Diversity in natural product families is governed by more than enzyme promiscuity alone: establishing control of the pacidamycin portfolio†
Sabine
Grüschow
,
Emma J.
Rackham
and
Rebecca J. M.
Goss
*
School of Chemistry, University of East Anglia, Norwich, UK. E-mail: r.goss@uea.ac.uk; Fax: +44 (0)1603 592-003; Tel: +44 (0)1603 593-766
First published on 11th August 2011
Abstract
As with many other antibiotics, pacidamycins are produced as a suite of related compounds. Unlike most other secondary metabolites, however, this diversity is not solely the result of the substrate promiscuity of the biosynthetic enzymes but also arises from a gene duplication event (Pac21, Pac21h) and control of the precursor pool (PhhA). We are demonstrating the ability to harness these three levels of control in order to direct the selective production of specific members of this family of metabolites in a “dial-a-molecule” fashion. Furthermore, PhhA is shown to be a phenylalanine 3-hydroxylase, the first of the iron- and tetrahydropterin-dependent aromatic amino acid hydroxylases to be characterised with this regioselectivity.
Introduction
Natural products have proven to be a matchless starting point for drug discovery; during the past three decades over 70% of antimicrobials and over 60% of chemotherapeutics entering clinical trials have been based on such compounds.1 However, many natural products occur as suites of structurally related compounds, and issues such as difficulty in cheaply and reliably purifying the desired natural product from a mixture of related compounds have to some extent prejudiced industry against these drug molecules. In the case of the pacidamycins (Fig. 1), members of the uridyl peptide class of antibiotics (UPAs) with activity against Pseudomonas aeruginosa, around 20 naturally occurring variants have been determined to date in the producing organism Streptomyces coeruleorubidus, rendering the purification of individual compounds a non-trivial exercise.2–4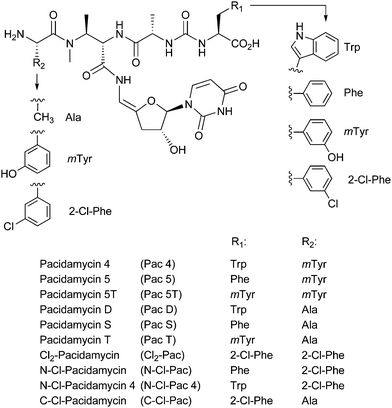 | ||
| Fig. 1 Structures of pacidamycins discussed in text. | ||
Diversity in natural product generation is thought to confer evolutionary advantage, providing the producing organism with the required degree of flexibility to rapidly adapt within changing environments. The generation of suites of related metabolites is typically attributed to the substrate promiscuity of the biosynthetic enzymes or by partial processing and premature release of a metabolite. In this paper, we demonstrate that the structural diversity of pacidamycins is enabled by three distinct mechanisms: substrate flexibility of biosynthetic enzymes, control of the precursor pool and a gene duplication and diversification event.
Results and discussion
Identification and characterisation of additional genes
We have recently identified the biosynthetic gene cluster responsible for the biosynthesis of the pacidamycins.5 The cluster comprises 22 genes, 8 of which are nonribosomal peptide synthetase (NRPS) components. Interestingly, no candidate gene for the formation of the non-proteinogenic amino acidmeta-tyrosine (mTyr) was identified in the cluster. Consistent with this observation, only the mTyr-less pacidamycins D and S (Fig. 1) are produced upon heterologous expression of the 22 core genes in Streptomyces lividansRG-4289. Furthermore, the full suite of pacidamycins was not restored upon supplementing mTyr to the medium suggesting that something other than precursor availability contributes to the structural diversity of the pacidamycins.5In silico analysis of genome scan data and additional sequencing revealed the presence of two adjacent genes, pac21h and phhA, that are likely to be involved in pacidamycin biosynthesis. The two genes are separated by a stretch of just over 400 bp. The gene product Pac21h shows homologies to AMP-forming enzymes and shows 84% sequence identity and 90% similarity on the amino acid level to the pacidamycin adenylation domain Pac21 that is part of the published core cluster. The phhAgene product is homologous to the catalytic domain of phenylalanine hydroxylases, and hence an excellent candidate for the formation of mTyr from phenylalanine. We postulated that the most likely function of Pac21 and Pac21h was the incorporation of alanine or meta-tyrosine, respectively, into the N-terminus of pacidamycin. Hence, Pac21 is likely to enable selective access to the “lettered” pacidamycins D, S and T, whereas Pac21h is likely to provide selective access to “numbered” pacidamycins 4, 5, and 5T (Fig. 1). A combination of gene disruption, genetic complementation and heterologous expression was employed to investigate the functional role of the three genesphhA, pac21, and pac21h and, furthermore, to explore the level of control over the pacidamycin profile that could be applied.
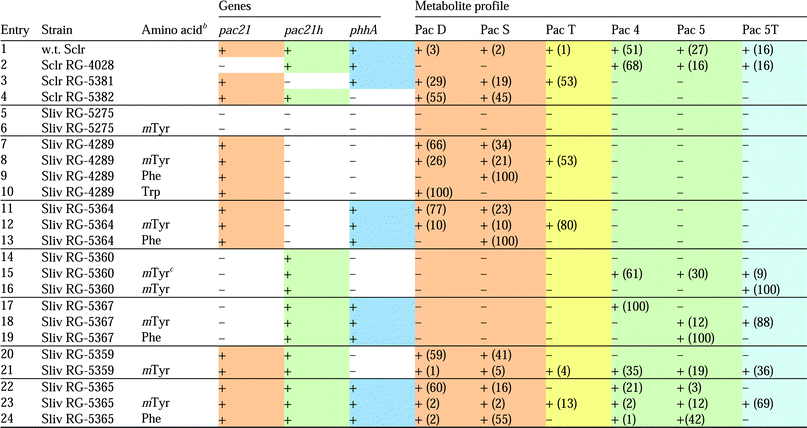
In a first set of experiments, we disrupted each of the three genes of interest in the pacidamycin producer S. coeruleorubidus using PCR targeted mutagenesis (Table 1, entries 2–4).6,7 Culture extracts of the resulting mutant strains were then analysed by LC-MS for their metabolite profile. In the wild type strain, pacidamycins 4, 5 and 5T are the dominant constituents (94% of total) whereas pacidamycins D, S and T are minor ones (Table 1, entry 1). As expected, the pac21 mutant RG-4028 only produced pacidamycins with N-terminal mTyr residue (pacidamycin 4, 5, 5T); the pac21h mutant strain RG-5381 produced exclusively pacidamycins with N-terminal Ala residue (pacidamycin D, S, T); and only mTyr-less pacidamycins (pacidamycin D and S) were detected in the phhA mutant RG-5382. These findings corroborate the function of phhA in generating the precursor pool of mTyr and demonstrate that pac21, lying within the isolated core cluster, is responsible for the incorporation of alanine into the N-terminal position. The homologous genepac21h, situated outside the biosynthetic gene cluster, is responsible for the incorporation of mTyr into the N-terminal position of pacidamycin.8 It is intriguing that pac21, although being an integral part of the cluster, is involved in the biosynthesis of the minor constituents of the pacidamycins, and that the make-up of the pacidamycin suite is dominated by the action of the remote pac21h. Furthermore, we observe the co-localisation of two genes of which one, phhA, supplies the substrate for the gene product, Pac21h, of the other. How evolutionary forces act to keep functionally coupled genes clustered together in genomes is still to be elucidated. It is also interesting to note that only one pac21homologue, npsP6, is located in the highly similar napsamycin/mureidomycin cluster.9 As expected, only mTyr is found in the N-terminal position of the antibiotic. A further UPA cluster is located in the Streptomyces roseosporusgenome.5,9 Interestingly, even though two pac21homologues are present in the genome, only one, SSGG_02994, lies within the UPA cluster. The second one, SSGG_05527 appears to be part of a putative vicenistatin-like gene cluster. Vicenistatin belongs to a group of macrolactam metabolites with antitumour activity.10,11 Thus, the S. roseosporusgenome appears to hold an evolutionary link between the UPA and the vicenistatin-like biosynthetic pathways.
| a In all cases, the 21 core genes are present, the absence (−) or presence (+) of a functional copy of phhA, pac21, or pac21h is indicated. The detection of pacidamycins was performed by LC-MS; numbers in brackets indicate the percentage of extracted ion peak area relative to all pacidamycins. b 5 mM final concentration in culture medium except where stated otherwise. c 1 mM final concentration. d Sclr S. coeruleorubidus; Sliv S. lividans. |
|---|
|
Dial-a-pacidamycin
Next we set out to use this knowledge to re-engineer pacidamycin profiles in the heterologous hostS. lividans. Two genetic backgrounds were created in this strain; S. lividansRG-4289 is isogenic to the previously reported RG-42885 and contains the full set of 22 pacidamycin biosynthesis genes but not pac21h or phhA (Table 1, entry 7). In S. lividans RG-5275 pac21 had been removed leaving only the 21 core genes. RG-5275 does not produce any pacidamycins; nor is pacidamycin production restored upon mTyr supplementation (Table 1, entry 5 and 6). Additional genes (pac21, pac21h, phhA) were introduced to RG-4289 and to RG-5275 using vector pIJ10257. This vector expresses genes from the constitutive promoter ermE* and is propagated through integration into the phage BT1 site.12 The pacidamycin profiles of the new S. lividans-derived strains were analysed as before by LC-MS.As described above, strain RG-4289 contains a functional copy of each of the 22 pacidamycin cluster genes and produces pacidamycins D and S in the absence of exogenously added mTyr, and pacidamycins D, S and T in the presence of exogenously added mTyr. Supplementing the culture medium with Phe, on the other hand, was enough to shift the RG-4289 profile exclusively to pacidamycin S; in the same manner addition of Trp provided solely pacidamycin D (Table 1, entries 7–10). It is also noteworthy in this context that increasing the amount of available mTyr increased the bias towards pacidamycins with mTyr as the C-terminal residue (Table 1, entries 15 and 16). These results suggest that the identity of the C-terminal pacidamycin residue is determined by substrate concentration. This is likely to be in part a reflection of the catalytic efficiency of the NRPS module Pac12. Its embedded adenylation domain is a relatively promiscuous enzyme that activates Trp, Phe or mTyr prior to incorporation into the diamino acyl urea fragment.13
When we attempted to provide mTyr in vivo through co-expression of phhA (strains RG-5364, RG-5365, RG-5367), only pacidamycins with Phe or Trp at the C-terminal position were produced (Pac D, S, 4, 5; Table 1, entries 11, 17, 22). Surprisingly though, pacidamycins with mTyr at the C-terminal position only appeared upon supplementation with mTyr (Pac 5, 5T; Table 1, entries 12, 18, 23). These findings implied that, in the heterologous hostS. lividans, the intracellular concentration of PhhA-derived mTyr is sufficient to be incorporated into the N-terminus of pacidamycin through the action of Pac21h (Table 1, entries 17, 19, 22, 24). However, it is not sufficient to compete with Trp and Phe as substrate for Pac12-mediated incorporation into the C-terminus (Table 1, entries 11, 13, 17, 19, 22, 24). As will be discussed below, the apparent low productivity of PhhA is likely to be due to ineffective expression of phhA from the ermE* promoter or due to poor co-factor recycling in the heterologous host. The former statement finds support from the fact that, when pac21h was expressed from the heterologous promoter, the usually dominant “numbered” pacidamycins 4 and 5 became less abundant than their counterparts pacidamycin D and S (Table 1, entries 1 and 22).
To summarise, we are now in a position to control the pacidamycin profile on the genetic level. An additional level of control is exerted through the exogenous supply of amino acids. In this way, we are able to dial into distinct sets of pacidamycins and even individual pacidamycins. For example, the profile of wild type S. coeruleorubidus was recreated with S. lividans RG-5359 or RG-5360 supplemented with mTyr (Table 1, entries 1, 21, 23). Exclusive production of pacidamycin 4, 5, or 5T (Table 1, entries 17, 19, 16) as well as exclusive production of pacidamycin D or S (Table 1, entries 10 and 9, 13) could also be achieved.
A new set of chlorinated pacidamycins
Having established control of production of the natural pacidamycins we explored whether an engineered system might confer advantage over the wild type in generation of analogues of pacidamycin through precursor directed biosynthesis. We have previously demonstrated that upon feeding, 2-Cl-Phe is incorporated into both the N- and C-terminal positions of pacidamycin in the wild type organism (Fig. 1).14 By feeding 2- Cl-Phe to the phenylalanine hydroxylase-deficient S. coeruleorubidus strain RG-5382 we were again able to generate a complementary set of chlorinated pacidamycins (Table 2, entries 8–10) with only the disubstituted compound Cl2-Pac being produced in both systems (Table 2, entry7). The mutasynthesis approach enabled us to increase the proportion of chlorinated to non-chlorinated pacidamycins from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 in the wild type strain to 3:2 in RG-5382.
:3 in the wild type strain to 3:2 in RG-5382.
| Entry | Compound | w.t. Sclr | Sclr RG-5382 |
|---|---|---|---|
| a Percentages were calculated from the peak areas of the corresponding extracted ion chromatograms relative to the total. Experiments were performed in triplicate, standard deviations are indicated. b R12-Cl-Phe, R2mTyr as described previously.14 | |||
| 1 | Pac D | 3.6 ± 1.2% | 25.5 ± 3.1% |
| 2 | Pac S | 1.6 ± 0.4% | 13.8 ± 1.5% |
| 3 | Pac T | n.d. | n.d. |
| 4 | Pac 4 | 36.6 ± 2.3% | n.d. |
| 5 | Pac 5 | 13.3 ± 0.9% | n.d. |
| 6 | Pac 5T | 19.2 ± 0.5% | n.d. |
| 7 | Cl2-Pac | 4.3 ± 0.3% | 38.3 ± 4.0% |
| 8 | N-Cl-Pac | n.d. | 6.6 ± 0.8% |
| 9 | N-Cl-Pac 4 | n.d. | 12.2 ± 1.0% |
| 10 | C-Cl-Pac | n.d. | 3.7 ± 0.5% |
| 11 | C-Cl-Pac 4b | 21.7 ± 2.8% | n.d. |
PhhA acts as a Phe 3-hydroxylasein vitro
In order to investigate the activity of the phenylalanine hydroxylase, we decided to test the in vitro activity of the PhhA. PhhA shows significant sequence identity (appr. 30%) to a small class of aromatic amino acid hydroxylases. In eukaryotes, these enzymes play a vital role in phenylalanine metabolism (phenylalanine 4-hydroxylase), and they catalyse the first step in the biosynthesis of serotonin (tryptophan 5-hydroxylase) and catecholamine (tyrosine 3-hydroxylase).15,16 In bacteria, this family of hydroxylases is less common and only three enzymes have been functionally characterised to the best of our knowledge.17–19 It has to be noted that, to date, no phenylalanine 3-hydroxylase has been characterised.20 Aromatic amino hydroxylases are non-heme iron proteins that require tetrahydropterin as a co-factor. In the current mechanistic understanding,15,21 molecular oxygen, the tetrahydropterin (PH4) co-factor and the amino acid substrate must all bind to the enzyme for any reaction to occur. O2 then reacts with the co-factor and the metal to form an intermediate μ-peroxypterin species (Scheme 1). Subsequent cleavage of the O–O bond furnishes the hydroxylating species Fe(IV)=O. After hydroxylation of the aromatic substrate the ferrous iron is restored and 4a-hydroxypterin and product are released from the enzyme.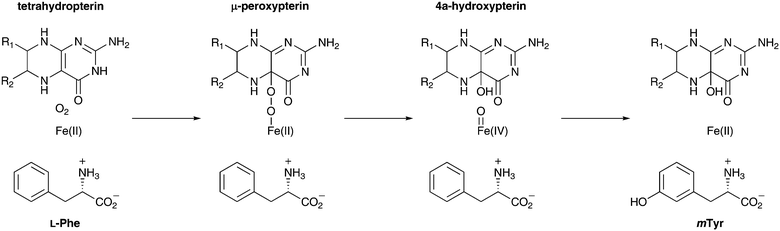 | ||
| Scheme 1 Hypothetical reaction mechanism of PhhA. | ||
S. coeruleorubidus PhhA was heterologously produced in E. coli as N-terminal His6-fusion protein. In order to ensure loading of the enzyme with ferrous iron, the cell lysis buffer was supplemented with ammonium iron(II) sulphate. Size exclusion chromatography showed PhhA to be a monomer in solution which is consistent with other bacterial hydroxylases. In vitro assays were performed in 50 mM HEPES buffer at pH 7.5 and contained 5 μM PhhA, 1 mM L-Phe, 0.1 mM 6,7-dimethyl-5,6,7,8-tetrahydropterin (DMPH4) and 84 U μl−1catalase. The concentration of molecular oxygen was not regulated. DMPH4 is a frequently used synthetic variant of natural pterins. Under these conditions we could observe the conversion of L-phenylalanine to mTyr (Fig. 2A). No para-tyrosine was detected, implying a high degree of regioselectivity. The presence of DMPH4 was absolutely required for activity. Reaction rates were assessed qualitatively using in situfluorescence detection. Changes in fluorescence during the course of the reaction are due to a combination of the consumption of DMPH4 and the production of mTyr. Non-enzymatic reactions contributed little to the measured rates as initial rates were directly proportional to the enzyme concentrations (Fig. S3†). At 0.5 μM PhhA, the fluorescence at 305 nm rapidly increased to reach a constant level after approximately 8 min indicating depletion of DMPH4 (Fig. 2B). Addition of dithiothreitol (DTT) did not affect the initial rate but did affect the position of the plateau level (Fig. 2B). This observation is consistent with the DTT-mediated co-factor recycling that occurs at a slower rate. Changing the co-factor from DMPH4 to tetrahydrobiopterin (BH4), which is the physiological cofactor for mammalian aromatic amino acid hydroxylases, had no observable effect on the initial rates (data not shown). This could indicate that neither pterin is the physiological co-factor for S. coeruleorubidus PhhA. This is a likely case as the chemical structure of pterins shows variation between species.22 Finally, the apparent Km value for L-Phe with constant DMPH4 is 0.9 ± 0.4 mM (Fig. S3†) which lies within reported values for mammalian and bacterial phenylalanine hydroxylases.18,23,24 The data obtained for the in vitro conversion of L-Phe to mTyr by S. coeruleorubidus PhhA suggest that the enzyme is fully active. It is therefore unlikely that the low in vivo productivity is due to the activity of PhhA itself. However, we cannot exclude factors influencing co-factor availability and recycling.
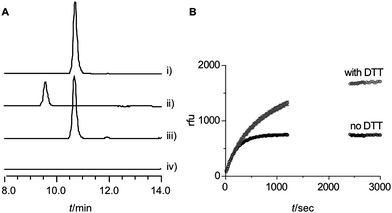 | ||
| Fig. 2 A: HPLC analysis of PhhA assay (ex 270 nm, em 305 nm). i) 0.1 mM mTyr standard, ii) 0.1 mM L-Tyr standard, iii) PhhA assay, for details see text, iv) as iii) with omission of DMPH4. Reactions were incubated at 28 °C for 3 h. B: Influence of DTT on composite reaction rate. | ||
Conclusions
The pacidamycins are important members of the uridyl peptide antibiotics, a class of compounds with a clinically unexploited mode of action against translocase I. The pacidamycins, as many other natural products, are biosynthesised as a complex series of metabolites. The structural variations within the pacidamycin family are mainly due to the identity of the N-terminal and C-terminal amino acid (Scheme 2). Our investigations demonstrate that three levels of control govern the generation of this suite of metabolites. Firstly, the activity of PhhA controls the supply of the non-proteinogenic amino acidmTyr. Unlike the biosynthesis of secondary metabolites, however, the biosynthesis of pacidamycins is not entirely dependent upon the presence of the non-proteinogenic amino acid. Secondly, the high degree of substrate promiscuity exhibited by the NRPS Pac12 allows for the incorporation of three amino acids into the C-terminus of the pacidamycin peptide. For mTyr to be able to successfully compete with Trp and Phe as a Pac12 substrate its concentration must be sufficiently high. Lastly, two orthologs of the AMP-forming enzyme involved in installing the N-terminal amino acid are present, namely Pac21 and Pac21h. The orthologs have probably arisen from a gene duplication event. Pac21 incorporates Ala whereas Pac21h incorporates mTyr. In contrast to Pac12, however, even relatively low concentrations of mTyr are sufficient to be efficiently processed through Pac21h. Furthermore, Pac21h appears to be the more efficient enzyme as the Pac21-dependent N-Ala substitution is only found in minor constituents of the pacidamycin suite.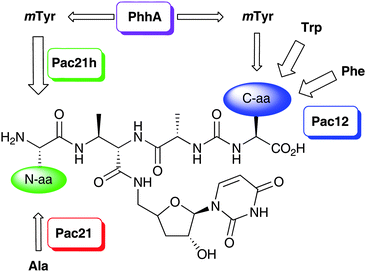 | ||
| Scheme 2 Origins of the structural diversity of pacidamycins. | ||
We have demonstrated ability to harness these three levels of control in order to direct the selective production of subsets of this family of metabolites in a “dial-a-molecule” fashion. We have utilised this approach also in combination with feeding experiments to enable the production of new pacidamycins.
Notes and references
- D. J. Newman, G. M. Cragg and K. M. Snader, J. Nat. Prod., 2003, 66, 1022 CrossRef CAS.
- R. H. Chen, A. M. Buko, D. N. Whittern and J. B. McAlpine, J. Antibiot., 1989, 42, 512 CAS.
- P. B. Fernandes, R. N. Swanson, D. J. Hardy, C. W. Hanson, L. Coen, R. R. Rasmussen and R. H. Chen, J. Antibiot., 1989, 42, 521 CAS.
- M. Winn, R. J. Goss, K. Kimura and T. D. Bugg, Nat. Prod. Rep., 2010, 27, 279 RSC.
- E. J. Rackham, S. Grüschow, A. E. Ragab, S. Dickens and R. J. M. Goss, ChemBioChem, 2010, 11, 1700 CrossRef CAS.
- B. Gust, G. L. Challis, K. Fowler, T. Kieser and K. F. Chater, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1541 CrossRef CAS.
- See Table S1 for primer sequences, Table S2 for vectors, and Table S3 for strains†.
- The same findings were reported in an independent study using purified enzymes: W. J. Zhang, I. Ntai, M. L. Bolla, S. J. Malcolmson, D. Kahne, N. L. Kelleher and C. T. Walsh, J. Am. Chem. Soc., 2011, 133, 5240 CrossRef CAS.
- L. Kaysser, X. Y. Tang, E. Wemakor, K. Sedding, S. Hennig, S. Siebenberg and B. Gust, ChemBioChem, 2011, 12, 477 CrossRef CAS.
- H. Jørgensen, K. F. Degnes, A. Dikiy, E. Fjærvik, G. Klinkenberg and S. B. Zotchev, Appl. Environ. Microbiol., 2010, 76, 283 CrossRef.
- Y. Ogasawara, K. Katayama, A. Minami, M. Otsuka, T. Eguchi and K. Kakinuma, Chem. Biol., 2004, 11, 79 CAS.
- H. J. Hong, M. I. Hutchings, L. M. Hill and M. J. Buttner, J. Biol. Chem., 2005, 280, 13055 CrossRef CAS.
- W. Zhang, B. Ostash and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16828 CrossRef CAS.
- A. E. Ragab, S. Grüschow, E. J. Rackham and R. J. Goss, Org. Biomol. Chem., 2010, 8, 3128 CAS.
- P. F. Fitzpatrick, Biochemistry, 2003, 42, 14083 CrossRef CAS.
- E. Olsson, K. Teigen, A. Martinez and V. R. Jensen, Adv. Inorg. Chem., 2010, 62, 437 CrossRef CAS.
- G. S. Zhao, T. H. Xia, J. Song and R. A. Jensen, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 1366 CrossRef CAS.
- D. W. Chen and P. A. Frey, J. Biol. Chem., 1998, 273, 25594 CrossRef CAS.
- H. K. S. Leiros, A. L. Pey, M. Innselset, E. Moe, I. Leiros, I. H. Steen and A. Martinez, J. Biol. Chem., 2007, 282, 21973 CrossRef CAS.
- During preparation of this manuscript an independent report on the pacidamycin PhhA enzyme has appeared: W. Zhang, B. D. Ames and C. T. Walsh, Biochemistry, 2011, 50, 5401 CrossRef CAS.
- A. J. Panay, M. Lee, C. Krebs, J. M. Bollinger and P. F. Fitzpatrick, Biochemistry, 2011, 50, 1928 CAS.
- A. Pribat, I. K. Blaby, A. Lara-Núñez, J. F. Gregory, V. de Crécy-Lagard and A. D. Hanson, J. Bacteriol., 2010, 192, 475 CrossRef CAS.
- S. C. Daubner, J. Melendez and P. F. Fitzpatrick, Biochemistry, 2000, 39, 9652 CrossRef CAS.
- S. C. Daubner, P. J. Hillas and P. F. Fitzpatrick, Arch. Biochem. Biophys., 1997, 348, 295 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental methods, supplementary Tables S1–S3, supplementary Figures S1–S3, LC-MS traces, MS/MS spectra. See DOI: 10.1039/c1sc00378j |
| This journal is © The Royal Society of Chemistry 2011 |