Self-assembly of new fluorescent Pd(II) and Pt(II) 2,7-diazapyrenium-based metallocycles and study of their inclusion complexes and [3]catenanes†‡
Victor
Blanco
,
Marcos D.
García
,
Carlos
Peinador
* and
José M.
Quintela
*
Departamento de Química Fundamental, Universidade da Coruña, Facultad de Ciencias, A Zapateira, s/n, 15008, La Coruña, Spain. Fax: +34 981167065; E-mail: carlos.peinador@udc.es
First published on 8th September 2011
Abstract
New fluorescent square-shaped metallocycles were self-assembled from N-monoalkyl-2,7-diazapyrenium derivatives 1 and 4 and PdII/PtII complexes. Using ligand 1, the metal-directed self-assembly process produced a single metallocycle, while the non-symmetrical salt 4 produced mixtures of regioisomeric PdII and PtII metallocycles upon complexation with the corresponding square-planar cis-complexes. Due to the improved π-deficient character of 1 and 4 compared to related bipyridinium-based ligands, complexation and catenation of the obtained metallocycles with selected electron-rich aromatic substrates produced the corresponding 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 inclusion complexes and [3]catenanes in a highly efficient fashion. This is particularly relevant for metallocycles derived from ligand 4, as complexation and catenation occurs in a regioselective fashion, generating only the supramolecules with the appropriate parallel arrangement of the diazapyrenium subunits in order to maximize the host–guest π-stacking interactions. The potential of the new metallocycles for optical signalling applications is illustrated by analyzing their absorption and emission behaviour upon complexation and catenation.
:2 inclusion complexes and [3]catenanes in a highly efficient fashion. This is particularly relevant for metallocycles derived from ligand 4, as complexation and catenation occurs in a regioselective fashion, generating only the supramolecules with the appropriate parallel arrangement of the diazapyrenium subunits in order to maximize the host–guest π-stacking interactions. The potential of the new metallocycles for optical signalling applications is illustrated by analyzing their absorption and emission behaviour upon complexation and catenation.
Introduction
Metal-directed self-assembly is one of the supramolecular synthetic methodologies that has attracted increasing attention in recent years,1 as it has proven its value in the construction of innumerable supramolecular entities, such as catenanes, rotaxanes and molecular knots.2 The success of this approach is based on two main features: i) the variety of geometries available in coordination chemistry and ii) the process occurring under thermodynamic control, which provides the system with reversibility and self-repairing capabilities. Therefore, the structure of a given supramolecular assembly obtained in this fashion is directly controlled by the chemical and geometrical information encoded within its building blocks.The combination of pyridyl ligands with PdII or PtII centres can be considered to be a paradigmatic example of such a strategy, resulting in the preparation of a great number of 2D and 3D structures (e.g. molecular triangles, squares, pentagons, hexagons, cubes or cages), which have shown important applications as hosts of a variety of substrates or as catalysts.3
Over the last few years, our research group has developed a modification of this methodology, with its most important feature being the use of bidentate ligands based on N-monoalkyl-4,4′-bipyridinium scaffolds instead of neutral bipyridine derivatives. The combination of these π-deficient ligands with PdII and PtII metal centres takes advantage of the thermodynamic control and high yields associated with metal-directed self-assembly and, on the other hand, ensures a superior π-deficient character of the resulting metallocycles. These can subsequently act as efficient hosts of aromatic substrates with π-donor properties by establishment of π–π interactions, which has resulted in the efficient synthesis of inclusion complexes, [2] and [3]catenanes and molecular knots.4,5
This strategy has also been exploited in the regioselective self-assembly of [3]catenanes and inclusion complexes, proving that π–π interactions are responsible for the observed regioselectivity.6
In this context, 2,7-diazapyrene is known to show a superior electron acceptor ability compared to 4,4′-bipyridine so, consequently, N-monoalkyl-2,7-diazapyrenium ligands have been used by our research group as improved π-deficient bidentate ligands in the synthesis of catenanes or as receptors for aromatic guests.7 These diazapyrenium-based hosts have proven to be very efficient, not only because of the stronger host–guest π⋯π interactions, but also as a result of the increased hydrophobic nature of the resulting metallocycle, especially in an aqueous environment – a fact that correlates with the extended aromatic surface of the heterocycle. Furthermore, the well-known luminescent properties of 2,7-diazapyrene and its alkylated derivatives are particularly attractive8 due to the potential of the resulting diazapyrene-containing metallocycles in the field of optical molecular recognition of relevant analytes,9,10 or as luminescent π-acceptor subunits involved in pseudorotaxane, rotaxane or catenane structures (acting as probes to reveal the occurrence of CT interactions, as well as to monitor structural changes induced by external stimuli).11
Continuing our investigations into PdII/PtII-directed self-assembly of supramolecular architectures based on N-monoalkyl-2,7-diazapyrenium units, we report herein the synthesis of two [3]catenanes assembled from the fluorescent PdII metallocycle 3 containing a symmetric bis(N-monoalkyl-2,7-diazapyrenium) ligand 1 (Scheme 1) and the regioselective catenation and recognition processes of new fluorescent dinuclear PdII and PtII metallocycles 6a,b/7a,b based on the non-symmetric N-monoalkyl-2,7-diazapyrenium ligand 4 (Scheme 1).12 Furthermore, the potential of these new metallocycles for the optical recognition of binding events is illustrated through the study of their absorption and emission behaviour upon complexation or catenation with electron-rich aromatic systems.
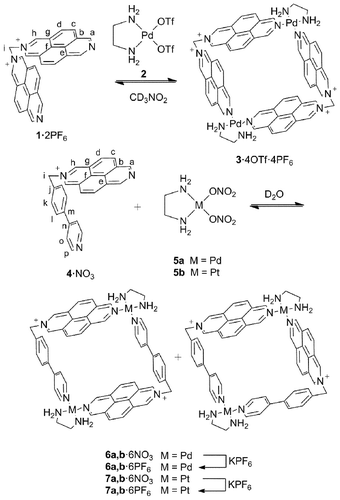 | ||
| Scheme 1 The synthesis of metallocycles 3, 6a,b and 7a,b. | ||
Results and discussion
Synthesis of ligands and self-assembly of metallocyles
Ligands 1 and 4 were obtained by N-alkylation of diazapyrene with dibromomethane or 4-(4′-chloromethylphenyl)pyridine as their hexafluorophosphate (1·2PF6 and 4·PF6) or nitrate (4·NO3) salts after counterion exchange. While ligand 4 was found to be thermally stable, 1 decomposes in aqueous solution (even at room temperature), restricting its use to organic media where it shows higher stability. Yellow crystals of 1·2PF6 suitable for single crystal X-ray diffraction studies were obtained by vapour diffusion of diethyl ether into an acetonitrile solution of the ligand.13 The crystal structure confirms the two diazapyrenium moieties bound to the same methylene group, with a NCN angle of 109° (see the supporting information‡).The addition of 1 equiv. of PdII complex 2 to a 2.5 mM solution of 1·2PF6 in CD3NO2 at room temperature gave rise to metallocycle 3·4OTf·4PF6 (Scheme 1). The 1H and 13C NMR spectra, together with the 2D (COSY, HMBC, HSQC) NMR experiments, provide good evidence for the formation of the supramolecule. Consequently, the 1H and 13C signals of positions a, b and e are shifted downfield as a result of the coordination of the ring to the metal centre (Table 1).
| a/a′ | b/b′ | c/c′ | d/d′ | e/e′ | f/f′ | g/g | h/h′ | i | j | k | l | m | n | o | p | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Hydrogen and carbon labels (a–p) are defined in Scheme 1. b The δ values are compared to those of the free ligand 1·2PF6, 4·PF6 or 4·NO3. c The δ values are compared to those of the free metallocycle. d Not assigned. e Only the data for (16)2⊂6a·6NO3 is presented; the values for the remaining inclusion complexes are similar to this. | |||||||||||||||||
| 3·4OTf·4PF6b | 1H | 0.30 | — | −0.22 | −0.08 | — | — | — | 0.07 | −0.02 | |||||||
| 13C | 0.5 | 2.8 | −1.6 | 2.5 | 0.8 | −1.9 | 0.1 | 1.8 | 0.2 | ||||||||
| 3(BPP34C10)2·4OTf·4PF6c | 1H | 0.28/0.11 | — | 0.24/0.17 | 0.20/0.29 | — | — | — | 0.29/0.20 | 0.00 | |||||||
| 13C | 0.5/0.5 | 0.6/0.2 | −0.9/−0.6 | −0.3/1.2 | −0.3/1.4 | 0.5/−0.8 | −0.1 | ||||||||||
| 6a,b·6PF6b | 1H | 0.30/0.12 | — | −0.14 | −0.03 | — | — | — | 0.01 | −0.11 | — | −0.21 | −0.36 | — | — | −0.34 | −0.08/0.06 |
| 13C | 0.3 | 2.4 | −1.2 | 2.4 | 1.0 | −1.4 | 0.1 | 1.5 | 0.5 | 0.7 | −0.4 | −0.5 | −1.5 | −1.7 | 0.9 | 5.6 | |
| 6a(BPP34C10)2·6PF6c | 1H | −0.31 | — | −0.36 | −0.31 | — | — | — | −0.07 | −0.1 | — | 0.29 | 0.09 | — | — | 0.14 | 0.5 |
| 13C | −0.5 | −0.4 | −0.6 | −0.4 | −1.4 | −1.1 | −1.1 | 0 | −0.4 | −0.1 | 0.9 | −0.2 | 0.4 | 0.1 | 0.7 | 0.6 | |
| 6a,b·6NO3b | 1H | 0.64/0.47 | — | 0.14 | 0.19 | — | — | — | 0.20 | 0.08 | — | −0.02 | −0.19 | — | — | −0.05 | 0.16/0.33 |
| 13C | 0.5 | 1.8 | −2 | 1.5 | 0.4 | −1.2 | −0.2 | 0.7 | 0 | 0.2 | −0.7 | −0.9 | −2 | −0.2 | 0.9 | 3.7 | |
| (16)2⊂6a·6NO3e | 1H | −0.28 | — | −0.53 | −0.69 | — | — | — | −0.45 | −0.25 | — | 0.27 | 0.39 | — | — | 0.48 | 0.59 |
| 13C | −0.7 | −0.7 | −0.8 | −1.2 | −1.5 | −2.5 | −0.9 | −1.7 | −0.2 | 0.5 | 0.5 | 0.4 | −0.1 | −0.2 | 0.4 | 0.6 |
| 3(9)2 | 6a(8)2 | |
|---|---|---|
| a In common: Refinement method, full-matrix least-squares on F2. Wavelength 0.71073 Å (Mo-Kα). Temperature 100(2) K. Absorption correction method: multi-scan.20 | ||
| Chemical formula | C74H76F15N8O22PPdS3 | C122H144F18N14O46Pd2S6 |
| Formula mass | 1947.98 | 3289.67 |
| Crystal system | triclinic | monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| a/Å | 12.518(5) | 12.0255(5) |
| b/Å | 18.378(5) | 20.4941(8) |
| c/Å | 20.119(5) | 28.0429(12) |
| α/° | 97.003(5) | 90 |
| β/° | 102.035(5) | 93.676(2) |
| γ/° | 107.893(5) | 90 |
| V/Å3 | 4221(2) | 6897(5) |
| Z | 2 | 2 |
| D c/Mg m−3 | 1.533 | 1.584 |
| μ/mm−1 | 0.422 | 0.467 |
| F(000) | 1992 | 3384 |
| θ limits | 1.74 to 28.41° | 2.06 to 28.37° |
| hkl limits | −16,16/−24,24/−26,26 | −16,16/−27,27/−37,37 |
| No. of reflections measured | 96651 | 155604 |
| No. of independent reflections | 21129 | 17198 |
| R int | 0.0717 | 0.0594 |
| R [I > 2σ(I)] | R 1 = 0.1162 | R 1 = 0.1064 |
| wR2 = 0.3326 | wR2 = 0.2566 | |
| R (all data) | R 1 = 0.1940 | R 1 = 0.1271 |
| wR2 = 0.3874 | wR2 = 0.2716 | |
| Goodness of fit on F2 | 1.299 | 1.119 |
Due to the non-symmetrical character of the diazapyrenium derivative 4·NO3, the addition of 1 equiv. of complex 5a to a 5 mM solution of this ligand in D2O at 60 °C14 resulted in the expected formation of two regioisomeric square-shaped metallocycles (6a,b·6NO3, Scheme 1) approximately in a 1:1 ratio, as can be seen from inspection of the 1D and 2D NMR spectra. Very similar results were obtained in CD3NO2 for metallocycles 6a,b·6PF6 (Scheme 1), which were obtained after counterion exchange of 6a,b·6NO3. In both cases, similar shifts to those described for 3·4OTf·4PF6 can be observed (Table 1). In contrast to related systems based on phenylenepyridinium or trans-1,2-bis-(pyridin-4-yl)ethylene motifs,6 where both isomers display the same chemical shifts for all of their corresponding 1H and 13C signals, different resonances associated with the nuclei at positions a and p can be observed for 6a and 6b (ΔδHa = 0.17 ppm and ΔδHp = −0.17 ppm)15 as a result of the higher shielding character of the diazapyrenium moiety when compared with the phenylpyridinium subunit. The composition of the mixture remained unchanged in the concentration ranges 2.5–0.25 mM (D2O) and 5.0–0.25 mM (CD3NO2) (see the supporting information‡).
In both systems, the diffusion coefficients obtained from the DOSY (Diffusion-Ordered NMR SpectroscopY)16 experiments for metallocycles 3·4OTf·4PF6 and 6a,b·6NO3 showed that each metallocycle is larger than its components as the resulting diffusion coefficients are lower than the values obtained for the free ligands in separate experiments. Moreover, the signals from the ligands and palladium complexes displayed the same diffusion coefficients, implying that the components of each metallocycle diffuse as a whole (see the supporting information‡).
Nevertheless, it should be considered that the known lability of the Pd–N bond in these compounds makes their isolation and accurate structural characterization difficult. The use of the more inert Pt–N bond should allow us to surpass those inconveniences because of its opportune inertness at low temperature and appropriate lability when the temperature is increased.17 Thus, a 1:1 mixture of both regioisomers could be obtained upon heating a 5 mM solution of 4·NO3 and the PtII complex 5b in water at 100 °C for 8 d (a higher temperature and reaction times were needed to assure the thermodynamic control over the self-assembly). The hexafluorophosphate salts of the PtII metallocycles 7a,b were isolated in 92% yield and characterized by NMR spectroscopy and ESI-HRMS. Mass spectrometry showed peaks resulting from the loss of between two and five hexafluorophosphate anions, with the experimental isotopic distribution of these fragments fitting very well with the theoretical calculation, and supporting therefore the proposed structure of the PtII squares 7a,b (see the supporting information‡).
Self-assembly of catenanes derived from ligand 1
Our catenation strategy involves the threading of crown ethers, BPP34C10 (8) and DN38C10 (9), with the π-deficient ligand 1·2PF6, followed by linkage of the resulting pseudorotaxanes with the metal complex 2, affording the [3]catenanes. Thus, some experiments were proposed to provide insights into the interactions between ligand 1·2PF6 and the cyclophanes employed, as were done in the corresponding previously reported bipyridinium system.4fThe 1H NMR spectra of equimolar solutions of ligand 1·2PF6 and cyclophanes 8 and 9 in CD3NO2 showed some changes when compared to those of the individual species. Thus, when the macrocycle used is BPP34C10, the singlet of the aromatic protons on the cyclophane is shifted to lower frequencies (Δδ = − 0.90 ppm) and, conversely, some of the aromatic signals of the ligand are slightly shifted upfield (ΔδHh = −0.10 ppm and ΔδHd = −0.12 ppm), suggesting the establishment of π-donor/π-acceptor interactions between the electron-rich cyclophane and the π-deficient ligand.
When DN38C10 was used, more pronounced shiftings, but with the same sign, are observed both for ligand (ΔδHh = − 0.14 ppm; ΔδHd = − 0.29 ppm) and cyclophane (ΔδH2 = − 0.54 ppm; ΔδH3 = − 1.04 ppm and ΔδH4 = − 1.30 ppm), which is a fact related to the higher donor character of naphthalene moieties that results in stronger interactions with the diazapyrenium moieties.
Moreover, after the addition of 1 equiv. of the corresponding cyclophane, the UV-vis spectra of ligand 1·2PF6 in CH3CN showed new intermolecular charge-transfer absorption bands, resulting in the change in the colour of the solutions to yellow (8) and red (9). Although in both cases the new band is partially hidden by an intense absorption band related to the diazapyrenium moiety (centered at 428 nm), it was possible to estimate the binding constant between 1·2PF6 and DN38C10 using the titration method (Ka = 4268 ± 124 M−1 in CH3CN). The titration curve fitted perfectly to the 1:1 binding isotherm (see the supporting information‡).18
The higher Ka value for the interaction between 1 and 9 in comparison to its bipyridinium analogue (Ka = 1292 ± 47 M−1 in CH3CN)4f corroborates the improved π-acceptor character of N-monoalkyldiazapyrenium derivatives.
The addition of 1 equiv. of cyclophane BPP34C10 (8) to a 2.5 mM solution of 1·2PF6 and (en)PdOTf2 (2) in CD3NO2 promoted the same colour change observed in the corresponding pseudorotaxane as a result of the charge-transfer absorption band.
The 1H NMR spectrum at room temperature showed well-defined signals, which were fully assigned by the 1H, 13C and 2D NMR experiments. The exchange between the inside and alongside hydroquinol rings of 8 is slow on the 1H NMR timescale, resulting in all of the –OCH2– groups being asynchronous. Moreover, the aromatic signal of the alongside dioxoaryl rings is characteristically shifted upfield (Δδ = −1.31 ppm). Furthermore, eight signals for the aromatic protons of the metallocycle (4 singlets for the α protons and 4 doublets for those in the γ position) and two different resonances for the ethylendiamine protons were observed; a splitting pattern consistent with a slow exchange between the diazapyrenium (DIAZ) systems orthogonal (DIAZort) to the inside dioxoaryl rings of 8 and the parallel (DIAZpar) to those. The protons of the parallel diazapyrenes are shifted upfield from those of the free metallocycle 3 as a result of the shielding effect of cyclophane 8 (ΔδHh′ = −0.20 ppm, ΔδHd′ = −0.29 ppm and ΔδHc′ = −0.17 ppm).
On the contrary, the protons of DIAZort are shifted downfield, suggesting [C–H⋯π] interactions between the aromatic rings of the diazapyrene units and the protons of the inside hydroquinol rings (ΔδHa = 0.28 ppm, ΔδHc = 0.24 ppm and ΔδHh = 0.29 ppm).
In the case of the self-assembly process involving cyclophane DN38C10 (9), the addition of 1 equiv. of 9 to a 2.5 mM equimolar solution of ligand 1·2PF6 and palladium complex 2 in CH3NO2 also produced a change in the colour of the solution from yellow to red, with the absorption spectrum showing a new charge-transfer centered at λmax = 512 nm.
The 1H NMR of this mixture in CD3NO2 at room temperature exhibited a high complexity that prevented its complete analysis. The reason is the high strength of the π⋯π interactions, resulting in all of the circumrotation movements being slow on the NMR timescale at room temperature, with the catenane subunits DIAZpar, DIAZort, inside NAPHin and alongside NAPHout naphthalene (NAPH) rings being non-equivalent. Moreover, the introduction of two 1,5-naphthalene units into the metallocycle results in the inequivalence of the 16 protons of DIAZpar and DIAZort and also of the 6 protons of the naphthalene rings. These facts, in addition to the possible orientations of the two π-donor components inside the metallocycle cavity, can explain the complexity of the spectrum.
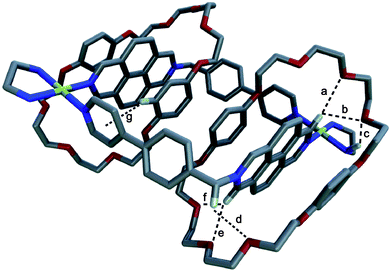
Fortunately, single crystals of catenane 3(DN38C10)2 suitable for single crystal X-ray crystallography were obtained by slow diffusion of diethyl ether into a solution of ligand 1·2PF6, (en)PdOTf2 and DN38C10 in acetonitrile (see Fig. 1 and Table 2). As shown in Fig. 2, the solid state structure displays the expected [3]catenane with the octacationic metallocycle interlocked by two molecules of cyclophane with a parallel π-stacking disposition of six aromatic systems: NAPHout/DIAZpar/NAPHin/NAPHin/DIAZpar/NAPHout, being the interplanar distances 3.35, 3.35 and 3.45 Å, respectively.19 The metallocycle in the P structure of 3(DN38C10)2·6OTf·2PF6 has a rhomboidal shape, with the Pd atoms exhibiting the typical square planar coordination geometry (Pd–N distances: 2.037 and 2.041 Å; N(DIAZ)–Pd–N(DIAZ) and N(en)-Pd–N(en) angles: 88° and 84°). With the N–CH2–N angle being 109.7°, in order to maintain the square planar geometry around the metal centres, both DIAZort units are twisted, being the angle defined by Pd–N and N+–CH2 vectors 13°. In contrast, the DIAZpar subunits keep their planar structure.
![Capped sticks projection of the crystal structure of catenane 3 (DN38C10)2·6OTf·2PF6 showing [C–H⋯O] (a–c, e–g) and [N–H⋯O] (d) bonds. [H⋯O] and [C⋯O] or [N⋯O] distances and [C–H⋯O] angles: a 2.21, 3.12 Å, 152°; b 2.22, 3.12 Å, 153°; c 2.91, 3.78 Å, 157°; d 2.54, 3.32 Å, 143°; e 2.52, 3.12 Å, 121°; f 2.50, 3.42 Å, 164°; g 2.37, 3.31 Å, 168°. Solvent molecules, counterions and remaining hydrogen atoms are omitted for clarity. Colour scheme: C, grey; O, red; N, blue; H, light grey; Pd, yellow.](/image/article/2011/SC/c1sc00508a/c1sc00508a-f1.gif) | ||
| Fig. 1 Capped sticks projection of the crystal structure of catenane 3 (DN38C10)2·6OTf·2PF6 showing [C–H⋯O] (a–c, e–g) and [N–H⋯O] (d) bonds. [H⋯O] and [C⋯O] or [N⋯O] distances and [C–H⋯O] angles: a 2.21, 3.12 Å, 152°; b 2.22, 3.12 Å, 153°; c 2.91, 3.78 Å, 157°; d 2.54, 3.32 Å, 143°; e 2.52, 3.12 Å, 121°; f 2.50, 3.42 Å, 164°; g 2.37, 3.31 Å, 168°. Solvent molecules, counterions and remaining hydrogen atoms are omitted for clarity. Colour scheme: C, grey; O, red; N, blue; H, light grey; Pd, yellow. | ||
![Observed (top) and theoretical (bottom) isotopic distribution for: (a) Fragment [7a(8)2 – 4PF6−]4+ (left, exp. m/z = 654.2035, theoretical m/z = 654.2044). (b) Fragment [7a(9)2 – 3PF6−]3+ (right, exp. m/z = 987.2800, theoretical m/z = 987.2816).](/image/article/2011/SC/c1sc00508a/c1sc00508a-f2.gif) | ||
| Fig. 2 Observed (top) and theoretical (bottom) isotopic distribution for: (a) Fragment [7a(8)2 – 4PF6−]4+ (left, exp. m/z = 654.2035, theoretical m/z = 654.2044). (b) Fragment [7a(9)2 – 3PF6−]3+ (right, exp. m/z = 987.2800, theoretical m/z = 987.2816). | ||
The NAPHin moieties are not completely eclipsed but displaced by 1.52 Å (distance between centroids = 3.77 Å). The angle between the mean plane of the metallocycle and the symmetry plane of NAPHin is 88° and the associated O–O vectors are tilted 66° again with regard to the plane of the metallocycle.
[C–H⋯π] interactions were detected between H–4 and H–8 (para position to the O) of the cyclophane and the pyridine rings of DIAZort ([H⋯π]/[C⋯π] distances and [C–H⋯π] angles: 2.70 Å, 3.48 Å, 140°; 2.78 Å; 3.59 Å and 143°).
As usual in these systems, the structure is also stabilized by means of [C–H⋯O] and [N–H⋯O] hydrogen bonds between the α–CH diazapyrenium, methylene and NH2 hydrogens with the oxygen atoms of the polyether chains of 9 (Fig. 2).
Regioselective self-assembly of inclusion complexes and catenanes from ligand 4
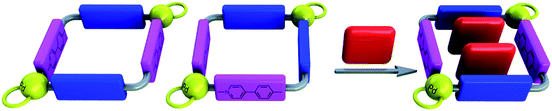
In recent times, our research group has turned its attention to the study of regioselective catenation processes using non-symmetrical ligands derived from 4,4-bipyridinium or trans-1,2-bis-(pyridin-4-yl)ethylene.6 As the regioselectivity observed in those systems was found to be promoted by π–π interactions, structures derived from the diazapyrenium-based ligand 4 are expected to be more efficient, displaying higher regioselectivities than those explored previously.Regioselective catenation
Although the combination of metallocycles 6a,b with BPP34C10 (8) could give rise to three reasonable catenane structures (two [3]catenanes derived from metallocycle 6a, depending on whether the internal dioxoaryl groups are parallel or perpendicular to the diazapyrenium moieties, and a third [3]catenane derived from 6b), only the square 6a presents the correct disposition of the π-acceptor moieties in order to maximize the most efficient π–π stacking interactions established with aromatic substrates, namely, those involving parallel diazapyrenium/phenylenepyridinium moieties.The 1H NMR spectrum, recorded upon the addition of 1 equiv. of BPP34C10 to a 5 mM solution of 6a,b·6PF6 in nitromethane, shows only one set of signals for both the macrocycle and the cyclophane, which is attributable to one of the potential [3]catenanes. The 1:2 integration ratio between the aromatic signals of the metallomacrocycle and 8 indicates that for each hexacationic square there are two encircling cyclophane rings, which is in keeping with the expected molecular structure of a [3]catenane. The diffusion coefficients obtained from the DOSY experiments of metallocycles 6a,b·6PF6 and catenane showed the latter being significantly larger than its components, with the signals of the metallocycle and cyclophane having the same diffusion coefficients, indicating therefore that these components diffuseing as a whole (see the supporting information‡). The protons and carbons of the diazapyrenes are shifted upfield (Table 1), with regard to those of the free metallocycle 6a,b, as a result of the shielding effect of cyclophane 8. In contrast, the protons and carbons in the pyridine ring of the 4-phenylpyridine (PHPY) moiety are shifted downfield (Table 1), suggesting weak [C–H⋯π] interactions between the pyridine and the protons of the inside hydroquinol rings (see also Fig. S52 in the supporting information‡). The sign and magnitude of these shifts clearly indicate that the hydroquinol rings inside the cavity are parallel to the 2,7-diazapyrenium systems and orthogonal to the phenylpyridines, resulting in the formation of only one of the two isomeric catenanes of 6a. Interestingly, the exchange between the inside and alongside hydroquinol rings in the [3]catenane is slow at room temperature. The aromatic signal of HQin is located at δ = 5.39 ppm (Δδ = −1.41 ppm), while the signal of HQout, assigned by an EXSY (EXchange SpectroscopY) correlation, is more shielded (δ = 3.44 ppm, Δδ = −3.36 ppm) (see the supporting information‡).
Analogous PtII catenanes could be obtained by heating a 5 mM solution of metallocycles 7a,b·6PF6 in CH3NO2 at 100 °C after adding a stoichiometric amount of cyclophane 8 or 9, again exploiting the “molecular lock” strategy (see Scheme 2).16 The 1H and 13C NMR spectra showed the presence of unique species, with spectroscopic characteristics very similar to those corresponding to the obtained PdII catenanes, suggesting the regioselective entanglement of metallocycle 7a to afford 7a(8)2·6PF6 and 7a(9)2·6PF6. Mass spectrometry supported the structure of the [3]catenane, showing peaks resulting from the loss of between three and five hexafluorophosphate anions. A comparison of the calculated isotopic distributions for the charged species and the experimental data confirmed the validity of both structures (Fig. 2).
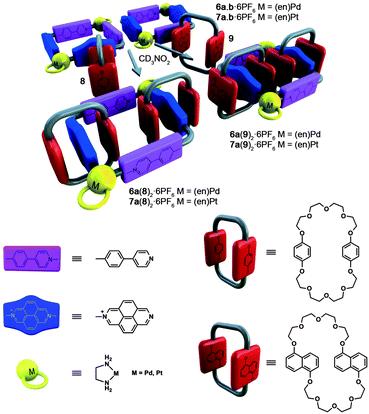 | ||
| Scheme 2 Regioselective self-assembly of catenanes 6a(8,9)2·6PF6 and 7a(8,9)2·6PF6. | ||
X-ray diffraction analysis of single crystals of catenane 6a(BPP34C10)2, obtained by a slow diffusion of diethyl ether into a solution of ligand 4·2PF6, 2, and 8 in nitromethane, shows the expected [3]catenane with the metallocycle interlocked by two molecules of cyclophane (Fig. 3 and Table 2).
 | ||
| Fig. 3 Capped sticks projection of the crystal structure of 6a(BPP34C10)2·6OTf. Solvent molecules, counterions and hydrogen atoms not involved in hydrogen bonding are omitted for clarity. Colour scheme as in Fig. 2. | ||
The metallomacrocycle has the usual rhomboid shape, with dimensions 16.49 × 12.94 Å2 (Pd–Pd and CH2–CH2 distances, respectively). The Pd–N distances are 2.018 and 2.032 Å and the N(DIAZ)–Pd–N(DIAZ) and N(en)–Pd–N(en) angles are 88° and 84°, respectively. The N–CH2–N angle is 110.7°. A π-stacking disposition of six aromatic systems can be observed: HQout/DIAZpar/HQin/HQin/DIAZpar/HQout, being the interplanar distances 3.41, 3.49 and 3.40 Å, respectively. As in 3(9)2, DIAZpar rings are not perfectly parallel to the inside nor alongside rings of the cyclophane, the corresponding angles between their planes being 5°, 5°, 0°.
HQin units are not completely eclipsed but are displaced in 1.89 Å (dcent = 3.89 Å) and they are almost orthogonal respect to the mean plane of the metallocycle (89°) while the associated O–O vectors are tilted by 61° with respect to this plane.
As in previously reported structures, further stabilization is achieved by means of [C–H⋯π] interactions between the hydrogens of HQint and the pyridine ring ([H⋯π] and [C⋯π] distances and [C–H⋯π] angle: 2.86 Å, 3.77 Å, 161°, respectively). [C–H⋯O] and [N–H⋯O] hydrogen bonds between the oxygen atoms of the polyether chains and the hydrogens of the methylene groups, amino groups or those in the α position to the pyridine nitrogens can also be found within the crystal lattice ([H⋯O] and [C⋯O] or [N⋯O] distances and [C–H⋯O] angles: a 2.80, 3.75 Å, 172°; b 2.70, 3.30 Å, 122°; c 2.14, 2.90 Å, 139°; d 2.60, 3.37 Å, 130°; e 2.50, 3.40 Å, 159°; f 2.54, 3.25 Å, 132°).
Regioselective self-assembly of inclusion complexes from metallocycles 6a,b
In addition to the study of the regioselective catenation processes described below, the self-assembly of inclusion complexes with a series of aromatic guests was also investigated.The addition of electron-rich aromatic guests 10–21 to a solution of 6a,b·6NO3 in D2O is expected to promote a partial or total reorganization of the “incorrect” metallocycle 6b·6NO3, yielding the 1:2 inclusion complexes of 6a·6NO3 (Table 3).6 Obviously, the mechanism requires (as in the case of catenanes), the dissociation of the N atoms from the palladium centres. The results are summarized in Table 3.
|
|
|
|||
|---|---|---|---|---|
|
|
|
|
||
|
a The number of equivalents is defined here as half the relation between the moles of guest and host, as the inclusion complexes and catenanes formed display a 1:2 stoichiometry.
b Data for inclusion complexes involving ligands containing bipyridinium or bis-(pyridin-4-yl)ethylene moieties taken from refs 6a and 6b, respectively.
c No complete reorganization towards one of the regioisomers was achieved, with the system showing a relative population of two isomers that was no longer dependent on the amount of guest.
|
||||
| BPP34C10 | 5 | 10 | 1 | |
| DN38C10 | 2 | 1 | ||

|
10 R = H | 7 | ||
| 11 R = (CH2)2O(CH2)2OH | 2 | |||

|
12 R = H | |||
| 13 R = (CH2)2O(CH2)2OH | 2 | |||

|
14 R = H | 2 | ||
| 15 R = (CH2)2O(CH2)2OH | 1 | 1 | 1 | |

|
16 R = H | 2 | 2 | 2 |
| 17 R = (CH2)2O(CH2)2OH | 2 | 3 | 1 | |

|
18 R = H | 2 | ||
| 19 R = (CH2)2O(CH2)2OH | 1 | 1 | ||

|
20 R = H | 1 | ||
| 21 R = (CH2)2O(CH2)2OH | 2 | 3 | 1 | |





Almost all of the substrates employed were able to promote a complete reorganization to the “correct” isomer. A comparison with similar macrocycles containing bipyridinium6a or bis-(pyridin-4-yl)ethylene6b shows that, in general, the reorganization process is easier with 6a,b as it is achieved with more substrates and/or fewer equivalents than the previously reported bipyridinium-based systems.
These results are a positive proof of its higher efficiency as receptors of the systems containing diazapyrenium moieties. In addition to their stronger π-acceptor character, their hydrophobicity dramatically enhances their ability to act as hosts of aromatic guests, due to the stronger hydrophobic interactions established.
Most of the spectroscopic characteristics of 6a(10–21)2·6NO3 are quite similar to those of the previously described catenanes, with the corresponding 1H and 13C NMR spectra of the different inclusion complexes showing the characteristic upfield shifts of the signals of the DIAZ systems and the slight downfield shifts of the signals of the phenylenepyridine moiety (Table 1), suggesting the expected insertion of the naphthalene/phenylene systems in a parallel fashion with regard to the DIAZs.
The DOSY experiment of a solution of 6a,b·6NO3 and 17 in D2O showed that the diffusion coefficient for the signals corresponding to both the host and guest are very similar, indicating that the complex diffuses as a whole, showing a stronger binding between the host and guest than in previous systems where the diffusion coefficient of the guest was intermediate between the host and the free guest.
UV-vis and fluorescence studies
In order to test the convenience of the diazapyrenium-containing metallocycles 3 and 6a,b/7a,b as π-deficient fluorescent probes for electron-rich aromatic substrates, the absorption and emission properties of ligands, derived metallocycles, inclusion complexes and catenanes were inspected.21Absorption spectra
The UV-vis absorption spectra of ligands 1·2PF6 and 4·X2 (X = NO3, PF6) and macrocycles 3·4OTf·4PF6, 6a,b·6NO3 and 7a,b·6PF6 show, either in organic or water solution,22 two intense structured absorption bands corresponding to electronic transitions to the first and second ππ* excited states, respectively, as is typical for these systems.23The λmax of absorption for ligand 4 (340 nm and 412 in H2O and CH3CN and 416 nm in CH3NO2) are lower than those of ligand 1·2PF6 (λmax = 427.5 nm in CH3CN and 430.5 nm in CH3NO2), indicating that the electronic transitions are less energetic in the latter. It is also worth noting that the absorption spectra of the metallocycles exhibit the characteristic bands of the ligand units.
In addition to the structured absorption bands that are characteristic of the diazapyrenium moieties, the UV-vis spectra of the inclusion complexes 6a(11)2 and 6a(15)2, as well as catenanes 7a(8)2 and 7a(9)2, show the charge-transfer bands related to π–π interactions (vide supra) in the range 370–550 nm (Fig. 4).
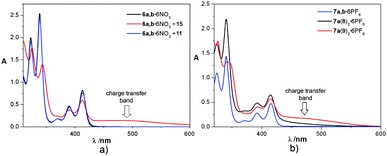 | ||
| Fig. 4 The UV-vis absorption spectra of: a) metallocycle 6a,b·6NO3 and inclusion complexes 6a(11)2·6NO3 and 6a(15)2·6NO3 (H2O); b) metallocycle 7a,b·6PF6 and catenanes 7a(8)2·6PF6 and 7a(9)2·6PF6 (CH3CN). | ||
Emission
The emission spectrum of ligand 4 displays a strong fluorescence band at room temperature, both in organic media (λmax = 431.5 nm in CH3CN and λmax = 428 nm in CH3NO2) or aqueous solution (λmax = 429 nm) (Fig. 5).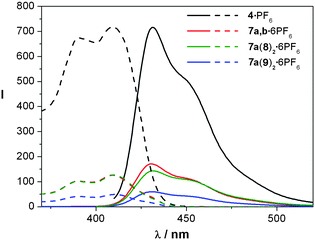 | ||
| Fig. 5 Emission (solid lines) and excitation spectra (dashed lines) (CH3NO2, 293 K) of ligand 4·PF6, metallocycle 7a,b·6PF6 and catenanes 7a(8)2·6PF6 and 7a(9)2·6PF6. | ||
In contrast, ligand 1 shows a much weaker fluorescence. This difference between the systems can be explained, as in other diazapyrenium derivatives, by an intersystem crossing to a excited triplet state, Tn, which allows a non-radiative decay to the ground triplet state, T1.23b As was observed in the UV-vis spectra, the emission band of this ligand is shifted to higher wavelengths (λmax = 460 nm) in comparison to that of 4.
When the fluorescence studies were carried out with the corresponding metallocycles, the resulting spectra showed the same diazapyrenium emission bands as those from the ligands, but with decreased intensity due to the quenching promoted by the metal centres, which induce an enhanced spin–orbit coupling, allowing efficient intersystem crossing.24
In the cases of catenanes 7a(8)2 and 7a(9)2, their excitation spectra match the corresponding absorption spectra. The intensity of the fluorescence is much lower than that of the corresponding metallocycles (Fig. 5). This fact is related to the presence of low-energy excited states linked to the charge-transfer processes, which provide fast non-radiative relaxation pathways from the excited energy levels of the diazapyrenium units.23a The emission intensity diminishes in proportion to the strength of the π-donor/π-acceptor interactions established between cyclophanes 8 and 9 and the diazapyrenium units of the metallocycles as charge-transfer processes are more intense. For this reason, the fluorescence of the catenanes derived from 9 is weaker than that exhibited by the interlocked structures, which incorporate cyclophane 8, as naphthalene moieties establish stronger π-interactions than the phenylene-derived systems. Similarly, this stronger fluorescence quenching effect was observed when substrates 11 and 15 were added to a solution of 6a,b·6NO3, rendering the corresponding inclusion complexes (Fig. 6 and the supporting information‡).25
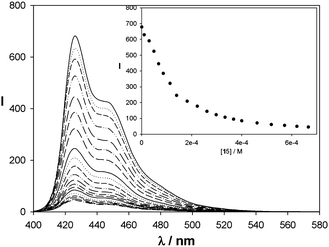 | ||
| Fig. 6 Fluorescence spectra (H2O, 293 K) of a solution of 6a,b·6NO3 (5.0 × 10−5 M) after the successive addition of aliquots of a solution of 6a,b·6NO3 (5.0 × 10−5 M) and 15 (2.0 × 10−3 M). Inset is the emission intensity at λ = 426 nm vs. concentration of 15. | ||
Conclusions
In summary, new PdII and PtII metallocycles displaying improved characteristics as π-acceptors and suitable spectroscopic features for optical recognition applications were prepared from diazapyrenium-containing ligands 1 and 4 using our well established metal-directed self-assembly protocol.Ligand 1 produced the square-shaped metallocycle 3 upon complexation with the palladium centre 2 in organic media. PdII [3]catenanes were prepared in nitromethane by the combination of 3 with the electron-rich cyclophanes BPP34C10 (8) and DN38C10 (9), as confirmed in solution using spectroscopic methods. In the case of 3(9)2·6OTf·2PF6, catenation was further corroborated in the solid state by X-ray diffraction analysis of a single crystal of the supramolecule.
On the other hand, the self-assembly of the non-symmetrical ligand 4 with PdII and PtII complexes yielded equimolecular isomeric mixtures of metallocycles (6a,b and 7a,b) in organic and aqueous media, differing on the relative disposition of the diazapyrenium rings (parallel or orthogonal). Regioselective catenation and complexation with electron-rich aromatic substrates was achieved using the PdII metallocyles. This is a result of the increased π-deficient nature of the diazapyrenium moieties (compared with the phenylenepyridinium subunits) that adopt a parallel arrangement within the metallocycles in order to maximize the π-stacking interactions. The identity of the PdII and PtII supramolecules prepared from ligand 4 was confirmed beyond doubt by means of spectroscopic and spectrometric techniques. The analysis of a single crystal obtained for catenane 6a(8)2·6OTf·2PF6 further confirmed the important role of π-stacking interactions on the regioselectivity of the recognition processes.
Finally, by studying the emission behaviour of the obtained metallocyles upon complexation or catenation with electron-rich aromatic guests, it was found that the fluorescence of the resulting supramolecular aggregates is quenched compared with that of the corresponding metallocycles as a result of the charge-transfer induced by the strong π-donor/π-acceptor interactions. The results open the door for the application of these diazapyrenium-containing PdII and PtII metallocycles as optical probes.
Acknowledgements
This research was supported by the Ministerio de Educación y Cultura and FEDER (CTQ2010-16484/BQU). M.D.G. thanks the Xunta de Galicia for financial support (‘programa Isidro Parga Pondal’).Notes and references
- (a) G. F. Swiegers and T. J. Malefetse, Chem. Rev., 2000, 100, 3483 CrossRef CAS; (b) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS; (c) P. H. Dinolfo and J. T. Hupp, Chem. Mater., 2001, 13, 3113 CrossRef CAS; (d) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef CAS; (e) P. J. Steel, Acc. Chem. Res., 2005, 38, 243 CrossRef CAS; (f) M. Fujita, Chem. Soc. Rev., 1998, 27, 417 RSC; (g) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853 CrossRef CAS; (h) B. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022 CrossRef CAS; (i) F. Würthner, C.-C. You and C. R. Saha-Möller, Chem. Soc. Rev., 2004, 33, 133 RSC; (j) J.-P. Collin, V. Heitz, S. Bonnet and J.-P. Sauvage, Inorg. Chem. Commun., 2005, 8, 1063 CrossRef CAS.
- (a) Molecular Catenanes, Rotaxanes and Knots, A Journey Through the World of Molecular Topology, ed. J.-P. Sauvage and C. Dietrich-Buchecker,Wiley-VCH, Weinheim, 1999 Search PubMed; (b) R. F. Carina, C. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1996, 118, 9110 CrossRef CAS; (c) C. P. McArdle, J. J. Vittal and R. J. Puddephatt, Angew. Chem., Int. Ed., 2000, 39, 3819 CrossRef CAS; (d) K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308 CrossRef CAS.
- (a) M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645 CrossRef CAS; (b) P. J. Stang and D. H. Cao, J. Am. Chem. Soc., 1994, 116, 4981 CrossRef CAS; (c) P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502 CrossRef CAS; (d) S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972 CrossRef CAS; (e) N. Takeda, K. Umemoto, K. Yamaguchi and M. Fujita, Nature, 1999, 398, 794 CrossRef CAS; (f) M. Tominaga, K. Suzuki, M. Kawano, T. Kusukawa, T. Ozeki, S. Sakamoto, K. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2004, 43, 5621 CrossRef CAS; (g) M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469 CrossRef CAS; (h) B. Olenyuk, J. A. Whiteford, A. Fechtenkötter and P. J. Stang, Nature, 1999, 398, 796 CrossRef CAS; (i) K. Ghosh, J. Hu, H. S. White and P. J. Stang, J. Am. Chem. Soc., 2009, 131, 6695 CrossRef CAS; (j) K. Suzuki, M. Tominaga, M. Kawano and M. Fujita, Chem. Commun., 2009, 1638 RSC; (k) J. K. Klosterman, Y. Yamauchi and M. Fujita, Chem. Soc. Rev., 2009, 38, 1714 RSC; (l) M. Yoshizawa, J. Nakagawa, K. Kumazawa, M. Nagao, M. Kawano, T. Ozeki and M. Fujita, Angew. Chem., Int. Ed., 2005, 44, 1810 CrossRef CAS; (m) K. Ono, M. Yoshizawa, T. Kato and M. Fujita, Chem. Commun., 2008, 2328 RSC; (n) T. Sawada, M. Yoshizawa, M.; S. Sato and M. Fujita, Nat. Chem., 2009, 1, 53 CrossRef; (o) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS; (p) M. Ferrer, A. Gutiérrez, M. Mounir, O. Rossell, E. Ruiz, A. Rang and M. Engeser, Inorg. Chem., 2007, 46, 3395 CrossRef CAS; (q) M. Ferrer, A. Pedrosa, L. Rodríguez, O. Rossell and M. Vilaseca, Inorg. Chem., 2010, 49, 9438 CrossRef CAS.
- (a) C. Peinador, E. Pia, V. Blanco, M. D. Garcia and J. M. Quintela, Org. Lett., 2010, 12, 1380 CrossRef CAS; (b) V. Blanco, M. D. García, C. Platas-Iglesias, C. Peinador and J. M. Quintela, Chem. Commun., 2010, 46, 6672 RSC; (c) C. Peinador, V. Blanco and J. M. Quintela, J. Am. Chem. Soc., 2009, 131, 920 CrossRef CAS; (d) V. Blanco, M. Chas, D. Abella, E. Pía, C. Platas-Iglesias, C. Peinador and J. M. Quintela, Org. Lett., 2008, 10, 409 CrossRef CAS; (e) D. Abella, V. Blanco, E. Pía, M. Chas, C. Platas-Iglesias, C. Peinador and J. M. Quintela, Chem. Commun., 2008, 2879 RSC; (f) V. Blanco, M. Chas, D. Abella, C. Peinador and J. M. Quintela, J. Am. Chem. Soc., 2007, 129, 13978 CrossRef CAS; (g) M. Chas, D. Abella, V. Blanco, E. Pía, G. Blanco, A. Fernández, C. Platas-Iglesias, C. Peinador and J. M. Quintela, Chem.–Eur. J., 2007, 13, 8572 CrossRef CAS; (h) M. Chas, E. Pia, R. Toba, C. Peinador and J. M. Quintela, Inorg. Chem., 2006, 45, 6117 CrossRef CAS; (i) R. S. Forgan, D. C. Friedman, C. L. Stern, C. J. Bruns and J. F. Stoddart, Chem. Commun., 2010, 46, 5861 RSC.
- A related approach, involving metal-directed self-assembly in combination with N-dialkylbipyridinium and other π-deficient motifs has been exploited by Liu: (a) Y. Liu, A. Bruneau, J. He and Z. Albliz, Org. Lett., 2008, 10, 765 CrossRef CAS; (b) G. Koshkakaryan, K. Parimal, J. He, X. Zhang, Z. Abliz, A. H. Flood and Y. Liu, Chem.–Eur. J., 2008, 14, 10211 CrossRef CAS.
- (a) V. Blanco, D. Abella, E. Pía, C. Platas-Iglesias, C. Peinador and J. M. Quintela, Inorg. Chem., 2009, 48, 4098 CrossRef CAS; (b) V. Blanco, A. Gutiérrez, C. Platas-Iglesias, C. Peinador and J. M. Quintela, J. Org. Chem., 2009, 74, 6577 CrossRef CAS.
- (a) V. Blanco, M. D. García, A. Terenzi, E. Pía, A. Fernández-Mato, C. Peinador and J. M. Quintela, Chem.–Eur. J., 2010, 16, 12373 CrossRef CAS; (b) M. Chas, V. Blanco, C. Peinador and J. M. Quintela, Org. Lett., 2007, 9, 675 CrossRef.
- H.-C. Becker, A. Broo and B. Nordén, J. Phys. Chem. A, 1997, 101, 8853 CrossRef CAS.
- A. K. Nair, P. P. Neelakandan and D. Ramaiah, Chem. Commun., 2009, 6352 RSC.
- S. Ghosh and P. S. Mukherjee, Organometallics, 2008, 27, 316 CrossRef CAS.
- For instance, see: D. W. Steuerman, H.-R. Tseng, A. J. Peters, A. H. Flood, J. O. Jeppesen, K. A. Nielsen, J. F. Stoddart and J. R. Heath, Angew. Chem., Int. Ed., 2004, 43, 6486 Search PubMed.
- The synthesis of ligand 1 and the self-assembly of metallocycle 3 was advanced in ref. 4c.
- Crystal data for 1·2PF6: C62H42F24N10P4, M = 1506.94, Triclinic, a = 13.067(5) Å, b = 14.272(5) Å, c = 17.273(5) Å, α = 67.392(5)°, β = 77.696(5)°, γ = 87.835(5)°, V = 2902.0(17) Å3, T = 100(2)K, space group P, Z = 2, μ(Mo-Kα) = 0.263 mm−1, 48225 reflections measured, 14379 independent reflections (Rint = 0.0647). The final R1 values were 0.0643 (I > 2σ(I)). The final wR(F2) values were 0.1725 (I > 2σ(I)). The final R1 values were 0.1278 (all data). The final wR(F2) values were 0.2228 (all data). The goodness of fit on F2 was 1.014.
- Although self-assembly of similar Pd(II) metallocycles proceeded at room temperature, heating became necessary to ensure the complete solubility of 1·NO3 in aqueous media. Metallocycles 6a,b·6NO3 are completely soluble at room temperature in D2O.
- Δδ between the δ of the same nucleus in each isomer.
- (a) N. Giuseppone, J.-L. Schmitt, L. Allouche and J.-M. Lehn, Angew. Chem., Int. Ed., 2008, 47, 2235 CrossRef CAS; (b) Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520 CrossRef CAS; (c) T. Megyes, H. Jude, T. Grósz, I. Bakó, R. Radnai, G. Tárkányi, G. Pálinkás and P. J. Stang, J. Am. Chem. Soc., 2005, 127, 10731 CrossRef CAS; (d) L. G. Longsworth, J. Phys. Chem., 1960, 64, 1914 CAS.
- This dual feature has been exploited by Fujita et al. by using the so-called “molecular lock” concept: (a) M. Fujita, F. Ibukuro, K. Yamaguchi and K. Ogura, J. Am. Chem. Soc., 1995, 117, 4175 CrossRef CAS; (b) C. J. Kuehl, S. D. Huang and P. J. Stang, J. Am. Chem. Soc., 2001, 123, 9634 CrossRef CAS.
- (a) K. A. Connors, Binding Constants, Wiley, New York, 1987 Search PubMed; (b) H.-J. Schneider, A. K. Yatsimirsky, Principles and Methods in Supramolecular Chemistry, John Wiley & Sons, New York, 2000 Search PubMed.
- It is noteworthy that DIAZpar rings are not perfectly parallel to either NAPHin or NAPHout, the angles between their planes being 3.0° and 2.5°, respectively.
- The refinement of 3(9)2 gives values of the indexes that are higher than usual due to the disorder associated with some of the triflate counterions and solvent molecules. However, this disorder does not involve the main structure of the catenane and, therefore, the discussion within the text, as can be observed from the ellipsoid representation included in the supporting information‡.
- Herein, we report preliminary results about the emission properties of these systems. Further studies on the optical characteristics and the application of these and other related structures are currently ongoing.
- When CH3NO2 is used, only the lowest energy band can be observed due to the strong absorption of this solvent below 360 nm.
- (a) V. Balzani, A. Credi, S. J. Langford, J. F. Stoddart and M. Venturi, Supramol. Chem., 2001, 13, 303 Search PubMed; (b) R. Ballardini, A. Credi, M. T. Gandolfini, C. Giansante, G. Marconi, S. Silvi and M. Venturi, Inorg. Chim. Acta, 2007, 360, 1072 CrossRef CAS.
- (a) H. Weissman, E. Shirman, T. Ben-Moshe, R. Cohen, G. Leitus, L. J. W. Shimon and B. Rybtchinsky, Inorg. Chem., 2007, 46, 4790 CrossRef CAS; (b) H. Yersin and J. Strasser, Coord. Chem. Rev., 2000, 208, 331 Search PubMed.
- At the concentrations that the fluorescence studies were carried out, some dissociation of the PdII metallocycles into free ligands is possible. Therefore, the quenching observed for catenanes 3(8)2 and 3(9)2 could have some contribution from the interaction of ligand 3 with cyclophanes 8 and 9. In the case of the inclusion complexes derived from metallocycle 6a,b·NO3 the fluorescence quenching can be attributed only to the formation of the inclusion complex, as the interaction between the guests and free ligand is very limited in this concentration range.
Footnotes |
| † Dedicated to the memory of Professor Rafael Suau. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures, 1H, 13C and 2D NMR data, titration data and X-ray crystallographic files (CIF). CCDC reference numbers 837131 and 837132. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00508a |
| This journal is © The Royal Society of Chemistry 2011 |