Thermoresponsive polymers in liquid chromatography
Irene
Tan
,
Farnoosh
Roohi†
and
Maria-Magdalena
Titirici
*
Max Planck Institute of Colloids & Interfaces, Wissenschaftspark Golm, Department Colloid, 14476, Potsdam, Germany. E-mail: Magdalena.Titirici@mpikg.mpg.de; Tel: + 49 (0)331-567-9508
First published on 19th October 2011
Abstract
In liquid chromatography, ‘reversed’ phase (RP) HPLC accounts for the vast majority of analyses concerning the separation of biomolecules. However, the typical usage of organic mobile phases as eluents associated with this technique has drawbacks in certain applications due to the possible denaturation of biocompounds, including other concerns related to the high cost of separations along with environment pollution. This disadvantage prompted the development of novel analytical methods in HPLC related to ‘greener’ stationary phases and process conditions. In this mini review, some advances in thermoresponsive liquid chromatography allowing separation processes on biomolecule mixtures using purely aqueous eluents under isocratic conditions are described.
 Irene Tan | Irene Tan was born in Penang, Malaysia in 1983. She graduated in 2005 with a BA degree in Applied Chemistry from the National University of Singapore. In 2008, she completed her MSc in Industrial Chemistry under a joint collaboration between Technical University of Munich and BASF SE, Germany, focusing on Thermoplastics. After, she conducted her studies in the field of ‘Thermoresponsive Polymers in Separation Science’ within Prof. Antonietti's group working together with Dr Maria-Magdalena Titirici at Max Planck Institute of Colloids and Interfaces, where she graduated in 2011 with a PhD in natural sciences. |
 Farnoosh Roohi | Farnoosh Roohi was born in Teheran, Iran in 1978. She finished her bachelor's degree in polymer engineering at the AmirKabir University, Teheran, Iran in 2002. After she moved to Berlin where she obtained a Master of Polymer Science at the Humboldt University in 2006. She continued her career with a PhD in natural sciences obtained in 2009 from Potsdam University. During her PhD she performed research at the Max-Planck Institute of Colloids and Interfaces in the group of Prof. Markus Antonietti working on thermo-responsive phases for chromatography together with Dr Maria-Magdalena Titirici. Presently Farnoosh Roohi works as a project manager in the research and development at Bayer Schering Pharma in Berlin. |
 Maria-Magdalena Titirici | Maria-Magdalena Titirici was born in Bucharest, Romania in 1977. She graduated in 1999 from the Department of Chemistry of Bucharest University. After, she conducted her PhD study first at the University of Mainz and later on at the University of Dortmund in the field of Molecularly Imprinted Polymers. In 2005 she joined Prof. Antonietti's group as a postdoctoral research at the Max-Planck Institute of Colloids and Interfaces. Currently Maria-Magdalena Titirici is leading the group of “Sustainable Materials for Energy Storage, Catalysis and Separation Science” in the same research institute and her scientific interests include green chemistry, carbon and mesoporous materials, energy storage, molecular recognition and development of novel stationary phases for chromatography. |
1. Introduction
The separation of large biomolecules is becoming increasingly important for life sciences. Biomolecules, such as peptides and proteins, are conventionally separated by reversed phase liquid chromatography (RPLC),1–6ion-exchange chromatography (IEC),7–9 hydrophobic interaction chromatography (HIC)10–12 or with the combination of more than one of the functions mentioned.RPLC is one of the most widely used chromatographic techniques for the separation, purification and the study of peptides. However, the use of organic solvents may result in the loss of bioactivity, and limit further applications of the separated molecules.13
Peptides and proteins have numerous functional groups, and they can possess either net positive or negative charges in varying solution pH. IEC separates proteins according to their net charges via electrostatic interactions, which are dependent on the composition of the mobile phase. Elution is in this case achieved by increasing the ionic strength of the solvent, thus allowing the analytes to unbind from the column surfaces.
In a conventional HIC system, separations are based on the surface hydrophobicity of proteins and peptides. Usually, they are performed using a starting mobile phase of very high salt concentration to promote a ‘salting out’ effect on the analytes, thus promoting hydrophobic binding to the column. Subsequently, this binding is reduced by lowering the salt concentration and thus decreasing the hydrophobic effect on the analytes. These processes, however, possibly induce alterations to the protein structures, resulting in decreased product yields and reduced bioactivity.
The limitations of conventional HPLC techniques used for biomolecule separation prompted the development of a new system based on an aqueous mobile phase which is low cost and more importantly does not lead to the loss of analyte bioactivity during the separation process.
However, the development of LC not only comprises an emerging number of separation techniques, but it has also undergone changes in column material (from glass to stainless steel), and stationary phase diameters (from large to small ∼4.6 mm). Interestingly, the evolution of column diameters offers versatile approaches in tackling separation challenges. Since their introduction in the 1980s, monolithic materials continue to be an important step towards fast chromatographic separations.14–16 In contrast to conventional particulate (3 to 10 μm spheres) packing materials, monolithic columns in HPLC have been known to exhibit superior mass transfer properties and faster kinetics. Such columns can be prepared either from organic polymers17–19 or from inorganic materials such as silica.20 Recently there is an increasing interest in the production of carbon based monoliths for chromatographic separations due to their high mechanical and chemical stability as well as their bipolar character.21,22 However this field is by far not yet as developed as silica or polymeric monoliths.
One advantage of the silica-based monoliths as a chromatographic support over polymeric ones is the possibility of fine tuning meso- and macroporosities using the sol–gel methodology developed by Nakanishi and Soga,20,23 based on the hydrolysis and polycondensation of tetramethoxysilane (TMOS) in the presence of polyethylene oxide (PEO) as a soft template. Thus, bimodal macro-mesoporous silica monoliths with well-defined pore sizes from 1 to 30 nm and different specific surface areas from 50 to 250 m2 g−1 can be easily produced.
However, one main drawback of silica-based supports is their low stability in basic environments. Especially in aqueous-based separations involving basic analytes, this can be crucial for the stability of the monolithic stationary phase with very thin pore walls due to the continuous hierarchical meso-macroporosity. In this respect, polymeric or carbon monoliths are interesting alternatives; however important developments are still required regarding tailoring of their porous structure, which is not as straightforward as for inorganic based materials. Up to date, polymeric and polymer composite-based stationary phases for chromatography are intensively used in a wide variety of applications; however, this is not the aim of the present review.
Among all the stationary phases, an emerging class of thermo-switchable polymer grafted onto silica chromatographic supports has been drawing a considerable amount of attention recently.24–26 Such thermoresponsive polymers have the capability to change their physico-chemical properties at a specific temperature, known as the lower critical solution temperature (LCST). In most cases, this transition happens in water and the polymer changes its conformation from expanded chains to collapsed coil, translated in practice to a change from hydrophilic to hydrophobic properties. Due to such temperature-responsive hydrophilic–hydrophobic surface property alterations, thermoresponsive polymer grafts have been used to generate temperature-sensitive stationary phases for size exclusion,27 hydrophobic,28 ionic29,30and affinity chromatography.30 These temperature sensitive stationary phases may be useful in method development as an extra tool to optimize the selectivity by adjusting the temperature rather than changing the mobile phase composition, and have the advantage that they can separate mixtures of biomolecules (large peptides, proteins) in a purely aqueous environment and under isocratic conditions. Therefore, the harsh conditions normally associated with protein separation employing the standard C18 columns (acetonitrile in low pH buffers as mobile phases) can be avoided. Acetonitrile, besides being relatively expensive, tends to lead to the total denaturation of proteins, while acidic environments are often destructive to the activities of many enzymes. Thus, the separation of biomolecules under a purely aqueous environment is an important achievement in this field. This could be particularly important especially for preparative scale/purification chromatography.
This special feature involving a simple temperature switch for the resolution of bioanalytes under ‘green’ conditions is useful as a tool in bioseparations, thus overcoming the limitations of conventional separation systems, especially when this technique could be upscaled for preparative chromatography. So far this was demonstrated only at an analytical scale.
Among thermoresponsive polymers, poly(N-isopropylacrylamide) (PNIPAAM) shows the sharpest phase transition in the class of N-alkylacrylamide polymers. PNIPAAM exhibits thermally reversible soluble-insoluble changes in an aqueous solution in response to temperature changes across a LCST at 32 °C.31 Below this LCST, the polymer chains show an expanded conformation in water due to strong hydration. Above its LCST, the PNIPAAM changes to compact forms via dehydration and intermolecular hydrogen bonding between the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and amine (N–H) groups.
O) and amine (N–H) groups.
Besides PNIPAAM, there are other polymers exhibiting similar behaviour in water as reviewed in several publications.32 Here, we will focus on two other systems, namely the structural isomers of PNIPAAM, poly-(2-oxazoline)s,33,34 as well as a system based on oligo(ethylene glycol) methacrylates.35–37 The advantage demonstrated with ethylene glycol-based polymers is that one can precisely finely tune the resultant polymer LCST by varying the ratio of hydrophilic side chains to hydrophobic polymer backbone.
This minireview will mainly describe our own investigations in the field of thermoresponsive stationary phases based on the different polymers grafted onto silica monolithic supports. Finally, a broader view and perspectives regarding this field will also be presented.
2. Thermoresponsive stationary phases
2.1 General principle
As previously mentioned, thermoresponsive polymers undergo physical changes in water when exposed to an external stimulus, in this case, heat. The temperature of this transition is known as the LCST, and this is when the polymer reversibly precipitates out of its aqueous solution in contrast to most polymers. The ability to exhibit such changes under easily controlled conditions can be exploited for many analytical techniques, especially in separation science.Such polymers could be covalently immobilized to the surface of silica supports (either beads or monoliths) via terminal functionality using the ‘grafting from’ or the ‘grafting to’38,39 procedures. Although the ‘grafting from’ method allows more evenly distributed chains and higher polymer grafting density, the latter method presents a straightforward and relatively efficient way to couple polymers onto supports. The polymers are grafted randomly onto these sites and the grafting density can be controlled by limiting the concentration of the polymer solution in contrast to the ‘grafting from’ approach in which polymerization is more difficult to control.
The silica gel surface can be firstly modified with suitable and accessible functional sites such as with (3-aminopropyl) triethoxysilane (APS) giving amino (–NH2) reactive surface groups to couple to the polymer's terminal functionalities such as carboxylate (–COOH), forming strong chemical bonds such as amide linkages. Thus, when a layer of thermoresponsive polymer is grafted onto pre-formed chromatographic support, the temperature-mediated properties could then be transferred to the stationary phases. The principle of thermoresponsive chromatography and the behaviour of a grafted polymer (e.g. PNIPAAM) below and above its LCST on a silica monolith column is illustrated in Fig. 1. At temperatures below the column LCST, the polymer chains show hydrated expanded conformation in water while the dehydrated compacted form is dominant when the surroundings are heated up (Fig. 1(a)). Turbidity measurements done on polymer-grafted particulate silica confirmed the polymer's behaviour when attached to a solid support (Fig. 1(b)).
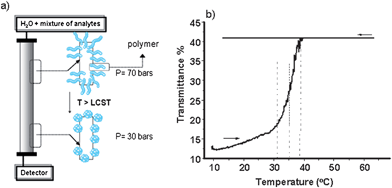 | ||
| Fig. 1 (a) Schematic representation of the concept of thermoresponsive chromatography; (b) Turbidity measurement of particulate silica modified with polymer (LCST = 33 °C) at 1 wt% in water. | ||
2.2 Poly(N-isopropylacrylamide)-based stationary phases
Heskins and Guillet31 established the LCST of PNIPAAM to be 32 °C as early as the 1960s. This temperature, being relatively close to body temperature, enables it to be widely explored for preparing switchable materials for biological applications.40 Another reason for its biomedical popularity is its insensitivity towards slight environmental changes such as pH or concentration. Thus, PNIPAAM is one of the most commonly known thermoresponsive polymers and it has not only been mainly exploited for drug delivery applications, but also for the preparation of smart stationary phases. Okano et al., one of the pioneers of thermoresponsive chromatography, proposed three types of PNIPAAM grafting methods on silica surfaces to prepare chromatographic stationary phases, including terminally,24,41–44 crosslinked,45,46 and polymer brush47,48 types.There are many possibilities to immobilize PNIPAAM polymers onto the silica supports. For example, using ‘grafting to’ techniques, PNIPAAM-grafted silica beads were activated by standard ester-amine coupling. Terminally carboxylated PNIPAAM was synthesized by semitelechelic polymerization, and as activated carboxyl groups, reacted with amino groups on silanized silica surfaces.28
The ‘grafting from’ method for PNIPAAM was also reported using a surface immobilized azo-initiator and crosslinker to prepare polymer layers employing conventional radical polymerization techniques.45,46 This method incorporates relatively large amounts of polymers onto the surfaces in comparison to the ‘grafting to’ method due to steric effects.
Recently, controlled free radical polymerization techniques have been extensively utilized to synthesize various polymers. A third ‘grafting from’ method allows the synthesis of high-density PNIPAAM brushes on silica-bead surfaces using a surface-immobilized atom transfer radical polymerization (ATRP) initiator.49
Since controlled polymerization techniques offer great advantages in comparison to classical radical polymerization such as controlled molecular weights, low polydispersities as well as the possibility of living polymerization which allows the possibility of re-initiation and the introduction of a new block at any stage of reaction, our group has employed RAFT polymerization50 in combination with the ‘grafting to’ technique to produce PNIPAAM-modified surfaces. Our approach was to obtain PNIPAAM containing carboxylic groups and graft it onto the solid support previously modified with amino groups (Fig. 2). The RAFT polymerization was done in the presence of a trithioester RAFT agent, 4-cyanopentanoic acid trithiododecane containing a terminal carboxylic group. Coupling of thermoresponsive polymers onto the monolithic silica was performed by standard amide bond formation between the activated carboxyl-terminated PNIPAAM and amino-modified silica.
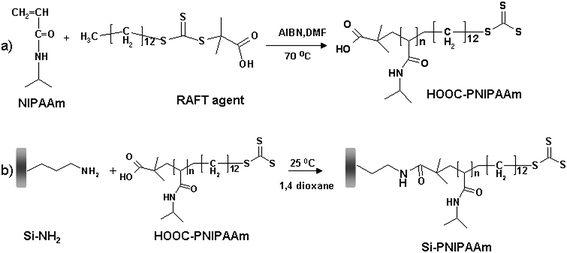 | ||
| Fig. 2 Strategy of grafting PNIPAAM onto silica supports using RAFT polymerization: (a) Polymerization of NIPAAM in the presence of 4-cyanopentanoic acid trithiododecane to produce a carboxyl-modified thermoresponsive polymer; (b) ‘In situ’ attachment of the carboxyl-modified polymer to the amino-modified silica support. | ||
The initial study and proof of concept for the separation of a mixture of steroids with varying log P values,51 and thus different hydrophobicities using PNIPAAM-based thermoresponsive chromatography, was done using mesoporous silica spheres52 as a stationary phase. Later, we switched to ‘in situ’ modification of bicontinuous meso-macroporous monolithic silica columns.53 The morphologies of the chromatographic supports used in our study are shown in Fig. 3. As already pointed out in the introduction, the use of bimodal porous monolithic materials allows efficient chromatographic separations due to improved mass transfer properties. This is clearly illustrated on the separation chromatograms showing the corresponding separation profiles of a mixture of steroids on two thermoresponsive columns based on PNIPAAM; one with mesoporous silica beads and the other one with silica monoliths. It has to be mentioned that for the above comparison, polymers with the same molecular weight were used and the grafting density of the polymers on the stationary support was within the same range (∼500 (μg m−2)/(chains nm−2) × 10−3). Although the stationary phases had the same chemical structure and composition, the performance of the monolithic column was more efficient on the separation of steroids as compared to the particle-packed column. While all the steroids could be well separated on the monolithic column, hydrocortisone, prednisolone and hydrocortisone acetate could not be efficiently resolved on the particulate column. The inefficiency could not be attributed to the lower hydrophobicity of the Si-PNIPAAM since the grafting densities are reported to be in the same range. One possible explanation could be due to the difference in the packing density of two columns. The density of the monolithic silica column was calculated to be about 0.3 g ml−1 and that of silica particle packed column was 0.58 g ml−1. Thus, the free volume available for the interactions between analytes and polymer chains in the case of the beaded column is much less compared to that of the monolithic column. Subsequently there would be fewer interactions between the polymer chains and analytes. Furthermore, it is necessary to mention that the plate numbers for the columns packed with silica beads are much lower than the thermoresponsive monolithic one.54 This indeed shows that the efficiency of the resultant monolithic column is much higher than for the column packed with silica beads. Moreover, the retention factors are also lower than in the case of the monolithic column.
 | ||
| Fig. 3 Electron micrographs of the chromatographic supports used for the immobilization of thermoresponsive polymers and their corresponding thermoresponsive elution profiles of an aqueous mixture of five steroids at 55 °C: (from left to right) (a) scanning electron micrograph, transmission electron micrograph and corresponding separation profile of mesoporous micrometre-sized silica beads; (b) scanning electron micrograph, transmission electron micrograph and corresponding separation profile on the continuous macro-mesoporous structure of a silica monolith; elution conditions: (1) hydrocortisone; (2) prednisolone; (3) dexamethasone; (4) hydrocortisone acetate; (5) testosterone; flow rate: 1 ml min−1; mobile phase: water. | ||
One important advantage of using RAFT polymerization in solution is the possibility of precise polymer characterization. Thus, the number average molecular weight of the polymer could be determined by size exclusion chromatography (SEC). It was found that the molecular weight of the grafted polymer can have a major influence on the performance of the column. This is due to the dependence of the polymer's temperature-response on its molecular weight, which is directly related to the hydrophobicity of the surface. On the other hand, the final mesopore size of the composite depends on the chain length and molecular weight of the grafted polymer.
Fig. 4 shows the effect of molecular weight of the grafted PNIPAAM on the separation performance above the column's LCST value. At lower molecular weights, the pore size remains similar to the original size. However the pore size decreased dramatically in the case of the composite obtained using high molecular weight PNIPAAM (Mn: 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1).54 While all steroids could be well separated in the case of silica grafted with medium and high molecular weight PNIPAAM, hydrocortisone and prednisolone could not be well separated on the lowest molecular weight column. It was proven that the thermo-responsivity of the PNIPAAM, meaning the rate at which hydrophilic-hydrophobic switch occurs, depends on the molecular weight;54 the lower the molecular weight, the less hydrophobic the polymer is. In this case its surface hydrophobicity is not high enough to distinguish between these two analytes as they have very similar log P values (1.47 and 1.62 respectively), and relatively close hydrophobicities.
800 g mol−1).54 While all steroids could be well separated in the case of silica grafted with medium and high molecular weight PNIPAAM, hydrocortisone and prednisolone could not be well separated on the lowest molecular weight column. It was proven that the thermo-responsivity of the PNIPAAM, meaning the rate at which hydrophilic-hydrophobic switch occurs, depends on the molecular weight;54 the lower the molecular weight, the less hydrophobic the polymer is. In this case its surface hydrophobicity is not high enough to distinguish between these two analytes as they have very similar log P values (1.47 and 1.62 respectively), and relatively close hydrophobicities.
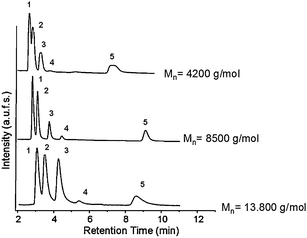 | ||
| Fig. 4 Elution profile of an aqueous mixture of five steroids on monolithic columns containing PNIPAAM chains with varying molecular weights; elution conditions: (1) hydrocortisone; (2) prednisolone; (3) dexamethasone; (4) hydrocortisone acetate; (5) testosterone; flow rate: 1 ml min−1; mobile phase: water; T = 45 °C. | ||
Therefore, the careful optimization of column properties plays a crucial role, contributing to efficient separation. This refers to the type of chromatographic support, polymer molecular weight and grafting density.
Lastly, the PNIPAAM-modified monolithic column was also compared to a commercially available monolithic RP18 column (Merck, Darmstadt) for the separation of the same mixture of steroids. According to the literature,55,56 a mixture of MeOH/water (50/50) for the RP18 column was used while the temperature of the column was set to 40 °C. For our column, we chose a pure aqueous solution at 55 °C such that the PNIPAAM chains will be in their collapsed state. Both separations were carried out under isocratic conditions. This comparison is presented in Fig. 5(a). While all five steroids could be well separated on the monolithic column modified with PNIPAAM, hydrocortisone and prednisolone eluted in a single peak on the RP18 column. These steroids, as mentioned before, show very close log P values (1.47 and 1.62, respectively) and presumably, the hydrophobicity of RP18 is too high for their discrimination. Furthermore even in the collapsed state, the PNIPAAM chains contain polar groups (N–H and CO) intramolecularly associated through hydrogen bonding, which together can be responsible for some polar interactions with the functional groups of hydrocortisone and prednisolone. The strong hydrophobic surface of the RP18 column is responsible for the longer elution time. The order of elution for all the steroids is similar on both columns, proving again that hydrophobic-hydrophobic interactions are mainly responsible for steroid separation on the PNIPAAM-based column.
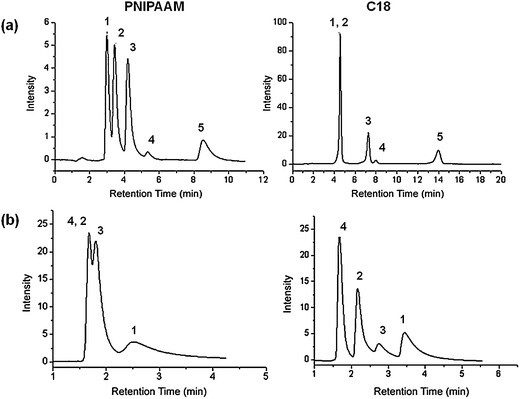 | ||
| Fig. 5 (a) Comparison between the performances of the column containing PNIPAAM and a monolithic RP18 column for the separation of a mixture of steroids: (1) hydrocortisone; (2) prednisolone; (3) dexamethasone; (4) hydrocortisone acetate; (5) testosterone; flow rate: 1 ml min−1; mobile phase: water; (b) elution profiles of an aqueous standard analyte mixture at varying temperatures on the column with PNIPAAM: (1) ethylbenzene; (2) phenol; (3) toluene; (4) uracil. | ||
In order to better elucidate the nature of the interactions involved in our thermoresponsive column, we also used a standard mixture of compounds that are normally involved for the characterization of RP chromatographic column56 properties. To evaluate the hydrophobicity of our columns we injected an aqueous mixture of uracil, phenol, toluene and ethylbenzene (Fig. 5(b)). Uracil is used in this case as the void marker. The retention of the two hydrocarbons, ethylbenzene and toluene, reflect the hydrophobicity of the column. According to the literature,55 the retention coefficients for ethylbenzene vary between 5 and 15 with RP18 columns and between 2 and 11 for RP8 columns. The selectivity coefficient α (ethylbenzene-toluene) amounts to about 1.75–1.82 for RP18 columns and 1.7 for RP8 columns. On the PNIPAAM column, we obtained a retention coefficient for ethylbenzene at 1.8 and a selectivity coefficient of ethylbenzene-toluene at 1.7. From these results we can deduce that above column LCST, our monolithic thermoresponsive column has hydrophobicity close to that of RP8 columns. At low temperatures, one would expect an inversion of elution for these analytes. However the elution order does not change, despite the separation efficiency being not very good. As a reference experiment, the chromatographic behavior of the starting silica monolith before PNIPAAM modification was also analyzed. No separation was accomplished at any of the previously mentioned temperatures. This clearly suggests that the successful separation of the steroid mixture and hydrocarbons is only due to the presence of polymer on the pore surfaces.
2.3 Poly(2-oxazoline)-based stationary phases
Another class of thermoresponsive polymer is represented by poly(2-oxazoline)s. Oxazolines are structural isomers of NIPAAM (Fig. 6); the N moiety of the former appears within the backbone chains instead when polymerized by ‘living’ cationic ring opening polymerization.57 The cloud point of each polymer varies with the differences in the extended alkyl chain of monomers, concentration of the polymer in solution, molecular weight and the addition of salts. Due to its biocompatibility, poly(2-oxazoline)s are widely studied for their potential use in drug delivery systems or thermoresponsive materials.58 It was found that by copolymerizing each different monomer, the LCST and the individual properties of each copolymer could be specifically tuned to a desired value in water. Therefore we copolymerized two monomers of 2-oxazolines each displaying different LCSTs: 2-N-propyl-2-oxazoline (NPOX) (23.8 °C) and 2-isopropyl-2-oxazoline (IPOX) (38.7 °C).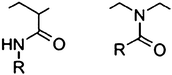 | ||
| Fig. 6 Structures of amide repeating units: N-alkyl acrylamide; N-acyl ethyleneimine (2-oxazoline). | ||
The copolymer P(IPOX-co-NPOX) with the comonomer units ratio of 32:15 ([IPOX]:[NPOX]) was synthesized with a molecular weight of approximately 10000 mol g−1. In order to couple the amino chain end of the polymer to the silica monolith, (3-isocyanatopropyl) triethoxysilane was used to modify the silica surface with cyano groups instead of amino. From elemental analyses, the amount of cyano groups immobilized onto the monolith was 504 μg m−2 and the grafting density from the first cycle polymer attachment was 0.029 chains nm−2. Turbidity measurements confirmed the LCST in water at 1 wt% to be 42 °C. Fig. 7 shows the elution of steroids in water at 55 °C.
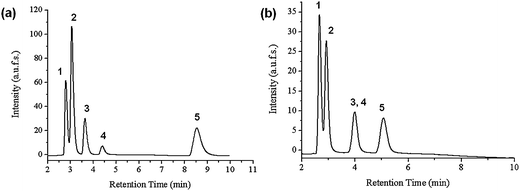 | ||
| Fig. 7 Elution profile of an aqueous mixture of five steroids at 55 °C in two different thermoresponsive columns (a) Si-PNIPAAM (b) Si-P(IPOX-co-NPOX): (1) hydrocortisone; (2) prednisolone; (3) dexamethasone; (4) hydrocortisone acetate; (5) testosterone; flow rate: 1 ml min−1; mobile phase: water. | ||
The column modified with P(IPOX-co-NPOX) at its optimal performance could only drive the separation of four peaks above the polymer's LCST (42 °C). Although all five steroids can be well separated on a Si-PNIPAAM with the same grafting density and molecular weight, dexamethasone and hydrocortisone acetate cannot be separated with Si-P(IPOX-co-NPOX). Though the log P values of hydrocortisone (1.47) and prednisolone (1.62) are more similar to each other than that of dexamethasone (1.83) and hydrocortisone acetate (2.45), the first two analytes can be separated, showing that the hydrophobicity of the Si-(PIPOX-co-NPOX) column is relatively high. Thus, it is suggested that the low hydrophobicity does not contribute to the poor elution of hydrocortisone acetate and dexamethasone. Comparing the chemical structures of these two thermoresponsive polymers (Fig. 2 and6) the main difference between these two structural isomers is the position of their N moieties. In PNIPAAM, they are situated in the side chains, whereas in poly(2-oxazoline)s, they are in the backbone. Thus, in the collapsed state (above the LCST), the N groups of poly(2-oxazoline)s are not easily available. Besides the main hydrophobic–hydrophobic interactions, the analytes also diffuse into the collapsed polymer chains, and by comparing both the chemical structures of hydrocortisone acetate and dexamethasone, the interactions between the amide bonding in the case of PNIPAAM play an important role for their separation. As amide bonding is not available in the collapsed poly(2-oxazoline)s, these two analytes eluted at the same time.
2.4 PEGylated stationary phases
Recently, oligo(ethylene glycol)-based thermoresponsive polymers have been proposed by the group of Lutz et al.36 (Fig. 8) as interesting alternatives to PNIPAAM. Indeed these polymers display reversible phase transitions in water and, in addition, are mainly composed of bioinert ethylene oxide units (i.e. poor hydrogen bond donors and highly hydrated acceptors). Moreover, these interesting macromolecules can be easily synthesized using commercially available monomers by surface-initiated atom transfer radical polymerization (ATRP) in the presence of initiator N-succimidyl 2-bromoisobutyrate.59 For instance, random copolymers 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA) and oligo(ethylene glycol) methacrylate (OEGMA) exhibit an LCST in water, which can be precisely adjusted by varying the comonomer composition (Fig. 8).37 The longer the hydrophilic OEGMA side chain, the higher the resultant polymer LCST.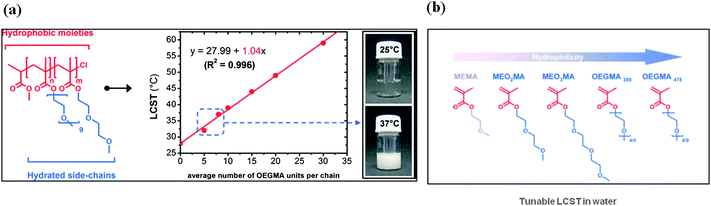 | ||
| Fig. 8 (a) Plots of the measured lower critical solution temperature (LCST) as a function of the theoretical average number of OEGMA units per chain for a series of P(MEO2MA-co- OEGMA) copolymers of various composition. (Hydrophobic and hydrophilic molecular regions in the copolymer are indicated in red and blue, respectively.) (b) Molecular structures of various oligo(ethylene glycol) methacrylates. (Hydrophobic and hydrophilic molecular regions are indicated in red and blue, respectively.) Taken with permission from J. F. Lutz, Journal of Polymer Science Part a-Polymer Chemistry, 2008, 46, 3459–3470. | ||
Thus, thermoresponsive P(MEO2MA-co-OEGMA) copolymers have been recently exploited for preparing a variety of smart biocompatible materials.60 In particular, it has been demonstrated lately that these polymers allow reversible control over bioadhesion.61,62
A series of P(MEO2MA-co-OEGMA) copolymers (composites a–f) with variable chain lengths and compositions were prepared as summarized in Table 1. Each of the resulting copolymer's properties such as the cloud point and the grafting density onto the monolithic column are shown in the table. The cloud points are observed to be retained (39 to 41 °C) when the comonomer compositions are kept the same ([OEGMA]:[MEO2MA] = 10:90) at varying molecular weights, proving that the LCSTs of P(MEO2MA-co-OEGMA) are not affected drastically by polymer chain length or concentration variations. The grafting of these copolymers and the successful application to bioseparation have been recently reported by Tan et al.63 The chromatographic performances of these P(MEO2MA-co-OEGMA)-grafted stationary phases were reported in aqueous media under isocratic conditions. Fig. 9(a) shows the elution profiles for the separation of the same mixture of steroids below and above the LCST of the column containing the composite e (Table 1, entry e). It can be observed that below the LCST of this column (33 °C), analytes hydrocortisone and prednisolone can be separated, while more hydrophobic analytes dexamethasone and hydrocortisone acetate are eluted in a single peak. Testosterone, which has a much higher log P51 value (3.32), is eluted in a separate peak from the others. Significantly, changing the temperature of the column above its LCST led to the separation of all five steroids. This, together with an increase in the retention time for the more hydrophobic analytes, seems to indicate that the driving forces for separation above LCST are hydrophobic–hydrophobic interactions between the analytes and the stationary phase. In that regard, the present materials behave similarly to PNIPAAM-modified columns52,53 as discussed previously. Nevertheless, at low temperatures, the P(MEO2MA-co-OEGMA)-grafted stationary phases allow a better separation of hydrophilic analytes with close log P values than their PNIPAAM counterparts.53 Therefore, this also suggested polar-type interactions as the driving force for separation below LCSTs, which is absent in PNIPAAM-based columns.
| [OEGMA]/[MEO2MA] | DPn th | M n | PDI | LCST/°C | Grafting density/μg m−2 | Grafting density/chains nm−2 | |
|---|---|---|---|---|---|---|---|
| a | 10:90 |
100 | 18100 |
1.28 | 41 | 233 | 0.00714 |
| b | 10:90 |
75 | 15690 |
1.41 | 38 | 402 | 0.01770 |
| c | 10:90 |
50 | 12310 |
1.30 | 39 | 371 | 0.02480 |
| d | 10:90 |
25 | 6220 | 1.36 | 40 | 305 | 0.03920 |
| e | 5:95 |
100 | 18120 |
1.66 | 33 | 250 | 0.00926 |
| f | 15:85 |
100 | 17040 |
1.41 | 43 | 235 | 0.00715 |
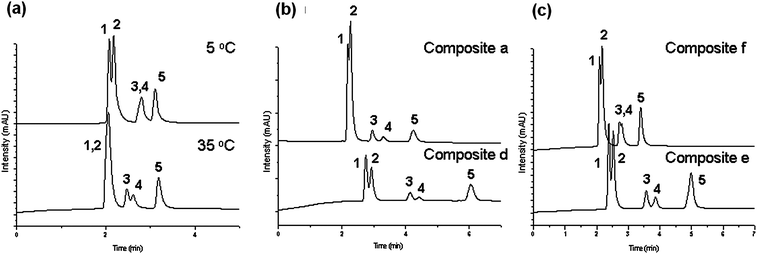 | ||
| Fig. 9 Elution profiles and changes in the retention times on five aqueous mixtures of steroids upon variation of (a) the temperature on composite e, (b) the molecular weight at 55 °C, and (c) the comonomer ratio at 35 °C; (1) hydrocortisone; (2) prednisolone; (3) dexamethasone; (4) hydrocortisone acetate; (5) testosterone; flow rate: 1 ml min−1; mobile phase: water. | ||
In addition, the influence of some macromolecular parameters (e.g. molecular weight or comonomer composition) on the separation of the steroid mixtures was investigated. For example, it was observed that the composites prepared with a polymer of high molecular weight (Table 1, entry a) require only a small grafting density (0.00714 chains nm−2) on the silica support to achieve reasonable separation of the five steroids at 55 °C. On the other hand, with a lower molecular weight polymer (Table 1, entry d), the grafting density had to be increased (0.0392 chains nm−2) to observe similar performance (Fig. 9(b)). Thus, to be able to achieve optimal control of the hydrophobicity of the column, leading to an efficient separation, higher molecular weight polymers are preferred because a hydrophobic column would require a lower grafting density. Over-grafting may lead to the blocking of mesopores, which are necessary for retention.
As already mentioned, one important advantage of the P(MEO2MA-co-OEGMA) copolymer is the possibility of tuning its thermo-sensitivity by adjusting its comonomer composition. In Fig. 9(c), the chromatograms for the separation of five steroids on two columns containing composites prepared with different co-monomer compositions and thus having different LCSTs are shown. For the polymer with a lower LCST (Table 1, entry e), separation of the more hydrophobic analytes dexamethasone and hydrocortisone acetate can be already achieved at lower temperatures (33 °C), while the composite with a higher LCST (Table 1, entry f) can only perform this separation above 43 °C with a worse resolution. Thus, we could show the hydrophilic–hydrophobic percentage of the P(MEO2MA-co-OEGMA) thermoresponsive polymer.
Lastly, P(MEO2MA-co-OEGMA)-grafted stationary phases were tested for protein chromatography. Previous attempts to separate proteins using thermoresponsive stationary phases employed PNIPAAM in combination with ion-exchange polymers such as acrylic acid, utilizing a combination of hydrophobic and ionic interactions.44 The most hydrophobic column (Table 1, entry e) was chosen to attempt the separation of two proteins with relatively close hydrophobicity and size, lysozyme and myoglobin. At the temperature below the LCST of column e (33 °C), the two proteins were shown to be eluted in a single peak. With an increase in the temperature, they were observed to achieve near-baseline separation (Fig. 10) in a relatively short elution time based only on simple polymers, in contrast to that of the PNIPAAM-acrylic acid analogue. Also, their relatively short elution time is in contrast to that of the pure PNIPAAM analogue, which showed extensive retention times.
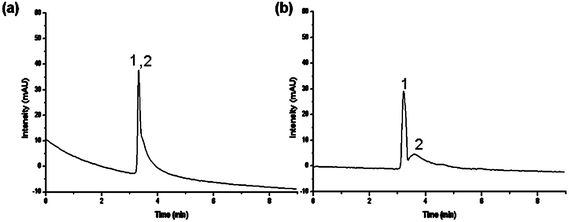 | ||
| Fig. 10 Elution profiles and changes in retention times with temperature variation on an aqueous mixture of two proteins, (1) lysozyme and (2) myoglobin, using composite e (Ds = 250 μg m−2); flow rate: 0.5 ml min−1; mobile phase: 0.1 M potassium phosphate buffer, elution: isocratic. | ||
A reason for this observation may be the fact that the non-linear poly(ethylene glycol) (PEG)-like-based polymer backbone is chemically inert, in contrast to the PNIPAAM analogues, which contains the amide bond, leading to some non-specific interactions and extensive retention on the column. The absence of such interactions thus led to the fast elution of proteins on the P(MEO2MA-co-OEGMA) columns. However, the broader peaks indicating tailing may also suggest the involvement of different types of interactions between proteins, instead of just hydrophobic–hydrophobic forces, as demonstrated in the case of steroid separation.
3. Conclusions
The chromatographic resolution of biomolecules is very important in various biological applications as it serves as a platform to fields such as drug or gene delivery, or in the selective recovery or purification of enzymes or proteins for medical studies. The crucial development of analytical methods for the separation and quantification of proteins and peptides in an aqueous environment using functional polymers on HPLC has been documented over the last 18 years by Kanazawa41 and co-workers. This work suggests a solvent gradient elution-like effect (RP18-like) which could be achieved by programming the temperature of the elution instead, thus avoiding environmentally unfriendly organic solvents. The use of temperature as a control factor in deciding the nature of the stationary phase prompted current developments on ‘intelligent’ chromatography.We explored temperature-sensitive chromatography using functional polymers which were efficiently ‘in situ’ ‘grafted to’ bimodal porous silica monoliths. We reported that PNIPAAM-grafted silica monoliths performed faster separation analyses on a mixture of steroids as compared to previous polymer-beads composite on the chromatograms (4 times faster).
Other related thermoresponsive polymers have also been investigated for similar separation analyses. Copolymers of P(IPOX-co-NPOX) and P(MEO2MA-co-OEGMA) have an advantage over homopolymer PNIPAAM as their cloud points could be adjusted linearly in water by varying comonomer compositions. In addition, it was reported that the PEGylated composite was able to selectively resolve both hydrophilic and hydrophobic steroids at optimal conditions, based on the selected elution temperature. Furthermore, the initial separation of a mixture of two proteins was achieved based on aqueous and isocratic elution with the P(MEO2MA-co-OEGMA)-grafted monolith, which is an improvement over the PNIPAAM-acrylic acid analogues normally used for this function. In addition, P(MEO2MA-co-OEGMA) is biocompatible and thus the scope of its applications can be further extended to biomedical technology.
Of course, as with other relatively new fields, much more is hoped to be accomplished here in the near future. The use of controlled polymerization techniques enables us to have control over molecular weight, but also allows a precise definition of the copolymer compositions. Therefore other blocks could be incorporated into the synthesis so that the interactions are precisely tuned between analyte mixture and the stationary phase. Particularly promising would be the grafting of thermoresponsive polymers with blocks comprising different LCSTs, and by applying a temperature gradient to achieve elution of both hydrophilic and hydrophobic analytes on the same stationary phase. In other words, the results discussed encompass the ability to operate in a multi-dimensional chromatographic system performed on a single column, all under ‘green’ aqueous media.
Notes and references
- J. G. Dorsey and K. A. Dill, Chem. Rev., 1989, 89, 331–346 CrossRef CAS.
- T. P. Hennessy, R. I. Boysen, M. I. Huber, K. K. Unger and M. T. W. Hearn, J. Chromatogr., A, 2003, 1009, 15–28 CrossRef CAS.
- G. J. Opiteck, K. C. Lewis, J. W. Jorgenson and R. J. Anderegg, Anal. Chem., 1997, 69, 1518–1524 CrossRef CAS.
- S. M. Staroverov and A. Y. Fadeev, J. Chromatogr., A, 1991, 544, 77–98 CrossRef CAS.
- J. Tang, M. Gao, C. Deng and X. Zhang, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 866, 123–132 CrossRef CAS.
- W. Z. Xiao and P. J. Oefner, Hum. Mutat., 2001, 17, 439–474 CrossRef CAS.
- H. Hara, H. Kobayashi, M. Maeda, A. Ueno and Y. Kobayashi, Anal. Chem., 2001, 73, 5590–5595 CrossRef CAS.
- E. Kimoto, H. Tanaka, T. Ohmoto and M. Choami, Anal. Biochem., 1993, 214, 38–44 CrossRef CAS.
- J. Kobayashi, A. Kikuchi, K. Sakai and T. Okano, Anal. Chem., 2001, 73, 2027–2033 CrossRef CAS.
- K. Adachi and T. Asakura, J. Chromatogr., Biomed. Appl., 1987, 419, 303–307 CrossRef CAS.
- T. Arakawa, Arch. Biochem. Biophys., 1986, 248, 101–105 CrossRef CAS.
- B. F. Roettger and M. R. Ladisch, Biotechnol. Adv., 1989, 7, 15–29 CrossRef CAS.
- S. Y. M. Lau, A. K. Taneja and R. S. Hodges, J. Chromatogr., A, 1984, 317, 129–140 CrossRef CAS.
- F. C. Leinweber and U. Tallarek, J. Chromatogr., A, 2003, 1006, 207–228 CrossRef CAS.
- B. Bidlingmaier, K. K. Unger and N. von Doehren, J. Chromatogr., A, 1999, 832, 11–16 CrossRef CAS.
- B. A. Grimes, R. Skudas, K. K. Unger and D. Lubda, J. Chromatogr., A, 2007, 1144, 14–29 CrossRef CAS.
- S. Hjerten, J. L. Liao and R. Zhang, J. Chromatogr., A, 1989, 473, 273–275 CrossRef CAS.
- F. Svec and J. M. J. Frechet, Anal. Chem., 1992, 64, 820–822 CrossRef CAS.
- Q. C. Wang, F. Svec and J. M. J. Frechet, Anal. Chem., 1993, 65(17), 2243–2248 CrossRef CAS.
- K. Nakanishi and N. Soga, J. Am. Ceram. Soc., 1991, 74, 2518–2530 CrossRef CAS.
- J. Hasegawa, K. Kanamori, K. Nakanishi and T. Hanada, C. R. Chim., 2010, 13, 207–211 CrossRef CAS.
- Z.-G. Shi, F. Chen, J. Xing and Y.-Q. Feng, J. Chromatogr., A, 2009, 1216, 5333–5339 CrossRef CAS.
- K. Nakanishi, H. Minakuchi, N. Soga and N. Tanaka, J. Sol-Gel Sci. Technol., 1997, 8, 547–552 CAS.
- H. Kanazawa, K. Yamamoto, Y. Matsushima, N. Takai, A. Kikuchi, Y. Sakurai and T. Okano, Anal. Chem., 1996, 68, 100–105 CrossRef CAS.
- H. Kanazawa, Anal. Bioanal. Chem., 2004, 378, 46–48 CrossRef CAS.
- H. Kanazawa, J. Sep. Sci., 2007, 30, 1646–1656 CrossRef CAS.
- K. Hosoya, E. Sawada, K. Kimata, T. Araki, N. Tanaka and J. M. J. Frechet, Macromolecules, 1994, 27, 3973–3976 CrossRef CAS.
- H. Kanazawa, Y. Kashiwase, K. Yamamoto, Y. Matsushima, A. Kikuchi, Y. Sakurai and T. Okano, Anal. Chem., 1997, 69, 823–830 CrossRef CAS.
- J. Kobayashi, A. Kikuchi, K. Sakai and T. Okano, Anal. Chem., 2001, 73, 2027–2033 CrossRef CAS.
- P. S. Stayton, T. Shimoboji, C. Long, A. Chilkoti, G. Chen, J. M. Harris and A. S. Hoffman, Nature, 1995, 378, 472–474 CrossRef CAS.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Chem., 1968, 2, 1441–1455 CrossRef CAS.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2011, 35, 45–93 CrossRef.
- R. Hoogenboom and H. Schlaad, Polymers, 2011, 3, 467–488 CrossRef CAS.
- C. Diehl, P. Cernoch, I. Zenke, H. Runge, R. Pitschke, J. Hartmann, B. Tiersch and H. Schlaad, Soft Matter, 2010, 6, 3784–3788 RSC.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J.-F. o. Lutz, Ã. z. r. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- J.-F. o. Lutz and A. Hoth, Macromolecules, 2005, 39, 893–896 CrossRef.
- H. W. Jarrett, J. Chromatogr., A, 1987, 405, 179–189 CrossRef CAS.
- V. A. Davankov, A. A. Kurganov and K. K. Unger, J. Chromatogr., A, 1990, 500, 519–530 CrossRef CAS.
- C.d. l. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- H. Kanazawa, Y. Kashiwase, K. Yamamoto, Y. Matsushima, A. Kikuchi, Y. Sakurai and T. Okano, Anal. Chem., 1997, 69, 823–830 CrossRef CAS.
- K. Yamamoto, H. Kanazawa, Y. Matsushima, N. Takai, A. Kikuchi and T. Okano, Chromatography, 2000, 21, 209–215 CAS.
- H. Kanazawa, T. Sunamoto, Y. Matsushima, A. Kikuchi and T. Okano, Anal. Chem., 2000, 72, 5961–5966 CrossRef CAS.
- H. Kanazawa, M. Nishikawa, A. Mizutani, C. Sakamoto, Y. Morita-Murase, Y. Nagata, A. Kikuchi and T. Okano, J. Chromatogr., A, 2008, 1191, 157–161 CrossRef CAS.
- T. Yakushiji, K. Sakai, A. Kikuchi, T. Aoyagi, Y. Sakurai and T. Okano, Anal. Chem., 1999, 71, 1125–1130 CrossRef CAS.
- H. Kanazawa, T. Sunamoto, E. Ayano, Y. Matsushima, A. Kikuchi and T. Okano, Anal. Sci., 2002, 18, 45–48 CrossRef CAS.
- K. Nagase, M. Kumazaki, H. Kanazawa, J. Kobayashi, A. Kikuci, Y. Akiyama, M. Annaka and T. Okano, ACS Appl. Mater. Interfaces, 2010, 2, 1247–1253 CAS.
- A. Mizutani, A. Kikuchi, M. Yamato, H. Kanazawa and T. Okano, Biomaterials, 2008, 29, 2073–2081 CrossRef CAS.
- G. Masci, L. Giacomelli and V. Crescenzi, Macromol. Rapid Commun., 2004, 25, 559–564 CrossRef CAS.
- F. O. Ganachaud, M. J. Monteiro, R. G. Gilbert, M.-A. Dourges, S. H. Thang and E. Rizzardo, Macromolecules, 2000, 33, 6738–6745 CrossRef CAS.
- A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525–616 CrossRef CAS.
- F. Roohi and M. Magdalena Titirici, New J. Chem., 2008, 32, 1409–1414 RSC.
- F. Roohi, M. Antonietti and M.-M. Titirici, J. Chromatogr., A, 2008, 1203, 160–167 CrossRef CAS.
- F. Roohi, Y. Fatoglu and M.-M. Titirici, Anal. Methods, 2009, 1, 52–58 RSC.
- K. K. Unger and E. Weber, A Guide to Practical HPLC, GIT VERLAG GMBH, Darmstadt, 1999 Search PubMed.
- H. Engelhardt, M. Arangio and T. Lobert, LC-GC, 1997, 15, 856–858 CAS , 860, 862, 864, 866.
- H. Uyama and S. Kobayashi, Chem. Lett., 1992, 1643–1646 CrossRef CAS.
- N. Adams and U. S. Schubert, Adv. Drug Delivery Rev., 2007, 59, 1504–1520 CrossRef CAS.
- J. Nicolas, V. S. Miguel, G. Mantovani and D. M. Haddleton, Chem. Commun., 2006, 4697–4699 RSC.
- M. Chanana, S. Jahn, R. Georgieva, J.-F. O. Lutz, H. Baumler and D. Wang, Chem. Mater., 2009, 21, 1906–1914 CrossRef CAS.
- E. Wischerhoff, K. Uhlig, A. Lankenau, H. G. Borner, A. Laschewsky, C. Duschl and J. F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 5666–5668 CrossRef CAS.
- E. Wischerhoff, S. Glatzel, K. Uhlig, A. Lankenau, J.-F. o. Lutz and A. Laschewsky, Langmuir, 2009, 25, 5949–5956 CrossRef CAS.
- I. Tan, Z. Zarafshani, J.-F. o. Lutz and M.-M. Titirici, ACS Appl. Mater. Interfaces, 2009, 1, 1869–1872 CAS.
Footnote |
| † Present address: Bayer Schering Pharma, 13353, Berlin. |
| This journal is © The Royal Society of Chemistry 2012 |