Metal ion responsive adhesion of vesicles by conformational switching of a non-covalent linker†
Siva Krishna
Mohan Nalluri
a,
Jelle B.
Bultema
b,
Egbert J.
Boekema
b and
Bart Jan
Ravoo
*a
aOrganic Chemistry Institute and Graduate School of Chemistry, Westfälische Wilhelms-Universität Münster, Corrensstrasse 40, 48149, Münster, Germany. E-mail: b.j.ravoo@uni-muenster.de
bDepartment of Biophysical Chemistry, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 7, 9747 AG, Groningen, The Netherlands
First published on 5th September 2011
Abstract
This contribution describes the metal ion responsive adhesion of vesicles induced by a conformational switch of a non-covalent linker molecule. A p-tert-butylbenzyl dimer with a flexible N,N′-bis(3-aminopropyl)ethylenediamine spacer was used as a non-covalent linker, which induces aggregation and adhesion (but not fusion) of host bilayer vesicles composed of amphiphilic β-cyclodextrins by the formation of hydrophobic inclusion complexes. The aggregation and adhesion of the vesicles in dilute aqueous solution was confirmed by isothermal titration calorimetry (ITC), optical density measurements at 600 nm (OD600), dynamic light scattering (DLS), ζ-potential measurements, cryogenic transmission electron microscopy (cryo-TEM) and fluorescence spectroscopy. However, in the presence of a divalent metal ion like Cu2+, the tetra-amine linker molecule forms a stable metal coordination complex and dramatically switches its conformation from linear to bent, which results in the dissociation of intervesicular complexes, and leads to the dispersion of vesicle clusters. This process is reversible in the presence of a strong metal ion chelator, such as EDTA, that scavenges the Cu2+ ion complexed by the linker. The linker molecule regains its linear conformation and triggers the re-aggregation of the vesicles. In contrast, conformational switching was inhibited by introducing a rigid N,N′-bis(3-aminopropyl)piperazine spacer in the non-covalent linker molecule and vesicles do not aggregate in the presence of a cyclic guest that can only bind intravesicularly. Thus, a metal ion regulated molecular switch can control the aggregation state of an organic colloidal solution.
Introduction
The recognition, adhesion and fusion of biological membranes is mediated by proteins and carbohydrates and, at least conceptually, cell–cell interactions can be mimicked by the interaction of liposomes, synthetic bilayer vesicles and polymersomes.1–3 Aggregation, adhesion and fusion of vesicles can be induced by changes of pH, temperature, ionic strength, etc., but also by metal ion binding and specific molecular recognition. It has been recently shown that complex biological cell adhesion molecules can be mimicked by rather simple synthetic complementary recognition units. Several groups have described the interactions of vesicles induced by electrostatic interactions,4–6hydrogen bonding7–11 and metal ion coordination.12–16 Among these, the metal ion induced interaction of vesicles has been particularly widely studied. Metal ion coordination at the surface of vesicles equipped with membrane-embedded ligands can occur either on the same vesicle (intravesicular binding) or between two different vesicles (intervesicular binding). Only intervesicular interaction induces aggregation, adhesion and, ultimately, fusion of vesicles. Such intervesicular interaction was observed when lecithin vesicles equipped with terpyridine ligands aggregated in the presence of Fe2+.12 Similarly, vesicles equipped with bipyridine ligands aggregate and fuse in the presence of Ni2+ or Co2+ through intervesicular coordination,13 whereas β-diketone ligands enhance the transportation of Eu3+ ions across the lipid bilayer through the formation of an intravesicular complex at the surface of the vesicles.14 It was also shown that the intervesicular interaction of membrane-bound Cu2+(iminodiacetate) complex and poly-(L-histidine) mediates the adhesion of vesicles.15 Furthermore, it was found that vesicles equipped with boronic acid specifically bind to vesicles containing a diol lipid, such as phosphatidylinositol.16 In all of these examples, the metal ion induces the aggregation and adhesion of vesicles and sometimes even leads to the fusion of vesicles. However, the reverse process, in which a metal ion regulates the dissociation of adhering vesicles (instead of the aggregation of vesicles), has not been studied yet.17 It would be of interest to develop a membrane mimetic system that is actually regulated, rather than simply triggered, by a metal ion.Herein, we report the metal ion responsive adhesion of vesicles induced by a conformational switching of a non-covalent linker molecule. It is shown that a flexible, non-covalent linker molecule induces the aggregation and adhesion of vesicles and that the aggregation can be reversed in the presence of a metal ion due to a change in the conformation of the linker molecule. The aggregation of vesicles can be re-induced with the removal of the metal ion by a chelator, such as EDTA. Thus, the non-covalent linker can be switched from an extended, intervesicular binding mode to a bent, intravesicular binding mode. We note that the artificial membrane adhesion described here bears a conceptual similarity to the adhesion of viruses to cell membranes, which essentially involves a pH dependent switch of a compact, inactive conformation of the fusion protein on the viral membrane to an extended conformation, which penetrates into the target cell membrane.18
The metal responsive model membrane system is based on bilayer vesicles composed of amphiphilic cyclodextrins (CDs 1a and 1b).19 In recent years, we have explored the size selective inclusion of guest molecules at the surface of such host vesicles.19–28 In particular, we have observed that the strong, divalent Cu2+ complex of adamantyl-functionalized ethylenediamine binds exclusively intravesicularly, whereas the weaker Ni2+ complex of adamantyl-functionalized ethylenediamine leads to intervesicular binding and the aggregation of vesicles.23 We have also demonstrated that the adhesion of CD vesicles can be mediated by a light-responsive linker.27 In this contribution, we report a homobifunctional p-tert-butylbenzyl dimer 2 with an N,N′-bis(3-aminopropyl)ethylenediamine spacer, which is used as a non-covalent linker to induce the aggregation and adhesion of host bilayer vesicles composed of amphiphilic β-CD 1b, provided that the linker is in an extended conformation. However, upon the addition of a suitable metal ion, like Cu2+, the linker switches from an extended to a bent conformation, which in turn triggers the dissociation of CD vesicles. This process is reversible in the presence of the strong metal chelator EDTA, which scavenges the metal ion and re-induces the aggregation of CD vesicles. Non-covalent linkers 3 (with a rigid N,N′-bis(3-aminopropyl)piperazine spacer) and 4 (with a permanent macrocyclic structure) were also investigated. The molecular structures of host and guest molecules 1–4 are shown in Scheme 1. The ion-responsive supramolecular system is illustrated in Fig. 1. The interaction of CD vesicles composed of 1a and 1b and guests 2–4 was investigated by isothermal titration calorimetry (ITC), optical density measurements at 600 nm (OD600), dynamic light scattering (DLS), ζ-potential measurements, cryogenic transmission electron microscopy (cryo-TEM) and fluorescence spectroscopy.
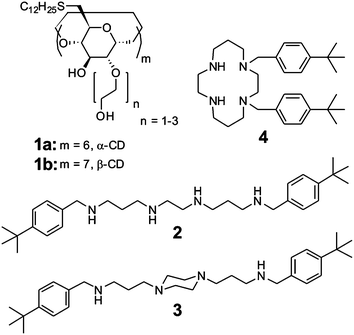 | ||
| Scheme 1 The structures of amphiphilic hosts 1a and 1b and divalent guests 2–4. | ||
 | ||
| Fig. 1 A schematic representation of the metal ion responsive supramolecular system, in which the aggregation and dispersion of CD vesicles is controlled by the reversible change in the conformation of a divalent guest molecule induced by metal ion coordination. | ||
Results and discussion
Amphiphilic α-CD 1a and β-CD 1b were synthesized as described previously.21 Unilamellar CD bilayer vesicles with a diameter of ca. 100 nm were prepared in a buffered aqueous solution by extrusion.21 Guest molecules 2 and 3 were prepared by the reaction of p-tert-butylbenzaldehyde with N,N′-bis(3-aminopropyl)ethylenediamine or N,N′-bis(3-aminopropyl)piperazine, respectively, followed by the reduction of imine derivatives with sodium borohydride. Guest molecule 4 was synthesized by the dialkylation of cyclamoxamide29 with p-tert-butylbenzyl bromide followed by hydrolysis under mild basic conditions. Details of the synthesis are reported in the electronic supplementary information (ESI†). The analytical and spectroscopic data for 2–4 are consistent with their molecular structure. Guest molecules 2 and 3 are homobifunctional non-covalent linker molecules that carry two identical hydrophobic supramolecular binding sites, p-tert-butylbenzyl groups, which are connected through the linear hydrophilic N,N′-bis(3-aminopropyl)ethylenediamine spacer and N,N′-bis(3-aminopropyl)piperazine spacer, respectively. Guest 4 is a cyclic homobifunctional molecule that carries two identical hydrophobic supramolecular binding sites, p-tert-butylbenzyl groups, which are connected to two adjacent nitrogens of cyclam. Each p-tert-butylbenzyl group of guest molecules 2–4 forms stable inclusion complexes with β-CD (Ka ∼ 1.3 × 104 M−1 for p-tert-butylbenzoate) and vesicles of β-CD 1b (Ka ∼ 6 × 103 M−1 for p-tert-butylbenzoate),20 but not with α-CD and vesicles of α-CD 1a because the cavity of α-CD is too small to host the p-tert-butylbenzyl group. As shown in Scheme 2, the N,N′-bis(3-aminopropyl)ethylenediamine spacer of guest 2 forms a highly stable tetradentate coordination complexes with suitable metal ions (Ka ∼ 1017 M−1 for the Cu2+ complex of N,N′-dibenzylated-bis(3-aminopropyl)ethylenediamine and Ka ∼ 1020 M−1 for the Cu2+ complex of triethylenetetramine).30,31 Due to the formation of a coordination complex, the conformation of 2 switches from linear (the free form in the absence of metal ions) to bent (the complex in the presence of metal ions). The X-ray structures of such complexes are available and show the bent conformation of the ligand and a square planar coordination of Cu2+.30 As a consequence, the average separation of the p-tert-butylbenzyl groups in guest 2 is dramatically reduced. In contrast, the piperazine ring of the rigid N,N′-bis(3-aminopropyl)piperazine spacer of guest 3 dominates in the chair conformation in the absence of metal ions and must convert to an energetically unfavorable boat or twist-boat confirmation when it is complexed with metal ions (Ka ∼ 1012 M−1 for Cu2+ complex of N,N′-dibenzylated-bis(3-aminopropyl)piperazine).31 Hence, guest 3 forms significantly less stable tetradentate coordination complexes than guest 2. On the other hand, the preorganization of macrocyclic guest 4 leads to the formation of strong and stable coordination complexes with suitable metal ions (Ka ∼ 1027 M−1 for the Cu2+ complex of cyclam).31,32 However, in this case, the conformation of ligand 4 does not change much upon coordination; it remains in a cyclic structure with both p-tert-butylbenzyl groups in neighbouring positions, irrespective of the metal ion coordination.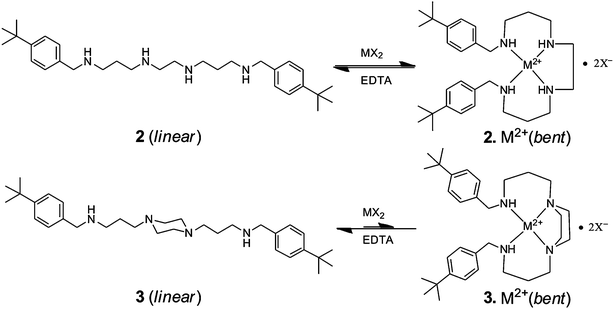 | ||
| Scheme 2 Conformational changes in the molecular structures of guest molecules 2 and 3 in the absence and presence of suitable metal ions (M2+). | ||
The interaction of guest 2 with β-CD in the absence and presence of CuCl2 was measured by using ITC. The results of these titrations are presented in Table 1 and the titration curves are shown in the ESI† (Figs S1 and S2). It can be seen from Table 1 that the affinity of guest 2 for β-CD is essentially independent of the coordination of Cu2+. The complexation is of stoichiometry 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 with equal affinity for both p-tert-butylphenyl groups. We note that these findings are in line with our previous measurements on Cu2+ complexes of adamantyl functionalized ethylenediamine ligands.23
:2 with equal affinity for both p-tert-butylphenyl groups. We note that these findings are in line with our previous measurements on Cu2+ complexes of adamantyl functionalized ethylenediamine ligands.23
| Host | Guest | n | K a [M−1] | ΔH [kJ mol−1] | ΔG [kJ mol−1] | ΔS [J K−1 mol−1] |
|---|---|---|---|---|---|---|
| β-CD | 2 | 2 | 8.1 × 103 | −8.28 | −22.3 | 47.1 |
| β-CD | 2.Cu2+ | 2 | 19.8 × 103 | −13.55 | −24.5 | 36.8 |
OD600, DLS and ζ-potential measurements, as well as cryo-TEM, were used to investigate the interaction of CD vesicles composed of 1b with guest molecule 2 in the absence and presence of divalent metal ions. The addition of as little as 6 μM of 2 to a dilute solution of vesicles of 1b (30 μM) at pH 9.0 resulted in a strong and time-dependent increase of OD600 from ca. 0.05 to ca. 0.35 (Fig. 2A, red trace). According to DLS, the average particle size increased from ca. 100 nm to more than 1000 nm (Fig. 2D, black and red traces, respectively). The rate and extent of the vesicle aggregation is dependent on the concentration of 2; fast and extensive aggregation occurs at a high concentration of 2, whereas slower and lesser aggregation occurs at low concentrations of 2 (Fig. 2A). The original OD600 was quickly recovered by the addition of excess host unmodified β-CD (750 μM) but not by the addition of excess unmodified α-CD (750 μM) (ESI Fig. S3†). Moreover, the addition of as much as 50 μM of guest 2 to a dilute solution of vesicles of α-CD 1a (instead of β-CD 1b) did not result in any significant changes in either OD600 or the average vesicle diameter (ESI Fig. S4†). These results indicate that guest 2 induces the aggregation of vesicles of β-CD 1b due to the formation of multiple intervesicular host–guest inclusion complexes. The cavity of α-CD, as well as amphiphilic α-CD 1a, is too small to form a stable inclusion complex with the p-tert-butylbenzyl groups of 2.
![The metal ion responsive aggregation of host vesicles of β-CD 1b by divalent guest 2. A) The aggregation of vesicles induced by guest 2. B) The dispersion of clusters of vesicles induced by Cu2+. C) The re-aggregation of vesicles induced by EDTA. D) The size distribution of vesicles of 1b according to DLS. Conditions: [1b] = 30 μM, [2] = 6 μM, [Cu2+] = 10 μM and [Na2H2EDTA] = 200 μM in 10 mM carbonate/bicarbonate buffer (pH 9.0). Guest 2 was added at 3 min, Cu2+ was added at 30 min and EDTA was added at 60 min, with the exception of one measurement, in which Cu2+ was added at 3 min and guest 2 was added at 5 min.](/image/article/2011/SC/c1sc00422k/c1sc00422k-f2.gif) | ||
| Fig. 2 The metal ion responsive aggregation of host vesicles of β-CD 1b by divalent guest 2. A) The aggregation of vesicles induced by guest 2. B) The dispersion of clusters of vesicles induced by Cu2+. C) The re-aggregation of vesicles induced by EDTA. D) The size distribution of vesicles of 1b according to DLS. Conditions: [1b] = 30 μM, [2] = 6 μM, [Cu2+] = 10 μM and [Na2H2EDTA] = 200 μM in 10 mM carbonate/bicarbonate buffer (pH 9.0). Guest 2 was added at 3 min, Cu2+ was added at 30 min and EDTA was added at 60 min, with the exception of one measurement, in which Cu2+ was added at 3 min and guest 2 was added at 5 min. | ||
The interaction of bifunctional guest 2 with vesicles of 1b was further investigated by cryo-TEM (Fig. 3). It was observed that vesicles of 1b are unilamellar and spherical in the absence of any guest. However, in the presence of guest 2, large and dense clusters of vesicles with extensive areas of close contact were observed. The aggregates contain dense multilamellar stacks. It is conceivable that multivalent intervesicular binding of guest 2 induces the adhesion, flattening and stacking of vesicles of 1b, similar to our observations for CD vesicles decorated with a zwitterion.26 It should be noted that for experimental reasons the concentration for the cryo-TEM experiments are ca. 50-fold higher than for the OD600 measurements.
![Cryo-TEM images of A) vesicles of 1b and B) clusters of vesicles of 1b in the presence of 2. The dark area at the bottom of image A shows the edge of the holey carbon film. Conditions: [1b] = 1 mM and [2] = 0.4 mM in 10 mM carbonate–bicarbonate buffer (pH 9.0).](/image/article/2011/SC/c1sc00422k/c1sc00422k-f3.gif) | ||
| Fig. 3 Cryo-TEM images of A) vesicles of 1b and B) clusters of vesicles of 1b in the presence of 2. The dark area at the bottom of image A shows the edge of the holey carbon film. Conditions: [1b] = 1 mM and [2] = 0.4 mM in 10 mM carbonate–bicarbonate buffer (pH 9.0). | ||
Strikingly, when 5 μM of Cu2+ is added to the aggregated vesicles of 1b, both the original OD600 (ca. 0.05) and the average vesicle diameter (ca. 150 nm) were quickly and almost completely recovered (Fig. 2B and 2D, blue trace). This observation can be attributed to the fact that guest 2 forms a stable metal coordination complex with Cu2+ and induces the switching of guest 2 from the linear to bent conformation (Scheme 2). We propose that the bent conformational isomer of guest 2 can only bind to the surface of one vesicle and intervesicular interaction is impossible. Moreover, in the bent conformation of guest 2, the p-tert-butylbenzyl groups are so close to each other that it is likely that only one of the groups binds to a CD cavity at the vesicle surface while the other group is exposed in the solution. This coordination-induced conformational change causes the dispersion of the vesicles. The effect of added Cu2+ at different concentrations (1–200 μM) on the aggregation of vesicles of 1b (30 μM) in the presence of guest 2 (6 μM) was also investigated. It was found that the rapid dispersion of vesicles occurs at all of the concentrations investigated, but a small fraction of larger aggregates inevitably persists (Fig. 2B). This finding suggests that a small number of intervesicular links remains intact in the presence of excess Cu2+. At a higher concentration of guest 2 (15 μM) in the aggregated vesicle solution, the addition of even 60 μM of Cu2+ did not lead to substantial changes in either OD600 or the average vesicle diameter (ESI Fig. S5†). This peculiar behaviour is likely due to the fact that at a high concentration (>10 μM) of guest 2 at the vesicle surface, a 2:1 intermolecular metal coordination takes place, which does not lead to the conformational switching of guest 2. We also performed measurements by changing the priority of additions of guest 2 and Cu2+ to vesicles of 1b. The addition of Cu2+ (10 μM) followed by the subsequent addition of guest 2 (6 μM) to vesicles of 1b did not change either OD600 (Fig. 2B, orange trace) or the average vesicle diameter (Fig. 2D, pink trace). This result confirms that guest 2 cannot induce intervesicular binding if it is coordinated with Cu2+. Furthermore, the metal ion induced conformational changes can be reversed in the presence of a strong metal chelator, such as EDTA. The addition of 200 μM of EDTA to the Cu2+ induced dispersion of vesicles of 1b in the presence of guest 2 caused re-aggregation, as indicated by an increase in both OD600 from ca. 0.05 to 0.35 (Fig. 2C, green trace) and the average vesicle diameter from ca. 150 nm to more than 1000 nm (Fig. 2D, green trace). It is likely that EDTA scavenges the Cu2+ ion and permits the relaxation of guest 2 from a bent to a linear conformation, which induces the re-aggregation of vesicles of 1b.
Altogether, the OD600, DLS and cryo-TEM data consistently indicate that guest 2 induces the rapid aggregation and adhesion of vesicles of 1b, which can be dispersed by the addition of Cu2+ ion unless the concentration of the guest 2 is high (>10 μM). The Cu2+ induced dispersion is reversible in the presence of EDTA, which can induce the re-aggregation of vesicles of 1b.
We further performed a “lipid mixing assay” to verify whether vesicle fusion occurs upon the addition of guest 2. In the fusion assay, 4.6% rhodamine B labeled vesicles of 1b and unlabeled vesicles of 1b were mixed at a ratio of 1:4, as previously described.26 As shown in Fig. 4, when guest 2 was added to the above mixture of vesicles of 1b, no increase in the fluorescence intensity was observed (red trace B). This indicates that no significant fusion of vesicles occurs because if vesicle fusion and mixing of the amphiphilic cyclodextrins occurs, the rhodamine B fluorescence should increase due to a relief of self-quenching upon the dilution of labeled cyclodextrin in the fused bilayer membrane.33 Also, after the addition of Cu2+, no increase in the fluorescence intensity was observed. Instead, a decrease in the fluorescence intensity was observed due to the quenching of rhodamine B fluorescence in the presence of Cu2+ at the vesicle surface (blue trace C). We have also previously noticed the quenching fluorescence of NBD cholesterol embedded in the membrane of CD vesicles in the presence of Cu2+ at the vesicle surface23 and we take this observation as additional evidence for the binding of the complex of guest 2 and Cu2+ at the CD vesicle surface. Upon subsequent addition of EDTA, the fluorescence intensity was recovered due to de-quenching by the removal of Cu2+ from the vesicle surface (green trace D). However, in this case, the fluorescence intensity is slightly higher than the fluorescence intensity of the starting vesicle mixture, which could indicate a small degree of fusion or exchange of labelled CD. A significantly higher fluorescence intensity was observed when the degree of rhodamine B labelling was reduced to 1.0% (corresponding to a 100% hypothetical fusion, brown trace F). A strong increase in fluorescence was observed upon the disruption of vesicles with a surfactant (pink trace E). These observations indicate no fusion of vesicles as a result of the interaction of guest 2 followed by Cu2+.
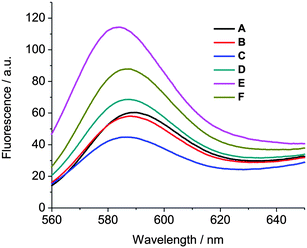 | ||
| Fig. 4 A fusion assay using rhodamine B fluorescence measurements. A) Unlabeled vesicles of 1b (24 μM) + 4.6% labeled vesicles of 1b (6 μM). B) Unlabeled vesicles of 1b (24 μM) + 4.6% labeled vesicles of 1b (6 μM) + 6 μM guest 2. C) Unlabeled vesicles of 1b (24 μM) + 4.6% labeled vesicles of 1b (6 μM) + 6 μM guest 2 + 6 μM Cu2+. D) Unlabeled vesicles of 1b (24 μM) + 4.6% labeled vesicles of 1b (6 μM) + 6 μM guest 2 + 6 μM Cu2+ + 200 μM EDTA. E) Unlabeled vesicles of 1b (24 μM) + 4.6% labeled vesicles of 1b (6 μM) + 6 μM guest 2 + 6 μM Cu2+ + 200 μM EDTA + 20 mM Triton-X 100. F) 1.0% labeled vesicles of 1b. | ||
Intrigued by the results observed for Cu2+, we further investigated the selectivity of divalent metal ions in the dissociation of vesicle clusters of 1b in the presence of guest 2. The addition of Hg2+, Ni2+, Zn2+ or Co2+ (14 μM in each case, which is more than twice the amount of guest 2) to clusters of vesicles of 1b (30 μM) in the presence of guest 2 (6 μM) resulted in a dramatic decrease of both OD600, from ca. 0.35 to ca. 0.05 (Fig. 5A), and the average vesicle diameter, from more than 1000 nm to ca. 150 nm (Fig. 5B). It is reasonable to assume that, similar to the effect shown by Cu2+, the tetramine spacer in guest 2 forms strong tetradentate coordination complexes with Hg2+, Ni2+, Zn2+ or Co2+, which induces the conformational switching of guest 2. It can be seen from Fig. 5A that the addition of Hg2+ or Ni2+ (similar to Cu2+) leads to a fast decrease of OD600 and the original OD600 is recovered within 10 min (black and green traces, respectively), whereas the addition of Zn2+ leads to a moderate decrease of OD600 and the minimum OD600 is obtained within 30 min (red trace) and the addition of Co2+ leads to a slow decrease of OD600 so that it takes over 60 min to reach the minimum OD600 (blue trace). This observation could reflect the slower ligand exchange of these metal ions. On the other hand, the addition of a wide range of metal ions (Mn2+, Fe2+, Ag+, Mg2+, K+, Ca2+, Cd2+ or Pb2+) to clusters of vesicles of 1b (30 μM) in the presence of guest 2 (6 μM) does not result in any significant changes in either OD600 or the average vesicle diameter (ESI Fig. S6†). The tetramine spacer present in guest 2 hardly coordinates to any of these metal ions. Consequently, guest 2 does not change its conformation and, hence, vesicle adhesion is not disrupted by these metal ions.
![The effect of divalent metal ions on the aggregation of vesicles of 1b in the presence of 2. A) Time-dependent measurement of OD600. B) The size distribution of vesicles of 1b according to DLS. Conditions: [1b] = 30 μM, [2] = 6 μM and [M2+] = 14 μM in 10 mM carbonate–bicarbonate buffer (pH 9.0). Guest 2 was added at 3 min and M2+ was added at 30 min.](/image/article/2011/SC/c1sc00422k/c1sc00422k-f5.gif) | ||
| Fig. 5 The effect of divalent metal ions on the aggregation of vesicles of 1b in the presence of 2. A) Time-dependent measurement of OD600. B) The size distribution of vesicles of 1b according to DLS. Conditions: [1b] = 30 μM, [2] = 6 μM and [M2+] = 14 μM in 10 mM carbonate–bicarbonate buffer (pH 9.0). Guest 2 was added at 3 min and M2+ was added at 30 min. | ||
ζ-Potential measurements were used to further understand the interaction of bifunctional guest 2 with vesicles of 1b. As shown in Table 2, the ζ-potential of vesicles of 1b in the absence of guest is ca. − 8.7 mV at pH = 9.0. The negative surface potential of the vesicles is due to presence of oligo(ethylene glycol) residues at the vesicle surface.21 The addition of guest 2 leads to the aggregation of vesicles of 1b and the ζ-potential increases from ca. −8.7 mV to ca. −5.2 mV. The small increase in ζ-potential is due to the presence of guest 2 on the surface of clusters of vesicles of 1b. The subsequent addition of Cu2+ causes the dispersion of vesicles of 1b and the resultant ζ-potential further increases from ca. −5.2 mV to ca. −3.1 mV. The small increase in the ζ-potential is due to the coordination of Cu2+ with guest 2 at the vesicle surface. The addition of excess of EDTA decreases the ζ-potential from ca. −3.1 mV to ca. −5.5 mV. This observation confirms the removal of Cu2+ (but not guest 2) from the vesicle surface by EDTA. Finally, the addition of divalent metal ions, like Hg2+, Ni2+ or Zn+2, also resulted in an increase in the ζ-potential from ca. −5.2 mV to ca. −3.2 mV in the case of Hg2+, ca. −3.8 mV in the case of Ni2+ or −4.1 mV in the case of Zn2+. In contrast, the addition of inactive metal ion Mg2+ did not change the ζ-potential (ca. −5.3 mV). Thus, the ζ-potential results are fully consistent with the results of the OD600 and DLS measurements. In addition we emphasize that the ζ-potential is slightly negative in every case (vesicles, vesicles with guest, vesicles with guest and metal ion). This observation once more confirms that the reversible adhesion of vesicles is mediated by specific molecular recognition, and not by unspecific electrostatic interactions.
| Sample | ζ-Potential/mV | |
|---|---|---|
| pH = 9.0 | pH = 4.0 | |
| 1b | −8.7 | −3.0 |
| 1b + 2 | −5.2 | +6.8 |
| 1b + 2 + Cu2+ | −3.1 | — |
| 1b + 2 + Cu2+ + EDTA | −5.5 | — |
| 1b + 2 + Hg2+ | −3.2 | — |
| 1b + 2 + Ni2+ | −3.8 | — |
| 1b + 2 + Zn2+ | −4.1 | — |
| 1b + 2 + Mg2+ | −5.3 | — |
The interaction of bifunctional guests 3 and 4 (instead of guest 2) with vesicles of β-CD 1b was also investigated by OD600 and DLS measurements. Similar to the observations for 2, the addition of 10 μM of 3 to a dilute solution of vesicles of 1b (30 μM) at pH 9.0 resulted in an increase of OD600 from ca. 0.05 to ca. 0.4 (Fig. 6A) and the average particle size increased from ca. 100 nm to more than 1000 nm (Fig. 6B, red trace). Unlike in the case of guest 2, the addition of metal ions, like Cu2+, Hg2+, Ni2+ or Zn+2, does not lead to any significant changes in either OD600 (Fig. 6A) or the average vesicle diameter (Fig. 6B). It is likely that due to the rigid spacer in guest 3, the metal complexes of guest 3 are energetically less favourable and guest 3 dominates in the chair form, even in the presence of metal ions. Hence, guest 3 persists in an extended conformation that induces the aggregation of vesicles of 1b due to intervesicular binding, even in the presence of metal ions. On the other hand, the addition of as much as 100 μM of cyclic guest 4 to a dilute solution of vesicles of 1b (30 μM at pH 9.0) does not result in any significant changes in either OD600 (Fig. 6A, green trace) or the average vesicle diameter (Fig. 6B, dark yellow trace). These observations indicate that both the p-tert-butylbenzyl groups of cyclic guest 4 bind to the surface of one vesicle and no intervesicular interaction is possible. Thus, the experiments with guests 3 and 4 are fully consistent with the observations made for guest 2: a metal ion induced conformational change resulting in a switch from intervesicular to intravesicular binding can occur in 2, but not in 3 and 4.
![The effect of different metal ions on the aggregation of vesicles of 1b in the presence of 3 and “inert” vesicles of 1b in the presence of 4. A) Time-dependent measurement of OD600. B) Size distribution of vesicles of 1b according to DLS. Conditions: [1b] = 30 μM, [3] = 10 μM, [4] = 100 μM and [M2+] = 14 μM in 10 mM carbonate–bicarbonate buffer (pH 9.0). Guest 3 (or 4) was added at 3 min and M2+ was added at 30 min, with the exception of one measurement, in which Cu2+ was added at 1 min and guest 3 was added at 3 min.](/image/article/2011/SC/c1sc00422k/c1sc00422k-f6.gif) | ||
| Fig. 6 The effect of different metal ions on the aggregation of vesicles of 1b in the presence of 3 and “inert” vesicles of 1b in the presence of 4. A) Time-dependent measurement of OD600. B) Size distribution of vesicles of 1b according to DLS. Conditions: [1b] = 30 μM, [3] = 10 μM, [4] = 100 μM and [M2+] = 14 μM in 10 mM carbonate–bicarbonate buffer (pH 9.0). Guest 3 (or 4) was added at 3 min and M2+ was added at 30 min, with the exception of one measurement, in which Cu2+ was added at 1 min and guest 3 was added at 3 min. | ||
Finally, we investigated the effect of pH on metal ion responsive vesicle adhesion. It is reasonable to assume that a lower pH should result in increased protonation and, hence, a lower metal affinity of the guest molecule. The interaction of bifunctional guest 2 with vesicles of 1b was measured in phosphate buffer at pH = 7.4. Similar to the observations at pH = 9.0, the addition of guest 2 to a dilute solution of vesicles of 1b at pH 7.4 leads to the aggregation of vesicles of 1b, as indicated by an increase in both OD600 from ca. 0.05 to ca. 0.2 and the average vesicle diameter from ca. 100 nm to more than 1000 nm. Unlike at pH 9.0, the addition of 16 μM of divalent metal ions (Cu2+, Hg2+ or Ni2+) does not lead to the complete recovery of either OD600 or the average vesicle diameter (ESI Fig. S7†). This observation can be explained from the fact that the secondary amines of guest 2 are partially protonated at pH 7.4 and thus guest 2 has a weaker affinity to metal ions, which prevents a switch of guest 2 from an extended linear to a bent conformation.
The interaction of guests 2–4 with vesicles of 1b was also measured in acetate buffer at pH 4.0. The addition of as much as 40 μM of guests 2–4 to a dilute solution of vesicles of 1b at pH 4.0 did not lead to any significant changes in either OD600 or the average vesicle diameter (ESI Fig. S8†). At pH 4.0, most of the secondary amines belonging to guests 2–4 are protonated and the surfaces of vesicles decorated with guests 2–4 are positively charged. The ζ-potential confirms the positive surface charge of vesicles after the addition of guest 2. As shown in Table 2, the ζ-potential of vesicles of 1b in the absence of any guest is ca. − 8.7 mV at pH = 9.0 and ca. − 3.0 mV at pH = 4.0. The addition of guest 2 increases the ζ-potential from ca. − 8.7 mV to ca. − 5.2 mV at pH 9.0 and from ca. − 3.0 mV to ca. + 6.8 mV at pH 4.0. These results show that even in the presence of guest 2 the vesicles are still negatively charged at pH 9.0, whereas the vesicles are positively charged at pH 4.0. The positively charged vesicles do not aggregate because of electrostatic repulsion.
Conclusion
A bifunctional p-tert-butylbenzyl dimer 2 with a flexible N,N′-bis(3-aminopropyl)ethylenediamine spacer was used as a non-covalent linker that induces aggregation and adhesion (but not fusion) of host bilayer vesicles composed of amphiphilic β-CDs 1b by the formation of hydrophobic inclusion complexes. Vesicle aggregation is due to intervesicular binding of guest 2, which extends in a linear conformation. However, in the presence of a suitable divalent metal ion (Cu2+, Hg2+, Ni2+, Zn2+ or Co2+) the linker molecule forms a stable tetradentate coordinate complex and dramatically switches its conformation from linear to bent, which brings the p-tert-butylbenzyl groups in close proximity and prevents intervesicular binding, which in turn causes the dispersion of the vesicle clusters. The aggregation of vesicles is re-obtained if the metal ion is sequestered by a strong chelator, such as EDTA. The aggregation of vesicles mediated by a bifunctional p-tert-butylbenzyl dimer 3 with a rigid N,N′-bis(3-aminopropyl)piperazine spacer is not disrupted by the addition of metal ions. Also, vesicles do not aggregate in the presence of cyclic guest 4, which can only bind intravesicularly.In summary, our findings consistently indicate that the adhesion of vesicles can be regulated by a conformational switch of a non-covalent linker from an intervesicular binding mode to an intravesicular binding mode. Thus, a molecular switch can control the aggregation state of an organic colloidal solution.34 In addition, ion-triggered supramolecular aggregation and dispersion of vesicles (or colloids) may be useful as sensor for the detection of metal ions based on turbidity experiments.
Materials and methods
Materials
All of the chemicals used in this study were purchased from Acros Organics (Schwerte, Germany) or Sigma-Aldrich (Taufkirchen, Germany) and used without further purification, unless otherwise noted. Cyclodextrins (CDs) were kindly donated by Wacker Chemie (Burghausen, Germany). All solvents were dried according to conventional methods before use. Commercially available CuCl2, NiCl2, ZnCl2, CoCl2, HgCl2, MnCl2, FeCl2, CaCl2, MgSO4, AgNO3, Cd(NO3)2 and Pb(OAC)2 metal salts were used for the measurements. All aqueous solutions were prepared in Milli-Q water.Synthesis
Amphiphilic α-CD 1a and β-CD 1b were synthesized as reported previously.21 Compounds 2–4 were synthesized as reported in the ESI†. The analytical and spectroscopic data for 2–4 are consistent with their molecular structure. The rhodamine B labeled 1b was synthesized as described in the literature.26 All reactions were carried out in oven-dried glassware and magnetically stirred under an inert gas atmosphere. Analytical TLC was performed on Merck silica gel 60 F254 plates. All compounds were visualized either by UV light or by dipping in basic permanganate solution. Column chromatography was carried out using silica gel 60 (230–400 mesh). 1H-NMR and 13C-NMR spectroscopic measurements were carried out using Bruker ARX 300MHz or 400MHz. Chemical shifts were referenced to internal standards CDCl3 (δ = 7.26 ppm for 1H and 77.0 ppm for 13C) or TMS (δ = 0.00 ppm for 1H and 13C). High-resolution mass spectrometry (HR-MS) was performed by using a MicroTof spectrometer (Bruker).Methods
Unilamellar CD bilayer vesicles of α-CD 1a and β-CD 1b were prepared by extrusion, as described previously.21 In short, several milligrams of 1a (or 1b) taken in approximately 1 ml of chloroform was dried by slow rotary evaporation to yield a thin film in a glass vial. The residual solvent was removed under high vacuum. 10 ml of buffered water was added and stirred overnight. The resulting suspension was repeatedly passed through a polycarbonate membrane with 100 nm pore size in a Liposofast manual extruder. For lipid mixing assays, an appropriate amount of rhodamine B labeled 1b was added to unlabeled 1b prior to the preparation of the vesicles.Isothermal titration calorimetry
ITC measurements were performed using a Nano-Isothermal Titration Calorimeter III (model CSC 5300; Calorimetry Sciences Corporation, London, Utah, USA). ITC measurements were performed in Milli-Q water. A 10 mM solution of β-CD host was titrated into a 0.5 mM solution of guest 2 in the absence and presence of 0.5 mM CuCl2 in 10 mM carbonate/bicarbonate buffer (pH 9.0). Twenty injections (10 μL) were performed with an interval of 300 s. The stirring rate was 300 rpm.UV-vis spectroscopy
Optical density measurements at 600 nm were carried out at in 1.5 ml disposable cuvettes with dimensions 12.5 × 12.5 × 45 mm and a 10 mm path length using a Uvikon 923 double-beam spectrophotometer. The optical density was measured at λ = 600 nm (OD600). Measurements were performed for 30–60 min, unless otherwise noted, with data points collected every 12 s. The freshly prepared vesicles and metal ion solutions were used for each measurement and the measurement procedure was as follows. For example, 1 ml solution of vesicles of 1a or 1b (30 μM) in buffered water were taken in a cuvette and OD600 was measured for 3 min. After 3 min, a few μl of 2-4 (2 mM, prepared in DMSO) were added to the solution in the cuvette (this addition was done with slight mixing within one interval of 12 s) and OD600 was measured for 30 min. After 30 min, a few μl of concentrated metal ion solution in Millipore water was added to the above solution and OD600 was measured for at least 30 min. The same measurements were performed with the other samples following the above procedure. Typical concentrations: [1a] = [1b] = 30 μM, [2] = [3] = [4] = 4–50 μM, [M2+] = 1–100 μM, [Na2H2EDTA] = 30–500 μM, and [α-CD] = [β-CD] = 750 μM.Fluorescence spectroscopy
Fluorescence measurements were carried out with an excitation wavelength of 540 nm by using a JASCO FP-6500 spectrofluorometer in 1 ml disposable cuvettes. Typical concentrations: [4.6% Rhodamine B labeled 1b] = 6 μM, [unlabeled 1b] = 24 μM, [2] = 6 μM, [Cu2+] = 6 μM, [Na2H2EDTA] = 200 μM and [Triton-X 100] = 20 mM in in 10 mM carbonate-bicarbonate buffer (pH 9.0). Freshly prepared vesicles and metal ion solutions were used for each measurement.Dynamic light scattering
DLS measurements were performed using a Malvern Nano-ZS instrument (Malvern Instruments) with low-volume disposable cuvettes kept at 25 °C. The average size of the free vesicles of 1a or 1b, aggregation and dispersion of the vesicles was measured after 30 min after mixing the corresponding components. Typical concentrations: [1a] = [1b] = 30 μM, [2] = [3] = [4] = 4–50 μM range, [MX2] = 1–100 μM and [Na2H2EDTA] = 30–500 μM.Similarly, ζ-potential measurements were also performed using a Malvern Nano-ZS instrument (Malvern Instruments) at 25 °C.
Cryogenic-Transmission Electron Microscopy
Samples for cryo-TEM were prepared by deposition of a few μL of the sample solution (15 min after the addition of the corresponding components) on glow-discharged holey carbon-coated grids (Quantifoil 3.5/1, Quantifoil Micro Tools, Jena, Germany). After blotting the excess liquid at 100% humidity and 22 °C, the grids were vitrified in liquid ethane (Vitrobot, FEI, Eindhoven, The Netherlands). The vitrified specimens were mounted in a liquid nitrogen cooled Gatan 626 cryo-holder (Gatan Inc., Pleasanton, USA) and inserted into the electron microscope. Low-dose images were recorded with a Gatan 4 K slow-scan CCD camera (Pleasanton, CA) on a Philips CM 120 electron microscope (FEI, Eindhoven, The Netherlands) equipped with a LaB6 tip operated at 120 kV. Typical concentrations: [1b] = 1 mM, [2] = 0.5 mM and [Cu2+] = 7.5 mM in 10 mM carbonate–bicarbonate buffer (pH 9.0).Acknowledgements
The authors are grateful for financial support from the Graduate School of Chemistry in Münster (fellowship to S. K. M. N.). We thank Stefan Klein, Jens Mohr and Benjamin Vonhören for their contributions to the optical density and DLS measurements.References
- J. Voskuhl and B. J. Ravoo, Chem. Soc. Rev., 2009, 38, 495–505 RSC.
- H. R. Marsden, I. Tomatsu and A. Kros, Chem. Soc. Rev., 2011, 40, 1572–1585 RSC.
- N. P. Kamat, J. S. Katz and D. A. Hammer, J. Phys. Chem. Lett., 2011, 2, 1612–1623 CrossRef CAS.
- D. Papahadjopoulos, S. Nir and N. Düzgünez, J. Bioenerg. Biomembr., 1990, 22, 157–179 CrossRef CAS.
- I. Tsogas, D. Tsiourvas, G. Nounesis and C. M. Paleos, Langmuir, 2005, 21, 5997–6001 CrossRef CAS.
- I. Tsogas, D. Tsiourvas, G. Nounesis and C. M. Paleos, Langmuir, 2006, 22, 11322–11328 CrossRef CAS.
- S. Chirovolu, S. Walker, J. Israelachvili, F. J. Schmitt, D. Leckband and J. A. Zasadzinski, Science, 1994, 264, 1753–1756 Search PubMed.
- V. Marchi-Artzner, T. Gulik-Krzywicki, M. A. Guedeau-Boudeville, C. Gosse, J. M. Sanderson, J. C. Dedieu and J. M. Lehn, Chem. Phys. Chem., 2001, 2, 367–376 CrossRef CAS.
- F. M. Menger and H. Zhang, J. Am. Chem. Soc., 2006, 128, 1414–1415 CrossRef CAS.
- M. Ma, A. Paredes and D. Bong, J. Am. Chem. Soc., 2008, 130, 14456–14458 CrossRef CAS.
- M. Ma, Y. Gong and D. Bong, J. Am. Chem. Soc., 2009, 131, 16919–16926 CrossRef CAS.
- E. C. Constable, W. Meier, C. Nardin and S. Mundwiler, Chem. Commun., 1999, 1483–1484 RSC.
- A. Richard, V. Marchi-Artzner, M. N. Lalloz, M. J. Brienne, F. Artzner, T. Gulik-Krzywicki, M. A. Guedeau-Boudeville and J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15279–15284 CrossRef CAS.
- V. Marchi-Artzner, M. J. Brienne, T. Gulik-Krzywicki, J. C. Dedieu and J. M. Lehn, Chem.–Eur. J., 2004, 10, 2342–2350 CrossRef CAS.
- S. J. Webb, L. Trembleau, R. J. Mart and X. Wang, Org. Biomol. Chem., 2005, 3, 3615–3617 CAS.
- A. Kashiwada, M. Tsuboi and K. Matsuda, Chem. Commun., 2009, 695–697 RSC.
- To the best of our knowledge, there is only one report that describes the disassembly of gold nanoparticle clusters in the presence of Pb2+. However, this system is not reversible. See: J. Liu and Y. Lu, J. Am. Chem. Soc., 2005, 127, 12677–12683 CrossRef CAS.
- S. C. Harrison, Nat. Struct. Mol. Biol., 2008, 15, 690–698 CAS.
- B. J. Ravoo and R. Darcy, Angew. Chem., Int. Ed., 2000, 39, 4324–4326 CrossRef CAS.
- B. J. Ravoo, J. C. Jacquier and G. Wenz, Angew. Chem., Int. Ed., 2003, 42, 2066–2070 CrossRef CAS.
- P. Falvey, C. W. Lim, R. Darcy, T. Revermann, U. Karst, M. Giesbers, A. T. M. Marcelis, A. Lazar, A. W. Coleman, D. N. Reinhoudt and B. J. Ravoo, Chem.–Eur. J., 2005, 11, 1171–1180 CrossRef CAS.
- C. W. Lim, B. J. Ravoo and D. N. Reinhoudt, Chem. Commun., 2005, 5627–5629 RSC.
- C. W. Lim, O. Crespo-Biel, M. C. A. Stuart, D. N. Reinhoudt, J. Huskens and B. J. Ravoo, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6986–6991 CrossRef CAS.
- F. Versluis, I. Tomatsu, S. Kehr, C. Fregonese, A. W. J. W. Tepper, M. C. A. Stuart, B. J. Ravoo, R. I. Koning and A. Kros, J. Am. Chem. Soc., 2009, 131, 13186–13187 CrossRef CAS.
- J. Voskuhl, M. C. A. Stuart and B. J. Ravoo, Chem.–Eur. J., 2010, 16, 2790–2796 CrossRef CAS.
- J. Voskuhl, T. Fenske, M. C. A. Stuart, B. Wibbeling, C. Schmuck and B. J. Ravoo, Chem.–Eur. J., 2010, 16, 8300–8306 CrossRef CAS.
- S. K. M. Nalluri and B. J. Ravoo, Angew. Chem., Int. Ed., 2010, 49, 5371–5374 CrossRef CAS.
- R. V. Vico, J. Voskuhl and B. J. Ravoo, Langmuir, 2011, 27, 1391–1397 CrossRef CAS.
- F. Bellouard, F. Chubur, N. Kervarec, L. Toupet, S. Triki, Y. Le Mest and H. Handel, J. Chem. Soc., Perkin Trans. 1, 1999, 1, 3499–3505 RSC.
- M. A. Bernardo, F. Pina, E. García-España, J. LaTorre, S. V. Luis, J. M. Llinares, J. A. Ramírez and C. Soriano, Inorg. Chem., 1998, 37, 3935–3942 CrossRef CAS.
- R. J. Motekaitis, A. E. Martell and R. A. Handcock, Coord. Chem. Rev., 1994, 133, 39–65 CrossRef.
- D. K. Cabbiness and D. W. Margerum, J. Am. Chem. Soc., 1969, 91, 6540–6541 CrossRef CAS.
- D. Hoekstra, T. De Boer, K. Klappe and J. Wilschut, Biochemistry, 1984, 23, 5675–5681 CrossRef CAS.
- J. M. Spruell and C. J. Hawker, Chem. Sci., 2011, 2, 18–26 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00422k |
| This journal is © The Royal Society of Chemistry 2011 |