Sulfur-containing amide-based [2]rotaxanes and molecular shuttles†
Andrea
Altieri
a,
Vincent
Aucagne
a,
Romen
Carrillo
a,
Guy J.
Clarkson
b,
Daniel M.
D'Souza
a,
Jennifer A.
Dunnett
b,
David A.
Leigh
*a and
Kathleen M.
Mullen
a
aSchool of Chemistry, University of Edinburgh, The King's Buildings, West Mains Road, Edinburgh, EH9 3JJ, United Kingdom. E-mail: David.Leigh@ed.ac.uk; Fax: +44 (0) 131-650-6453; Tel: +44 (0) 131-650-4721
bDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, United Kingdom
First published on 14th July 2011
Abstract
We report on the synthesis, structure and properties of [2]rotaxanes prepared by the assembly of benzylic amide macrocycles around a series of amide and sulfide-/sulfoxide-/sulfone-containing threads. The efficacy of rotaxane formation is related to the hydrogen bond accepting properties of the various sulfur-containing functional groups in the thread, with the highest yields (up to 63% with a rigid vinyl spacer in the template site) obtained for sulfoxide rotaxanes. X-Ray crystallography of a sulfoxide rotaxane, 5, shows that the macrocycle adopts a highly symmetrical chair-like conformation in the solid state, with short hydrogen bonds between the macrocycle isophthalamide NH-protons and the amide carbonyl and sulfoxide S–O of the thread. In contrast, in the X-ray crystal structures of the analogous sulfide (4) and sulfone (6) rotaxanes, the macrocycle adopts boat-like conformations with long intercomponent NH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO and NH⋯S hydrogen bonds (in addition to several intercomponent amide–amide hydrogen bonds). Taking advantage of the different hydrogen bonding modes of the sulfur-based functional groups, a switchable molecular shuttle was prepared in which the oxidation level of sulfur determines the position of the macrocycle on the thread.
SO and NH⋯S hydrogen bonds (in addition to several intercomponent amide–amide hydrogen bonds). Taking advantage of the different hydrogen bonding modes of the sulfur-based functional groups, a switchable molecular shuttle was prepared in which the oxidation level of sulfur determines the position of the macrocycle on the thread.
Introduction
Since the hydrogen bond directed synthesis of the first amide [2]catenanes1 and rotaxanes,2,3 significant improvements have been made in the efficiency of their preparation and their structural diversity.4–14 Although the majority of hydrogen bond accepting groups used in the construction of benzylic amide macrocycle-based catenane and rotaxanes have been either amide4–6 or ester4,7 functionalities, recently attention has turned to the use of alternative hydrogen bond accepting moieties in the thread, such as squaraines,8 phenolates,9ureas,10pyridones,11 nitrones,12 azodicarboxamides13 and ion-pair14 motifs. The highly polarised S–O bond of sulfoxides is an attractive moiety for inclusion in interlocked structures, not only because of its superior hydrogen bonding acceptor properties (compared to amides15) but also due to the relative ease by which the sulfoxide moiety can be both oxidised and reduced, enabling the hydrogen bond properties of the functional group to be significantly varied. Here we exploit these features in the synthesis of a range of simple sulfide-, sulfoxide- and sulfone-containing [2]rotaxanes, and in the construction of a bistable molecular shuttle in which the oxidation level of the sulfur atom alters the position of the macrocycle on the thread.Results and discussion
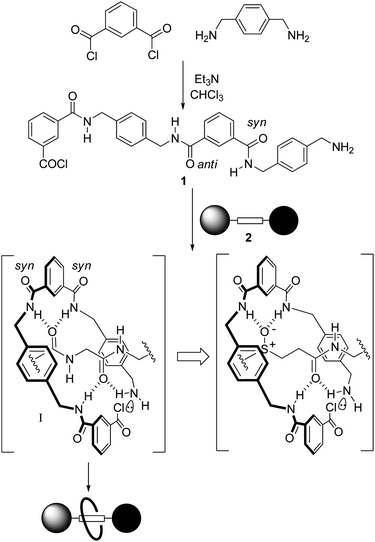
Simple dipeptide,5fumaramide4 ((E)-NHCOCHCHCONH–) and succinamide4 (–NHCOCH2CH2CONH–) motifs are effective templates for rotaxane formation because their hydrogen bond accepting carbonyl groups are well positioned to direct the assembly of a benzylic amide macrocycle around the template site via five-component ‘clipping’ reactions (Scheme 1). These reactions produce interlocked architectures because multipoint hydrogen bonding between the open-chain precursor 1 (which in the absence of a suitable template preferentially adopts a linear syn-anti conformation) and the thread 2 promotes a conformational change that brings the reactive end groups close together (Scheme 1, I), leading to rapid cyclisation of 1 about the axle.3–5 Given the geometric requirements of the multipoint hydrogen bonded intermediate I, we were intrigued as to whether it would be possible to exchange one of the sp2-hybridised amide groups in the template for an sp3-hybridised sulfur atom in any or all of its oxidation levels (sulfide, sulfoxide, sulfone), each of which exhibit different hydrogen bond accepting characteristics.16
An appropriate sulfide thread 3 was synthesised in four steps from commercially available 3-mercapto-propionic acid (see Supporting Information† and Scheme 2). Oxidation of 3 with m-chloroperoxybenzoic acid (MCPBA) could be used to afford either the sulfoxide (4) or sulfone (5) threads in good yields (Scheme 2, step ii). The three threads (3–5) were each subjected to rotaxane-forming conditions5 using equimolar amounts of isophthaloyl dichloride and p-xylylenediamine in CHCl3 in the presence of Et3N, pleasingly yielding the desired [2]rotaxane, 6–8, in each case. The sulfoxide rotaxane 7 was isolated in 43% yield (cf. 62% and 50% for glycylglycine5 and succinamide threads4 using similar rotaxane-forming protocols) whereas the yields of the sulfide 6 (12%) and sulfone 8 (10%) rotaxanes were considerably lower, presumably due to the modest hydrogen bond acceptor ability of those functional groups.
![Synthesis of sulfur-containing hydrogen bonded [2]rotaxanes 6–8. Reagents and conditions: (i) diphenylethanol (1.0 equiv.), bis(chlorodibutyltin)oxide (cat.), toluene, reflux, 8 h, 88% (3); (ii) MCPBA, CHCl3, −20 °C, 90 min, 70% (4, 1.0 equiv. MCPBA), 98% (5, from 4: 1.0 additional equiv. MCPBA); (iii) isophthaloyl dichloride (five-fold excess), p-xylylenediamine (five-fold excess), Et3N (ten-fold excess), CHCl3, RT, 4 h, 12% (6), 43% (7), 10% (8); (iv) MCPBA (1.0 equiv.), CHCl3, −20 °C to RT, 90 min., 95%; (v) MCPBA (1.0 equiv.), CHCl3, −20 °C to RT, 90 min., 97%; (vi) Lawesson's reagent (1.0 equiv.), THF, −20 °C to RT, 1 h, 87%.](/image/article/2011/SC/c1sc00335f/c1sc00335f-s2.gif) | ||
| Scheme 2 Synthesis of sulfur-containing hydrogen bonded [2]rotaxanes 6–8. Reagents and conditions: (i) diphenylethanol (1.0 equiv.), bis(chlorodibutyltin)oxide (cat.), toluene, reflux, 8 h, 88% (3); (ii) MCPBA, CHCl3, −20 °C, 90 min, 70% (4, 1.0 equiv. MCPBA), 98% (5, from 4: 1.0 additional equiv. MCPBA); (iii) isophthaloyl dichloride (five-fold excess), p-xylylenediamine (five-fold excess), Et3N (ten-fold excess), CHCl3, RT, 4 h, 12% (6), 43% (7), 10% (8); (iv) MCPBA (1.0 equiv.), CHCl3, −20 °C to RT, 90 min., 95%; (v) MCPBA (1.0 equiv.), CHCl3, −20 °C to RT, 90 min., 97%; (vi) Lawesson's reagent (1.0 equiv.), THF, −20 °C to RT, 1 h, 87%. | ||
Small crystals of each of the three sulfur-containing rotaxanes 6–8 were obtained by slow diffusion of diethyl ether into saturated solutions of the rotaxanes in methanol (6), dichloromethane (7) or chloroform (8) and their solid state structures determined by X-ray crystallography (Fig. 1–3).17 The crystal structure of the sulfoxide–amide [2]rotaxane 7 (Fig. 1) has features reminiscent of the solid state structures of various bis-amide thread (dipeptide,5fumaramide,4succinamide4) rotaxanes. The benzylic amide macrocycle adopts a low energy chair-like conformation with each isophthalamide group hydrogen bonding to the thread sulfoxide or carbonyl group through four short (2.12–2.56 Å NH⋯O) hydrogen bonds. The isophthalamide–sulfoxide hydrogen binding is sufficiently strong that it overcomes the problem of shape of the sp3-hybridised sulfoxide group, twisting the conformation of the thread in order to ensure the necessary relative positioning of the hydrogen bonding groups.
![X-Ray crystal structure of sulfoxide [2]rotaxane 7. Hydrogen bond lengths (Å): O42–H2N = 2.38; O42–H11N = 2.56; O45–H29N = 2.12; O45–H20N = 2.31. Hydrogen bond angles (°): N2–H2N–O42 = 127.3; N11–H11N–O42 = 142.2; N20–H20N–O45 = 148.7; N29–H29N–O45 = 171.7.](/image/article/2011/SC/c1sc00335f/c1sc00335f-f1.gif) | ||
| Fig. 1 X-Ray crystal structure of sulfoxide [2]rotaxane 7. Hydrogen bond lengths (Å): O42–H2N = 2.38; O42–H11N = 2.56; O45–H29N = 2.12; O45–H20N = 2.31. Hydrogen bond angles (°): N2–H2N–O42 = 127.3; N11–H11N–O42 = 142.2; N20–H20N–O45 = 148.7; N29–H29N–O45 = 171.7. | ||
![X-Ray crystal structure of sulfide [2]rotaxane 6.17Hydrogen bond lengths (Å): S1–H4N = 2.77; O3–H5N = 2.18; O5–H1N = 2.21; O5–H2N = 2.30. Hydrogen bond angles (°): N1–H1N–O5 = 160.6; N2–H2N–O5 = 172.1; N4–H4N–S1 = 151.6; N5–H5N–O3 = 151.3.](/image/article/2011/SC/c1sc00335f/c1sc00335f-f2.gif) | ||
| Fig. 2 X-Ray crystal structure of sulfide [2]rotaxane 6.17Hydrogen bond lengths (Å): S1–H4N = 2.77; O3–H5N = 2.18; O5–H1N = 2.21; O5–H2N = 2.30. Hydrogen bond angles (°): N1–H1N–O5 = 160.6; N2–H2N–O5 = 172.1; N4–H4N–S1 = 151.6; N5–H5N–O3 = 151.3. | ||
![X-Ray crystal structure of sulfone [2]rotaxane 8.17Hydrogen bond lengths (Å): O38–H2N = 2.15; O10–H42N = 1.97; O41–H20N = 2.07, O41–H29N = 2.22. Hydrogen bond angles (°) N2–H2N–O38 = 143.3; N42–H42N–O10 = 153.5, N20–H20N–O41 = 159.6; N29–H29N–O41 = 175.1.](/image/article/2011/SC/c1sc00335f/c1sc00335f-f3.gif) | ||
| Fig. 3 X-Ray crystal structure of sulfone [2]rotaxane 8.17Hydrogen bond lengths (Å): O38–H2N = 2.15; O10–H42N = 1.97; O41–H20N = 2.07, O41–H29N = 2.22. Hydrogen bond angles (°) N2–H2N–O38 = 143.3; N42–H42N–O10 = 153.5, N20–H20N–O41 = 159.6; N29–H29N–O41 = 175.1. | ||
In contrast, in the solid state structures of the sulfide–amide (6) and sulfone–amide (8) rotaxanes (Fig. 2 and 3, respectively), the macrocycle adopts relatively high energy conformations that maximise the number of intercomponent amide–amide hydrogen bonds (presumably because amide–sulfide and amide–sulfone hydrogen bonds are intrinsically weak).17 The strength of the amide–amide hydrogen bonding distorts the macrocycle into boat-like conformations with only a single (2.77 Å NH⋯S (6); 2.15 Å NH⋯OS(O) (8)) intercomponent amide–sulfide/sulfone hydrogen bond in each case (together with three intercomponent amide–amide hydrogen bonds).
The difference in hydrogen bonding behavior of these motifs suggested that it might be possible to modulate the strength of the intercomponent binding in such rotaxanes by chemically switching the oxidation level of the sulfur-based functional group. Using MCPBA, the sulfide rotaxane 6 could be oxidised to either the sulfoxide 7 or sulfone 8rotaxanes, depending on the stoichiometry of the peroxy acid used, and the sulfoxide rotaxane 7 similarly converted into sulfone 8 (Scheme 3). However, although sulfoxide 7 could be reduced to sulfide 6 using an excess of Lawesson's reagent ((CH3OC6H4PS2)2),18 chemical reduction of the sulfone rotaxane 8 could not be achieved. The inability to reduce sulfone 8, as well as the modest template ability of the flexible threads, led us to consider the use of a more rigid vinyl sulfoxide-based template. This could allow the substitution of a sulfoxide or sulfone functionality by Michael addition of a nucleophile and subsequent elimination of the sulfoxide/sulfone fragment.19 If the nucleophile used is the thiolate analogue of the rotaxane stopper then this Michael–retro-Michael sequence could be employed to afford the ‘reduced’ sulfide–amide rotaxane (Scheme 3, inset).20
![Synthesis of vinyl sulfur-based hydrogen bonded [2]rotaxanes 9–12. Reagents and conditions: (i) isophthaloyl dichloride (five-fold excess), p-xylylenediamine (five-fold excess), Et3N (ten-fold excess), CHCl3, RT, 3 h, 63% (11); (ii) MCPBA (1.5 equiv.), CH2Cl2, RT, 18 h, 83% (12); (iii) Ph2CHCH2CH2SNa (10 equiv.), DMF, 120 °C, 15 h, 85% (10); (iv) Ph2CHCH2CH2SNa (10 equiv.), DMF, 120 °C, 16 h, 92% (10). Inset: mechanism for the Michael–retro-Michael substitution of the sulfoxide/sulfone stopper group.](/image/article/2011/SC/c1sc00335f/c1sc00335f-s3.gif) | ||
| Scheme 3 Synthesis of vinyl sulfur-based hydrogen bonded [2]rotaxanes 9–12. Reagents and conditions: (i) isophthaloyl dichloride (five-fold excess), p-xylylenediamine (five-fold excess), Et3N (ten-fold excess), CHCl3, RT, 3 h, 63% (11); (ii) MCPBA (1.5 equiv.), CH2Cl2, RT, 18 h, 83% (12); (iii) Ph2CHCH2CH2SNa (10 equiv.), DMF, 120 °C, 15 h, 85% (10); (iv) Ph2CHCH2CH2SNa (10 equiv.), DMF, 120 °C, 16 h, 92% (10). Inset: mechanism for the Michael–retro-Michael substitution of the sulfoxide/sulfone stopper group. | ||
Vinyl sulfoxide–amide thread 9 was prepared in three steps from commercially available building blocks (see the Supporting Information†). The bulky diphenylmethyl group of 9 is positioned further away from the sulfur centre in comparison to threads 3–5 in order to minimise steric interactions between the macrocycle and the stopper. Rotaxane formation using the standard clipping protocol afforded the vinyl sulfoxide rotaxane 11 in 63% yield (Scheme 3), significantly higher than for the flexible sulfoxide thread (4) and similar to that of other well-positioned hydrogen bond acceptors on rigid threads (fumaramide,4bis-nitrone,12azodicarboxamide13).
Oxidation of 11 with MCPBA generated the vinyl sulfone rotaxane 12. Treatment of either 11 or 12 with excess sodium 3,3-diphenylpropan-1-thiolate in hot DMF allowed complete and clean conversion to the vinyl sulfide [2]rotaxane 10. Only a single diastereomer (E) was apparent by 1H and 13C NMR spectroscopy. Presumably Michael addition of the thiolate stopper on the α-β-unsaturated amides of 11 and 12 generates the corresponding amide enolates (Scheme 3, inset), stabilised through hydrogen bonds with the amide macrocycle. Subsequent β-elimination forms the vinyl sulfide rotaxane 10.
The significant difference in binding modes of the amide–sulfur group binding sites led us to incorporate the vinyl sulfur hydrogen bonding motif into a more complex structure in which chemical oxidation/reduction of the sulfur moiety could be used to induced shuttling of the macrocycle between two different sites of the rotaxane thread.21,22 Various external stimuli, including pH,23 metal ions,24 photochemistry,25,26 electrochemistry,27 temperature28 and the reversible formation (and breaking) of C–C bonds,29 have been used to induce shuttling in bistable amide rotaxane switches. However, the use of chemical (rather than electrochemical) oxidation and reduction to induce shuttling in rotaxanes remains rare.13 Here the design of the rotaxane thread consists of both an oxidation-level-tuneable sulfur-based binding site and a flexible succinamide group (Scheme 4). The macrocycle preferentially binds to the sulfoxide moiety over the succinamide station due to the superior hydrogen bonding strength of the S–O bond and the rigidity of the vinyl spacer between the amide and sulfoxide hydrogen bond accepting groups.26 Chemical reduction or oxidation to the corresponding sulfide or sulfone switches off the strong hydrogen bonding to the sulfur functional group and the macrocycle relocates to the succinamide binding site.
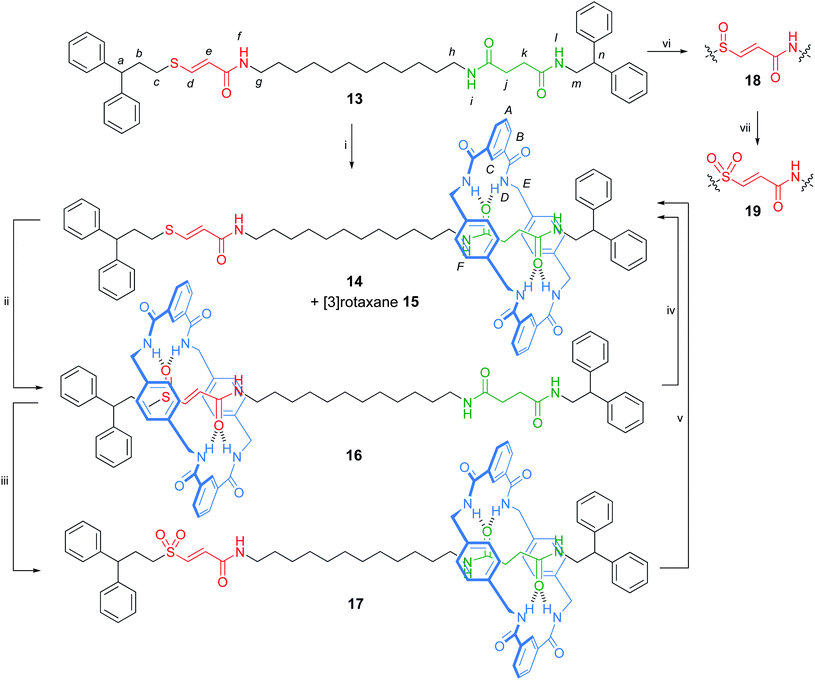 | ||
| Scheme 4 Synthesis and switching of a sulfur–succinamide molecular shuttle. Reagents and conditions: (i) isophthaloyl dichloride (five-fold excess), p-xylylenediamine (five-fold excess), Et3N (ten-fold excess), CHCl3, RT, 23% 14 + 8% 15; (ii) MCPBA (1.0 equiv.), CH2Cl2, −78 °C to RT, 2 h, 90%; (iii) MCPBA (2.0 equiv.), CH2Cl2, −78 °C to RT, 2 h, 92%; (iv) Ph2CHCH2CH2SNa (10 equiv.), DMF, 120 °C, 16 h, 93%; (v) Ph2CHCH2CH2SNa (10 equiv.), DMF, 120 °C, 18 h, 90%; (vi) MCPBA (1.0 equiv.), CH2Cl2, −78 °C to RT, 2 h, 93%; (vii) MCPBA (2.0 equiv.), CH2Cl2, −78 °C to RT, 2 h, 88%. | ||
The synthesis of a vinyl sulfide–succinamide thread, 13, was achieved in four steps from commercially available starting materials (see the Supporting Information†). Formation of the interlocked architecture using the multi-component clipping reaction furnished a mixture of [2]- and [3]-rotaxanes, 14 and 15, which were separated by flash column chromatography. [2]Rotaxane 14 could be smoothly and selectively oxidised to the corresponding vinyl sulfoxide 16 or vinyl sulfone 17rotaxane, depending upon the stoichiometry of oxidant used. The corresponding sulfoxide and sulfone threads 18 and 19 were prepared by oxidising 13 with MCPBA (Scheme 4).
Comparison of the 1H NMR spectra of the three oxidation states of the vinyl sulfur rotaxanes 14, 16 and 17 in CDCl3 with the corresponding free threads allowed the net position of the macrocycle to be determined in each case. With the vinyl sulfide 14 and vinyl sulfone 17rotaxanes, the chemical shift of the signal corresponding to the ethylene bridge of the succinamide station are significantly shifted upfield (Fig. 4b and 4f, respectively) due to shielding from the xylylene rings of the macrocycle, whereas signals from the vinyl sulfide and sulfone moieties do not undergo large shifts between thread and rotaxane, consistent with the macrocycle being predominantly located over the succinamide binding site. In contrast to rotaxanes 14 and 17, the resonances of sulfoxide rotaxane 16 are broad at room temperature in CDCl3, indicative of slow dynamic processes, presumably a result of stronger intercomponent hydrogen bonding in the sulfoxide shuttle. At 55 °C the resonances are comparable in sharpness to the other rotaxanes at room temperature (Fig. 4d). Modest shielding of the protons of the succinamide station (Δδ 0.2 ppm) occurs in sulfoxide rotaxane 16 (Fig. 4d), accompanied by a much larger shift in the vinyl sulfoxide protons (Δδ 0.7 ppm) indicating that the macrocycle is preferentially located around the vinyl sulfoxide–amide station. Furthermore, the xylylene aromatic protons of the macrocycle (HF) appear as a diastereotopic set in 16 (Fig. 4d) indicating that the macrocycle is held in a well-expressed chiral environment, i.e. in the vicinity of the asymmetric sulfoxide group.
![1H NMR spectra (400 MHz, CDCl3, 25 °C unless otherwise indicated) of (a) sulfide thread 13, (b) sulfide [2]rotaxane 14, (c) sulfoxide thread 18 (55 °C), (d) sulfoxide [2]rotaxane 16 (55 °C), (e) sulfone thread 19, (f) sulfone [2]rotaxane 17. The assignments correspond to the lettering shown in Scheme 4.](/image/article/2011/SC/c1sc00335f/c1sc00335f-f4.gif) | ||
| Fig. 4 1H NMR spectra (400 MHz, CDCl3, 25 °C unless otherwise indicated) of (a) sulfide thread 13, (b) sulfide [2]rotaxane 14, (c) sulfoxide thread 18 (55 °C), (d) sulfoxide [2]rotaxane 16 (55 °C), (e) sulfone thread 19, (f) sulfone [2]rotaxane 17. The assignments correspond to the lettering shown in Scheme 4. | ||
The sulfide-, sulfoxide- and sulfone-containing rotaxanes 14, 16 and 17 could be interconverted by oxidation and Michael–retro-Michael reactions in excellent yields (Scheme 4). Accordingly, the vinyl sulfur–amide system is a chemically-switchable molecular shuttle that could prove useful as a component of more complex molecular machines.
Conclusions
The diverse hydrogen bond accepting properties of sulfide-, sulfoxide- and sulfone-based amide threads can be effectively introduced into hydrogen bonded amide-based [2]rotaxanes through five component ‘clipping’ reactions. The weaker nature of amide–sulfide and amide–sulfone hydrogen bond interactions means the corresponding [2]rotaxanes are formed in lower yields than the amide–sulfoxide template and maximise amide–amide macrocycle-thread hydrogen bonding motifs in the solid state at the expense of lower energy macrocycle conformations. The sufide, sulfoxide and sulfone rotaxanes can be interconverted through oxidation, reduction and Michael–retro-Michael addition/elimination processes. As well as adding to the chemical diversity of templates available for assembling amide-macrocycle rotaxanes, these motifs can be used in the synthesis and operation of chemically-responsive bistable molecular shuttles in which the oxidation state of the vinyl sulfur group determines the position of the macrocycle in the rotaxane. The manipulation of hydrogen bonding strengths of functional groups in this way may prove useful for the construction of more complex synthetic molecular machine systems.22Experimental section
General method for the preparation of sulfur-based amide [2]rotaxanes
The sulfur-containing thread (3, 4 or 5, 1.0 equiv.) and Et3N (ten-fold excess) was dissolved in anhydrous CHCl3 and stirred vigorously while solutions of the amine (five-fold excess) and the acid chloride (five-fold excess) in anhydrous CHCl3 were added over 3 h using motor-driven syringe pumps. The reaction was filtered and the solvent removed under reduced pressure. The crude material was then subjected to column chromatography (silica gel, CHCl3/MeOH as eluent) to give the [2]rotaxane 6, 7 or 8 in 12, 43 or 10% yield, respectively. Further details are given in the Supporting Information.†Acknowledgements
The authors thank Dr J. K. Y. Wong for the X-ray crystallography. This work was supported by the ERC and the EPSRC. We thank the Deutsche Akademia der Naturforscher Leopoldina (BMBF LPD 9901/8-166; D.M.D'S.), the Peter und Traudl Engelhorn-Stiftung (D.M.D'S.) and the Fundación Ramón Areces (R.C.) for postdoctoral fellowships.Notes and references
- C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303–5311 CrossRef CAS; F. Vögtle, S. Meier and R. Hoss, Angew. Chem., Int. Ed. Engl., 1992, 31, 1619–1622 CrossRef; A. G. Johnston, D. A. Leigh, R. J. Pritchard and M. D. Deegan, Angew. Chem., Int. Ed. Engl., 1995, 34, 1209–1212 CrossRef.
- F. Vögtle, M. Händel, S. Meier, S. Ottens-Hildebrandt, F. Ott and T. Schmidt, Liebigs Ann., 1995, 739–743 CrossRef.
- A. G. Johnston, D. A. Leigh, A. Murphy, J. P. Smart and M. D. Deegan, J. Am. Chem. Soc., 1996, 118, 10662–10663 CrossRef CAS.
- F. G. Gatti, D. A. Leigh, S. A. Nepogodiev, A. M. Z. Slawin, S. J. Teat and J. K. Y. Wong, J. Am. Chem. Soc., 2001, 123, 5983–5989 CrossRef CAS.
- D. A. Leigh, A. Murphy, J. P. Smart and A. M. Z. Slawin, Angew. Chem., Int. Ed. Engl., 1997, 36, 728–732 CrossRef CAS.
- A. S. Lane, D. A. Leigh and A. Murphy, J. Am. Chem. Soc., 1997, 119, 11092–11093 CrossRef CAS; W. Clegg, C. Gimenez-Saiz, D. A. Leigh, A. Murphy, A. M. Z. Slawin and S. J. Teat, J. Am. Chem. Soc., 1999, 121, 4124–4129 CrossRef; V. Bermudez, N. Capron, T. Gase, F. G. Gatti, F. Kajzar, D. A. Leigh, F. Zerbetto and S. Zhang, Nature, 2000, 406, 608–611 CrossRef; J. S. Hannam, T. J. Kidd, D. A. Leigh and A. J. Wilson, Org. Lett., 2003, 5, 1907–1910 CrossRef; P. Linnartz, S. Bitter and C. A. Schalley, Eur. J. Org. Chem., 2003, 24, 4819–4829 CrossRef; C. A. Schalley, W. Reckien, S. Peyerimhoff, B. Baytekin and F. Vögtle, Chem.–Eur. J., 2004, 10, 4777–4789 CrossRef; D. A. Leigh, A. Venturini, A. J. Wilson, J. K. Y. Wong and F. Zerbetto, Chem.–Eur. J., 2004, 10, 4960–4969 CrossRef; D. A. Leigh, M.Á. F. Morales, E. M. Pérez, J. K. Y. Wong, C. G. Saiz, A. M. Z. Slawin, A. J. Carmichael, D. M. Haddleton, A. M. Brouwer, W. J. Buma, G. W. H. Wurpel, S. León and F. Zerbetto, Angew. Chem., Int. Ed., 2005, 44, 3062–3067 CrossRef; E. R. Kay and D. A. Leigh, Top. Curr. Chem., 2005, 262, 133–177 CrossRef; M. N. Chatterjee, E. R. Kay and D. A. Leigh, J. Am. Chem. Soc., 2006, 128, 4058–4073 CrossRef; D. González Cabrera, B. D. Koivisto and D. A. Leigh, Chem. Commun., 2007, 4218–4220 RSC; J. Berná, G. Bottari, D. A. Leigh and E. M. Pérez, Pure Appl. Chem., 2007, 79, 39–54 CrossRef; J. J. Gassensmith, L. Barr, J. M. Baumes, A. Paek, A. Nguyen and B. D. Smith, Org. Lett., 2008, 10, 3343–3346 CrossRef; M. Alvarez-Pérez, S. M. Goldup, D. A. Leigh and A. M. Z. Slawin, J. Am. Chem. Soc., 2008, 130, 1836–1838 CrossRef.
- X. Fradera, M. Marquez, B. D. Smith, M. Orozco and F. J. Luque, J. Org. Chem., 2003, 68, 4663–4673 CrossRef CAS.
- E. Arunkumar, C. C. Forbes, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 3288–3289 CrossRef CAS; E. Arunkumar, C. C. Forbes and B. D. Smith, Eur. J. Org. Chem., 2005, 4051–4059 CrossRef; E. Arunkumar, N. Fu and B. D. Smith, Chem.–Eur. J., 2006, 12, 4684–4690 CrossRef; S. Xiao, N. Fu, K. Peckham and B. D. Smith, Org. Lett., 2010, 12, 140–143 CrossRef.
- P. Ghosh, O. Mermagen and C. A. Schalley, Chem. Commun., 2002, 2628–2629 RSC.
- Y. L. Huang, W. C. Hung, C. C. Lai, Y. H. Liu, S. M. Peng and S. H. Chiu, Angew. Chem., Int. Ed., 2007, 46, 6629–6633 CrossRef CAS.
- A. Vidonne and D. Philp, Tetrahedron, 2008, 64, 8464–8475 CrossRef CAS.
- D. M. D'Souza, D. A. Leigh, L. Mottier, K. M. Mullen, F. Paolucci, S. J. Teat and S. Zhang, J. Am. Chem. Soc., 2010, 132, 9465–9470 CrossRef CAS.
- J. Berná, M. Alajarín and R.-A. Orenes, J. Am. Chem. Soc., 2010, 132, 10741–10747 CrossRef.
- J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469–12476 CrossRef CAS; M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul and A. R. Cowley, J. Am. Chem. Soc., 2004, 126, 15364–15365 CrossRef; M. D. Lankshear and P. D. Beer, Coord. Chem. Rev., 2006, 250, 3142–3160 CrossRef; M. R. Sambrook, P. D. Beer, M. D. Lankshear, R. F. Ludlow and J. A. Wisner, Org. Biomol. Chem., 2006, 4, 1529–1538 Search PubMed; M. D. Lankshear and P. D. Beer, Acc. Chem. Res., 2007, 40, 657–668 CrossRef; K. M. Mullen and P. D. Beer, Chem. Soc. Rev., 2009, 38, 1701–1713 RSC.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS.
- We also investigated the effect of replacing both amide groups of the succinamide and fumaramide template motifs with sulfoxides. Such bis-sulfoxide threads (both meso- and chiral bis-sulfoxide diastereoisomers) did not form rotaxanes in the five-component ‘clipping’ reaction. This may be due to the relative orientation adopted by the hydrogen bond accepting groups on the thread or the steric and conformational consequences of replacing both trigonal sp2-hybridised carbonyl groups with tetrahedral sp3-hybridised sulfoxides.
- F. Biscarini, M. Cavallini, D. A. Leigh, S. León, S. J. Teat, J. K. Y. Wong and F. Zerbetto, J. Am. Chem. Soc., 2002, 124, 225–233 CrossRef CAS.
- M. Madesclaire, Tetrahedron, 1988, 44, 6537–6580 CrossRef CAS; H. Bartsch and T. Erker, Tetrahedron Lett., 1992, 33, 199–200 CrossRef.
- P. N. Confalone, E. M. Huie and N. G. Patel, Tetrahedron Lett., 1983, 24, 5563–5566 CrossRef CAS; J. S. O'Donnell and A. L. Schwan, Tetrahedron Lett., 2003, 44, 6293–6296 CrossRef; F. Chery, P. Rollin, O. De Lucchi and S. Cossu, Synthesis, 2001, 286–292 CrossRef; E. Cabianca, F. Chery, P. Rollin, S. Cossu and O. De Lucchi, Synlett, 2001, 1962–1964 CrossRef; F. Chery, M. Desroses, A. Tatibouet, O. De Lucchi and P. Rollin, Tetrahedron, 2003, 59, 4563–4572 CrossRef.
- For other rotaxane systems in which the substitution of stopper groups can occur without the ring being able to dethread, see: S. J. Rowan and J. F. Stoddart, J. Am. Chem. Soc., 2000, 122, 164–165 CrossRef CAS; D. W. Zehnder and D. B. Smithrud, Org. Lett., 2001, 3, 2485–2487 CrossRef; S.-H. Chiu, S. J. Rowan, S. J. Cantrill, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem.–Eur. J., 2002, 8, 5170–5183 CrossRef; O.Š. Miljanić, W. R. Dichtel, I. Aprahamian, R. D. Rohde, H. D. Agnew, J. R. Heath and J. F. Stoddart, QSAR Comb. Sci., 2007, 26, 1165–1174 Search PubMed; J. M. Spruell, W. R. Dichtel, J. R. Heath and J. F. Stoddart, Chem.–Eur. J., 2008, 14, 4168–4177 CrossRef; A. Fernandes, A. Viterisi, F. Coutrot, S. Potok, D. A. Leigh, V. Aucagne and S. Papot, Angew. Chem., Int. Ed., 2009, 48, 6443–6447 CrossRef.
- Molecular Catenanes, Rotaxanes and Knots: A Journey Through the World of Molecular Topology, ed. J.-P. Sauvage and C. Dietrich-Buchecker, Wiley-VCH, Weinheim, Germany, 1999 RSC; M. Venturi, A. Credi and V. Balzani, Molecular Devices and Machines - A Journey into the Nanoworld, Wiley-VCH, Weinheim, Germany, 2003 RSC; H. Tian and Q. C. Wang, Chem. Soc. Rev., 2006, 35, 361–374 RSC.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS.
- C. M. Keaveney and D. A. Leigh, Angew. Chem., Int. Ed., 2004, 43, 1222–1224 CrossRef CAS; D. A. Leigh and A. R. Thomson, Org. Lett., 2006, 8, 5377–5379 CrossRef.
- D. S. Marlin, D. González Cabrera, D. A. Leigh and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2006, 45, 77–83 CrossRef CAS; D. S. Marlin, D. González Cabrera, D. A. Leigh and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2006, 45, 1385–1390 CrossRef.
- A. M. Brouwer, C. Frochot, F. G. Gatti, D. A. Leigh, L. Mottier, F. Paolucci, S. Roffia and G. W. H. Wurpel, Science, 2001, 291, 2124–2128 CrossRef CAS; G. W. H. Wurpel, A. M. Brouwer, I. H. M. van Stokkum, A. Farran and D. A. Leigh, J. Am. Chem. Soc., 2001, 123, 11327–11328 CrossRef; M. R. Panman, P. Bodis, D. J. Shaw, B. H. Bakker, A. C. Newton, E. R. Kay, A. M. Brouwer, W. J. Buma, D. A. Leigh and S. Woutersen, Science, 2010, 328, 1255–1258 CrossRef.
- A. Altieri, G. Bottari, F. Dehez, D. A. Leigh, J. K. Y. Wong and F. Zerbetto, Angew. Chem., Int. Ed., 2003, 42, 2296–2300 CrossRef CAS.
- A. Altieri, F. G. Gatti, E. R. Kay, D. A. Leigh, D. Martel, F. Paolucci, A. M. Z. Slawin and J. K. Y. Wong, J. Am. Chem. Soc., 2003, 125, 8644–8654 CrossRef CAS; G. Fioravanti, N. Haraszkiewicz, E. R. Kay, S. M. Mendoza, C. Bruno, M. Marcaccio, P. G. Wiering, F. Paolucci, P. Rudolf, A. M. Brouwer and D. A. Leigh, J. Am. Chem. Soc., 2008, 130, 2593–2601 CrossRef.
- G. Bottari, F. Dehez, D. A. Leigh, P. J. Nash, E. M. Pérez, J. K. Y. Wong and F. Zerbetto, Angew. Chem., Int. Ed., 2003, 42, 5886–5889 CrossRef CAS.
- D. A. Leigh and E. M. Pérez, Chem. Commun., 2004, 2262–2263 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of all compounds. CCDC reference numbers 828037 (6), 829630 (7) and 828038 (8). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00335f |
| This journal is © The Royal Society of Chemistry 2011 |