discovery, which allows rapid screening to identify the best-performing molecules for further investigation. Finally, construction of ODF sensors is simple, since the sensor sequences are prepared by automated synthesis directly on the solid support, thus avoiding complicated coating or fabrication procedures required in other gas-sensing approaches.
Experimental section
Monomer and library syntheses
The syntheses of the Y, E and D monomer deoxyribosides (as α anomers) were perfomed as described previously.29 The porphyrin nucleoside was prepared as reported;30 here the α anomer of the free nucleoside was used (see ESI†). The four monomers were combined to yield 256 tetramer-length ODF sequences in all combinations. The ODF library was constructed on 130 μm amine-functionalized polyethylene glycol-polystyrene (PEG-PS) beads using standard sequence-tagging procedures31 and split-and-mix methods28 previously reported. Zn(II) complexation of the porphyrin-containing sequences was performed after the bead-supported library was built, by incubating with excess zinc(II) triflate in ethanol at 25 °C for 12 h. Deprotection of the phosphate protecting groups on all beads was performed in concentrated ammonia solution (12 h at 55 °C) to yield the library used for screening. See Supporting Information (ESI†) for details.Analyte preparation
5 mL of each gas in a gas-tight syringe was injected into a 50 mL round-bottom flask which had been previously flushed with dry argon gas, yielding a tenfold dilution. A volume of 35 μL of diluted gas was then injected into a screw-cap quartz cuvette, which has a volume of 3.5 mL of atmospheric air with a silicone rubber septum, yielding a further 100-fold dilution and yielding a final concentration of approximately 1000 ppm of gas inside the quartz imaging cuvette. Details are given in the ESI.†Screening, image processing and decoding
For initial screening, the bead-based sensor library was exposed to approximately 1% of each toxic gas in air in a closed quartz fluorescence cell as described above. Fluorescence was monitored under an epifluorescence microscope with excitation at 340–380 nm, and all visible emission was observed (long-pass filter, >420 nm). Digital images were taken before and after exposure to the gas for 2, 7, and 15 min in the chamber at room temperature. Changes in response to gases were monitored by constructing 50% gray-based difference maps of the beads by merging the inverse of the image before exposure with the image taken after 15 min exposure using 50% transparency. Beads that showed the strongest responses were picked up and sequence- decoded by electron-capture gas chromatography.24 Details and examples are shown in the ESI.†Oligodeoxyfluoroside synthesis
The 15 sensor ODFs selected from the initial screening were individually resynthesized on an Applied Biosystems 394 DNA/RNA synthesizer on a 1 μmole scale and possessing a 3′-phosphate group for characterization off of the beads. Standard β-cyanoethyl phosphoramidite chemistry was employed for coupling, but with an extended coupling time (999 s) for the synthesized nucleotides. Overall coupling efficiencies exceeded 70%. Zn(II) complexation for porphyrin-containing sequences was performed after solid-phase ODF synthesis as described above. Standard automated DNA synthesis techniques were used except that 50/50 of cleavable controlled pore glass (CPG) and non-cleavable PEG-PS beads were included in the column, so that sensor beads and corresponding ODFs on standard CPG were made at the same time. This allowed the characterization of the ODFs off of the beads while making the corresponding sensors on beads in the same synthesis. Oligomers for off-bead characterization were deprotected and cleaved from the solid support with concentrated ammonia solution (55 °C, 12 h). They were purified by reverse-phase HPLC, and characterized by HRMS or MALDI-MS (see ESI†).Cross-screening and chemometric analysis
Cross-screening experiments were performed under an epifluorescence microscope with a 4X objective and fluorescence images were taken before and after exposure to each analyte. The emission was then quantified as RGBL (red, green, blue, luma) values with Adobe Photoshop. Average RGBL values were determined from a 16 × 16-pixel box in the center of each bead. Color/intensity changes after exposure, expressed in ΔR, ΔG, ΔB and ΔL, were calculated and averaged (five beads per analyte for a given sensor). A set of quantitative color-change profiles was then constructed by plotting the ΔR, ΔG, ΔB and ΔL values for each analyte. Standard deviations were determined to evaluate the accuracy and reproducibility of the responses. To analyze the response data quantitatively, principal component analysis (PCA) and agglomerative hierarchical clustering (AHC) analyses were performed with XLSTAT software (Addinsoft) using the ΔR, ΔG, ΔB and ΔL values as input.Acknowledgements
We thank the National Institutes of Health (GM067201) for support. C.K.K. acknowledges the Croucher Foundation for a postdoctoral fellowship. F.S. was supported by the Swiss National Science Foundation.Notes and references
- (a) J. M. Sedlak and K. F. Blurton, Talanta, 1976, 23, 445–448 CrossRef CAS; (b) R. Redondo, V. Cunha Machado, M. M. Baeza, J. Lafuente and D. Gabriel, Anal. Bioanal. Chem., 2008, 391, 789–798 CrossRef CAS.
- (a) G. Grubert, M. Stockenhuber, O. P. Tkachenko and M. Wark, Chem. Mater., 2002, 14, 2458–2466 CrossRef CAS; (b) J. Huang, N. Matsunaga, K. Shimanoe, N. Yamazoe and T. Kunitake, Chem. Mater., 2005, 17, 3513–3518 CrossRef CAS; (c) A. Martucci, Chem. Mater., 2010, 22, 3407–3417 CrossRef; (d) F. Gyger, M. Hübner, C. Feldmann, N. Barsan and U. Weimar, Chem. Mater., 2010, 22, 4821–4827 CrossRef CAS.
- (a) D. R. Kauffman and A. Star, Angew. Chem., Int. Ed., 2008, 47, 6550–6570 CrossRef CAS.
- (a) E. Bekyarova, I. Kalinina, M. E. Itkis, L. Beer, N. Cabrera and R. C. Haddon, J. Am. Chem. Soc., 2007, 129, 10700–10706 CrossRef CAS; (b) C. Y. Lee and M. S. Strano, J. Am. Chem. Soc., 2008, 130, 1766–1773 CrossRef CAS; (c) S. Mubeen, T. Zhang, N. Chartuprayoon, Y. Rheem, A. Mulchandani, N. V. Myung and M. A. Deshusses, Anal. Chem., 2010, 82, 250–257 CrossRef CAS; (d) E. Bekyarova, I. Kalinina, X. Sun, T. Shastry, K. Worsley, X. Chi, M. E. Itkis and R. C. Haddon, Adv. Mater., 2010, 22, 848–852 CrossRef CAS.
- (a) G. Eranna, B. C. Joshi, D. P. Runthala and R. P. Gupta, Crit. Rev. Solid State Mat. Sci., 2004, 29, 111–188 CrossRef CAS; (b) K. J. Choi and H. W. Jang, Sensors, 2010, 10, 4083–4099 CrossRef CAS.
- M. Righettoni, A. Tricoli and S. E. Pratsinis, Anal. Chem., 2010, 82, 3581–3587 CrossRef CAS.
- (a) F. Naso, F. Babudri, D. Colangiuli, G. M. Farinola, F. Quaranta, R. Rella, R. Tafuro and L. Valli, J. Am. Chem. Soc., 2003, 125, 9055–9061 CrossRef CAS; (b) J. Tian, T. Arbatan, X. Li and W. Shen, Chem. Commun., 2010, 46, 4734–4736 RSC.
- (a) M. Albrecht and G. Koten, Adv. Mater., 1999, 11, 171–174 CrossRef CAS; (b) D. Benito-Garagorri, M. Puchberger, K. Mereiter and K. Kirchner, Angew. Chem., Int. Ed., 2008, 47, 9142–9145 CrossRef CAS; (c) J. Esteban, J. V. Vicente Ros-Lis, R. Martinez-Manez, M. Marcos, M. Moragues, J. Soto and F. Sancenon, Angew. Chem., Int. Ed., 2010, 49, 4934–4937 CrossRef CAS.
- H. S. Mader and O. S. Wolfbeis, Anal. Chem., 2010, 82, 5002–5004 CrossRef CAS.
- K. Persaud and G. Dodd, Nature, 1982, 299, 352–355 CrossRef CAS.
- (a) A. P. Umali, K. J. Albert, N. S. Lewis, C. L. Schauer, G. A. Sotzing, S. E. Stitzel, T. P. Vaid and D. R. Walt, Chem. Rev., 2000, 100, 2595–2696 CrossRef; (b) P. C. Jurs, G. A. Bakken and H. E. McClelland, Chem. Rev., 2000, 100, 2649–2678 CrossRef CAS; (c) A. T. Wright and E. V. Anslyn, Chem. Soc. Rev., 2006, 35, 14–28 RSC; (d) B. E. Collins, A. T. Wright and E. V. Anslyn, Top. Curr. Chem., 2007, 277, 181–218 CrossRef CAS; (e) E. V. Anslyn, Curr. Opin. Chem. Biol., 2010, 14, 685–692 CrossRef.
- (a) M. A. Palacios, Z. Wang, V. A. Montes, G. V. Zyryanov, B. J. Hausch, K. Jursíková and P. Jr. Anzenbacher, Chem. Commun., 2007, 3708–3710 RSC; (b) M. A. Palacios, Z. Wang, V. A. Montes, G. V. Zyryanov and P. Jr. Anzenbacher, J. Am. Chem. Soc., 2008, 130, 10307–10314 CrossRef CAS; (c) Z. Wang, M. A. Palacios and P. Jr. Anzenbacher, Anal. Chem., 2008, 80, 7451–7459 CrossRef CAS; (d) Y. Liu, M. A. Palacios and P. Jr. Anzenbacher, Chem. Commun., 2010, 46, 1860–1862 RSC; (e) R. L. Phillips, O. R. Miranda, C.-C. You, V. M. Rotello and U. H. F. Bunz, Angew. Chem., Int. Ed., 2008, 47, 2590–2594 CrossRef CAS.
- (a) N. T. Greene, S. L. Morgan and K. D. Shimizu, Chem. Commun., 2004, 1172–1173 RSC; (b) N. A. Rakow, A. Sen, M. C. Janzen, J. B. Ponder and K. S. Suslick, Angew. Chem., Int. Ed., 2005, 44, 4528–4532 CrossRef CAS; (c) C. Zhang and K. S. Suslick, J. Am. Chem. Soc., 2005, 127, 11548–11549 CrossRef CAS; (d) H. S. Hewage and E. V. Anslyn, J. Am. Chem. Soc., 2009, 131, 13099–13106 CrossRef CAS.
- (a) S. C. McCleskey, M. J. Griffin, S. E. Schneider, J. T. McDevitt and E. V. Anslyn, J. Am. Chem. Soc., 2003, 125, 1114–1115 CrossRef CAS; (b) A. T. Wright, M. J. Griffin, Z. Zhong, S. C. McCleskey, E. V. Anslyn and J. T. McDevitt, Angew. Chem., Int. Ed., 2005, 44, 6375–6378 CrossRef CAS; (c) A. T. Wright, E. V. Anslyn and J. T. McDevitt, J. Am. Chem. Soc., 2005, 127, 17405–17411 CrossRef CAS; (d) L. Baldini, A. J. Wilson, J. Hong and A. D. Hamilton, J. Am. Chem. Soc., 2004, 126, 5656–5657 CrossRef CAS; (e) L. A. Joyce, G. M. da Cruz, V. M. Lynch, S. Sorey and E. V. Anslyn, J. Am. Chem. Soc., 2009, 131, 13125–13131 CrossRef; (f) S. Rochat, J. Gao, X. Qian, F. Zaubitzer and K. Severin, Chem.–Eur. J., 2010, 16, 104–113 CrossRef CAS; (g) T. Takeuchi, J. Montenegro, A. Hennig and S. Matile, Chem. Sci., 2011, 2, 303–307 RSC.
- (a) R. L. Phillips, O. R. Miranda, C.-C. You, V. M. Rotello and U. H. F. Bunz, Angew. Chem., Int. Ed., 2008, 47, 2590–2594 CrossRef CAS; (b) A. Bajaj, O. R. Miranda, I.-B. Kim, R. L. Phillips, D. J. Jerry, H. F. Uwe, U. H. F. Bunz and V. M. Rotello, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10912–10916 CrossRef CAS; (c) A. Bajaj, O. R. Miranda, I.-B. Kim, R. L. Phillips, D. J. Jerry, U. H. F. Bunz and V. M. Rotello, J. Am. Chem. Soc., 2010, 132, 1018–1022 CrossRef CAS.
- (a) P. Jr Anzenbacher, Y. Liu and M. E. Kozelkova, Curr. Opin. Chem. Biol., 2010, 14, 693–704 CrossRef; (b) P. Jr. Anzenbacher, P. Lubal, P. Buček, M. A. Palaciosa and M. E. Kozelkova, Chem. Soc. Rev., 2010, 39, 3954–3979 RSC.
- S. H. Lim, L. Feng, J. W. Kemling, C. J. Musto and K. S. Suslick, Nat. Chem., 2009, 1, 562–567 CrossRef CAS.
- F. Samain, S. Ghosh, Y. N. Teo and E. T. Kool, Angew. Chem., Int. Ed., 2010, 49, 7025–7029 CrossRef CAS.
- (a) A. Cuppoletti, Y. Cho, J. S. Park, S. Strassler and E. T. Kool, Bioconjugate Chem., 2005, 16, 528–534 CrossRef CAS; (b) J. N. Wilson, J. Gao and E. T. Kool, Tetrahedron, 2007, 63, 3427–3433 CrossRef CAS; (c) J. N. Wilson, Y. Cho, S. Tan, A. Cuppoletti and E. T. Kool, ChemBioChem, 2008, 9, 279–285 CrossRef CAS.
- F. Samain, N. Dai and E. T. Kool, Chem.–Eur. J., 2011, 17, 174–183 CrossRef CAS.
- S. S. Tan, Y. N. Teo and E. T. Kool, Org. Lett., 2010, 12, 4820–4823 CrossRef CAS.
- (a) A. Cuppoletti, Y. Cho, J. S. Park, C. Strassler and E. T. Kool, Bioconjugate Chem., 2005, 16, 528–534 CrossRef CAS; (b) Y. N. Teo and E. T. Kool, Bioconjugate Chem., 2009, 20, 2371–2380 CrossRef CAS.
- K. M. Kadish, K. M. Smith, R. Guilard, The Porphyrin Handbook; Academic Press: Boston, MA, 2000; Vols. 1–10 Search PubMed.
- D. James, S. M. Scott, Z. Ali and W. T. O'Hare, Microchim. Acta, 2004, 149, 1–17 CrossRef.
- B. Timmer, W. Olthius and A. van den Berg, Sens. Actuators, B, 2005, 107, 666–677 CrossRef.
- J. White, K. Truesdell, L. B. Williams, M. S. AtKisson and J. S. Kauer, PLoS Biol., 2008, 6, e9 Search PubMed.
- L. Feng, C. J. Musto, J. W. Kemling, S. H. Lim and K. S. Suslick, Chem. Commun., 2010, 46, 2037–2039 RSC.
- J. Gao, S. Watanabe and E. T. Kool, J. Am. Chem. Soc., 2004, 126, 12748–12749 CrossRef CAS.
- (a) R. X.-F. Ren, N. C. Chaudhuri, P. L. Paris, S. IV Rumney and E. T. Kool, J. Am. Chem. Soc., 1996, 118, 7671–1678 CrossRef CAS; (b) J. Gao, C. Strassler, D. Tahmassebi and E. T. Kool, J. Am. Chem. Soc., 2002, 124, 11590–11591 CrossRef CAS.
- H. Morales-Rojas and E. T. Kool, Org. Lett., 2002, 4, 4377–4380 CrossRef CAS.
- M. H. J. Ohlmeyer, M. Swanson, L. W. Dillard, J. C. Reader, G. Asouline, R. Kobayashi, M. Wigler and W. C Still, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 10922–10926 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data; details of experimental methods; color-change plots and scattering data. See DOI: 10.1039/c1sc00301a |
| This journal is © The Royal Society of Chemistry 2011 |