NHC-carbene cyclopentadienyl iron based catalyst for a general and efficient hydrosilylation of imines†‡
Luis C. Misal
Castro
,
Jean-Baptiste
Sortais
* and
Christophe
Darcel
*
UMR 6226 CNRS-UR1, “Sciences Chimiques de Rennes”, Team “Catalysis and Organometallics”, Université de Rennes 1, Campus de Beaulieu, 35042 RENNES cedex, France. E-mail: jean-baptiste.sortais@univ-rennes1.fr; christophe.darcel@univ-rennes1.fr; Fax: +33 2 23 23 69 39
First published on 31st October 2011
Abstract
A general and efficient hydrosilylation of imines catalysed by a well defined NHC-carbene cyclopentadienyl iron complex has been developed. Both aldimines and ketimines are converted to the corresponding amines under mild conditions, and under visible light activation.
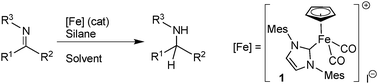
The development of synthetic strategies towards amines has attracted the interest of chemists due to the significance and omnipresence of these compounds in the field of natural products, pharmaceutical and agronomical compounds.1 Among the direct and efficient synthesis of amines, imine catalyzed-reduction methodologies2 (such as hydrogenation,2hydrogen transfer3 or hydrosilylation)4 were developed using transition metals such as ruthenium, iridium, rhodium and are some of the most ubiquitous protocols in organic synthesis. Besides, due to high natural abundance, benign environmental impact, and low cost, iron has emerged as an interesting potential surrogate in catalytic processes for precious transition metals. Interestingly, the last decade has seen an impressive rise of its use as the catalyst and efficient processes are now able to compete with other metal-catalyzed ones.5 In the reduction area,6 several groups have described iron catalyzed hydrogenation6,7 or hydrogen transfer.8 For the reduction of ketimines, some very recent impressive results were made in particular in asymmetric series.9 In addition to these well established catalytic reductions with H2 or transfer hydrogen reagents, hydrosilanes are useful reducing agents and iron catalyzed hydrosilylation of alkenes, aldehydes and ketones were also intensively studied.10 On the other hand, more surprisingly, the apparently simple iron-catalyzed hydrosilylation of aldimines and ketimines is still a challenging transformation as to date, only few isolated examples of iron-catalyzed hydrosilylation of imines were reported.11
During our recent studies on iron-catalyzed transformations,12 and particularly on hydrosilylation reactions,13 we have developed an efficient well-defined complex, [CpFe(IMes)(CO)2]I, for catalytic hydrosilylation of aldehydes, ketones, and amides.14 Herein, we describe the first general hydrosilylation of both aldimines and ketimines involving this iron complex used as the pre-catalyst.
We started our investigation by the reduction of 4-methyl-N-(4-methylbenzylidene)aniline as the model substrate with the iron complex 1 as the pre-catalyst (Scheme 1) under various conditions (Table 1 and Tables S1 and S2 in the ESI‡). Among a variety of silanes, optimal conversions were obtained with 2 equiv. of PhSiH3 under solvent-free conditions at 70 °C for 16 h using 5 mol% of complex 1 (Table 1, entries 1–4). It is noteworthy that the role of visible light irradiation is crucial to obtain a full conversion (entries 4 and 5). Notably, the temperature can be reduced to 30 °C and the pre-catalyst loading to 2 mol%, and we found, eventually, that the reduction occurred with a full conversion at only 30 °C after 30 h with 2 mol% of the pre-catalyst 1 (entry 7).
| Entry | Silane | Solvent | 1 [mol%] | T/°C | Time/h | Conv. [%]b |
|---|---|---|---|---|---|---|
| a Typical conditions: aldimine (0.5 mmol), silane (2 equiv.), complex 1 and solvent were stirred upon visible light irradiation (using 24 watt compact fluorescent lamp); the reaction is then hydrolysed with MeOH and NaOH(aq.) 2 M. b Determined by 1H NMR after extraction with Et2O. c No light activation. | ||||||
| 1 | PhSiH3 | THF | 5 | 70 | 16 | 51 |
| 2 | PhSiH3 | Toluene | 5 | 70 | 16 | 44 |
| 3 | PhSiH3 | Dioxane | 5 | 70 | 16 | 71 |
| 4 | PhSiH3 | Neat | 5 | 70 | 16 | 95 |
| 5c | PhSiH3 | Neat | 5 | 70 | 16 | 24 |
| 6 | PhSiH3 | Neat | 2 | 30 | 16 | 60 |
| 7 | PhSiH3 | Neat | 2 | 30 | 30 | >97 |
| 8 | Ph2SiH2 | Neat | 2 | 30 | 60 | 23 |
With an optimized catalytic system in hand, we then explored the scope of the reaction with several aldimines (Table 2). We have found that the electronic effect at the 4 position on the aniline moieties was limited (entries 1–4). However, for the p-iodo substituted derivative (entry 4), the reaction is complete at 50 °C after 24 h. It is noteworthy that, in some cases, due to miscibility issues, a small amount of toluene was added in order to solubilize all the reactants. When the benzylidene moieties bear both electron donating or withdrawing substituents (entries 4–10), the corresponding amines were isolated with good to high yields.
| Entry | Substrate | Isolated yield (%) | |
|---|---|---|---|
| a Typical procedure: aldimine (0.5 mmol), PhSiH3 (1 mmol), and complex 1 (2 mol%) were stirred at 30 °C without solvent upon visible light irradiation for 30 h, the reaction is then hydrolysed with MeOH and NaOH(aq.) 2 M. b 50 °C, 24 h. c 0.2 mL of toluene as the solvent. d 0.1 mL of toluene as the solvent. e Complex 1 (5 mol%), CH2Cl2 (1 mL). f Conversion by 1H NMR. | |||
| 1 |
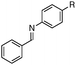
|
R = H | 91 |
| 2 | R = Me | 95 | |
| 3 | R = OMe | 95 | |
| 4b,c | R = I | 95 | |
| 5 |
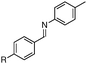
|
R = Me | 95 |
| 6d | R = OMe | 95 | |
| 7d | R = NMe2 | 95 | |
| 8d | R = Br | 92 | |
| 9d | R = Cl | 95 | |
| 10e | R = COCH3 | 71 | |
| 11e | R = CO2CH3 | 30f | |
| 12b,c |
|
94 | |
| 13b,d |
|
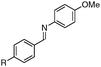
R = CN | 10f |
| 14e | R = NO2 | 15f | |
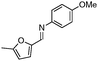
| 15b,d |
|
95 | |
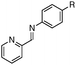
| 16b,d |
|
R = OMe | 95 |
| 17 | R = Br | 53 | |
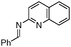
| 18 |
|
63 | |
| 19c |
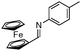
|
83 | |
| 20c |
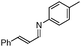
|
36 | |
| 21 |
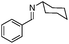
|
26 | |
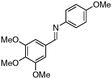
Notably, no catalytic dehalogenation occurred during this reaction (entries 4, 8 and 9). More strikingly, functional carbonyl groups such as ketones and esters were not altered under these catalytic conditions (entries 10 and 11), although the conversion of the ester derivative was moderate (30%), no product resulting from the reduction of the ester group was detected. Surprisingly, low conversions were observed for the hydrosilylation of 4-nitro- and 4-cyanobenzylidene derivatives but no reduction of the nitro or cyano15 group was observed, and 80% of the starting material was recovered in the case of the cyano derivative (entries 13 and 14). More interestingly, heteroaromatic imines (entries 15–18), which are potentially good bidentate ligands for the iron centre, were reduced with good to excellent yields. Likewise, 4-methyl-N-(ferrocenylmethylidene)-aniline was totally reduced and led to the corresponding amine with good yield (entry 19). Finally, the reduction of 4-methyl-N-cinnamylideneaniline afforded selectively the amine resulting from the 1,2-reduction in 36% yield16 (entry 20).
Given the very good activity of the catalytic system for aldimines, we investigated its potential for reduction of ketimines, with 4-methyl-N-[1-(4-methylphenyl)ethylidene]-aniline as the model substrate. Compared with aldimines, 90% conversion was reached under slightly harsher conditions, 24 h at 100 °C under neat conditions with 5 mol% of pre-catalyst, and 2 equiv. of PhSiH3 as the silane were needed (Table 3, entry 6). Notably, when performing in THF, toluene or dioxane, or with lower catalyst loading, the conversion dramatically decreased (Table 3, entries 1–3, 7).
| Entry | Silane | Solvent | 1 [mol%] | T/°C | Time/h | Conv. [%]b |
|---|---|---|---|---|---|---|
| a Typical conditions: ketimine (0.5 mmol), silane (2 equiv.), pre-catalyst 1 and solvent were stirred upon visible light irradiation; the reaction is then hydrolysed with MeOH and NaOH(aq.) 2 M. b Determined by 1H NMR after extraction with Et2O. | ||||||
| 1 | PhSiH3 | THF | 5 | 70 | 16 | 9 |
| 2 | PhSiH3 | Toluene | 5 | 70 | 16 | 11 |
| 3 | PhSiH3 | Dioxane | 5 | 70 | 16 | 9 |
| 4 | PhSiH3 | Neat | 5 | 70 | 16 | 49 |
| 5 | PhSiH3 | Neat | 5 | 100 | 16 | 75 |
| 6 | PhSiH3 | Neat | 5 | 100 | 24 | 90 |
| 7 | PhSiH3 | Neat | 2 | 100 | 16 | 18 |
The scope of the reaction was thus surveyed. With several ketimine derivates from substituted acetophenone and 4-substituted anilines (Table 4, entries 1–8), the corresponding amines where obtained in good to high yields (83–95%). The reaction proceeds well even with ortho-substituted phenylethylidene aniline or naphthylethylidene toluidine (Table 4, entries 8 9, and 11), which shows a low impact of the steric hindrance on to the reaction. The ferrocenyl imine derivative was also converted into the corresponding amine, but with a moderate yield (Table 4, entry 10). More strikingly, the reaction could be performed with N-alkylimines and aliphatic ketimines under the same conditions with good yields (Table 4, entries 12 and 13).
| Entry | Substrate | Isolated yield (%) | |
|---|---|---|---|
| a Typical procedure: ketimine (0.5 mmol), PhSiH3 (1 mmol), and pre-catalyst 1 (5 mol%) were stirred at 100 °C without solvent upon visible light irradiation for 24 h, the reaction is then hydrolysed with MeOH and NaOH(aq.) 2M. b 0.1 mL of toluene as solvent. c 0.2 mL of toluene as the solvent. | |||
| 1b |
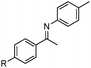
|
R = H | 95 |
| 2 | R = Me | 90 | |
| 3c | R = OMe | 83 | |
| 4b | R = Br | 90 | |
| 5b |
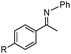
|
R = F | 90 |
| 6b | R = CF3 | 91 | |
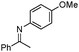
| 7b |
|
93 | |
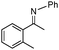
| 8b |
|
91 | |
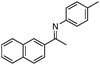
| 9b |
|
87 | |
| 10b |
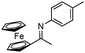
|
57 | |
| 11b |
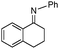
|
78 | |
| 12b |
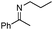
|
86 | |
| 13b |
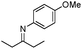
|
95 | |
In summary, we have developed the first general and efficient catalytic system for the hydrosilylation of both aldimines and ketimines using a well defined iron complex as the pre-catalyst which gives access to various amines. Compared to other transition metals used as catalysts in hydrosilylation reactions, the activity of this iron catalyst is competitive for the reduction of aldimines. Moreover, it is noteworthy that several functional groups easy to reduce such as ketone, ester and alkene are tolerated with this system. Further investigations into the reactivity of such complexes and especially directed towards the mechanism of this reaction are currently underway in our laboratory.
CNRS, Rennes Métropole, Ministère de la Recherche et l'Enseignement Supérieur are acknowledged for financial support, and the Ministère des affaires étrangères and the Fundacion Gran Mariscal de Ayacucho for a grant to L.C.M.C.
Notes and references
- (a) R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparation, Wiley-VHC, New York, 1989 Search PubMed; (b) S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, 2004 Search PubMed; (c) K. S. Hayes, Appl. Catal., A, 2001, 221, 187–195 CrossRef CAS.
- (a) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS; (b) N. Fleury-Brégeot, V. de la Fuente, S. Castillon and C. Claver, ChemCatChem, 2010, 2, 1346–1371 Search PubMed; (c) Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elservier, Wiley-VCH, Weinheim, 2007 Search PubMed; (d) Transition Metals for Organic Synthesis, ed. M. Beller and C. Bolm, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed; (e) A. Fabrello, A. Bachelier, M. Urrutigoïty and P. Kalck, Coord. Chem. Rev., 2010, 254, 273–287 CrossRef CAS.
- (a) S. Gladiali and E. Alberico, in Transition Metals for Organic Synthesis, ed. M. Beller and C. Bolm, Wiley-VCH, Weinheim, 2004, vol. 2, p. 145 Search PubMed; (b) S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS.
- (a) O. Riant, N. Mostefaï and J. Courmarcel, Synthesis, 2004, 2943–2958 CrossRef CAS; (b) J.-F. Carpentier and V. Bette, Curr. Org. Chem., 2002, 6, 913–936 CrossRef CAS.
- (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS; (b) B. Plietker, in Iron Catalysis in Organic Chemistry, ed. Bernd Plietker, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS; (d) A. Correa, O. Garcia Mancheno and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108–1117 RSC; (e) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS; (f) W. M. Czaplik, M. Mayer, J. Cvengros and A. Jacobi von Wangelin, ChemSusChem, 2009, 2, 396–417 CrossRef CAS; (g) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS.
- Representative review on iron-catalyzed reduction: (a) K. Junge, K. Schröder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC; (b) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282–2291 RSC.
- Selected papers on iron-catalyzed hydrogenation: (a) C. Bianchini, A. Meli, M. Peruzzini, P. Frediani, C. Bohanna, M. A. Esteruelas and L. A. Oro, Organometallics, 1992, 11, 138–145 CrossRef CAS; (b) S. Enthaler, B. Hagemann, G. Erre, K. Junge and M. Beller, Chem.–Asian J., 2006, 1, 598–604 CrossRef; (c) C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816–5817 CrossRef CAS; (d) C. Sui-Seng, F. Freutel, A. J. Lough and R. H. Morris, Angew. Chem., Int. Ed., 2008, 47, 940–943 CrossRef CAS.
- Selected papers on iron catalyzed transfer hydrogenation: (a) S. Enthaler, G. Erre, M. K. Tse, K. Junge and M. Beller, Tetrahedron Lett., 2006, 47, 8095–8099 CrossRef CAS; (b) C. Bianchini, E. Farnetti, M. Graziani, M. Peruzzini and A. Polo, Organometallics, 1993, 12, 3753–3761 CrossRef CAS; (c) A. Mikhailine, A. J. Louggh and R. H. Morris, J. Am. Chem. Soc., 2009, 131, 1394–1395 CrossRef CAS; (d) Buchard, H. Heuclin, A. Auffrant, X. F. Le Goff and P. Le Floch, Dalton Trans., 2009, 1659–1667 RSC; (e) N. Meyer, A. J. Lough and R. H. Morris, Chem.–Eur. J., 2009, 15, 5605–5610 CrossRef CAS; (f) A. Naik, T. Maji and O. Reiser, Chem. Commun., 2010, 46, 4475–4477 RSC.
- (a) S. Zhou, S. Fleischer, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5120–5124 CrossRef CAS; (b) S. Zhou, S. Fleischer, K. Junge, S. Das, D. Addis and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8121–8125 CrossRef CAS.
- Representative examples of iron-catalyzed hydrosilylation: (a) S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794–13807 CrossRef; (b) H. Nishiyama and A. Furuta, Chem. Commun., 2007, 760–762 RSC; (c) N. S. Shaikh, K. Junge and M. Beller, Org. Lett., 2007, 9, 5429–5432 CrossRef CAS; (d) N. S. Shaikh, S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 2497–2501 CrossRef CAS; (e) A. M. Tondreau, E. Lobkovsky and P. J. Chirik, Org. Lett., 2008, 10, 2789–2792 CrossRef CAS; (f) B. K. Langlotz, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2008, 47, 4670–4674 CrossRef CAS; (g) A. M. Tondreau, J. M. Darmon, B. M. Wile, S. K. Floyd, E. Lobkovsky and P. J. Chirik, Organometallics, 2009, 28, 3928–3940 CrossRef CAS; (h) T. Inagaki, L. T. Phong, A. Furuta, J.-I. Ito and H. Nishiyama, Chem.–Eur. J., 2010, 16, 3090–3096 CrossRef CAS.
- Only two examples dealing with the hydrosilylation of the aldimine PhCH = NPh were mentioned: (a) Ref. 10b ; (b) S. Zhou, K. Junge, D. Addis, S. Das and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 9507–9510 CrossRef CAS . Furthermore, an elegant iron-catalyzed reductive amination of aldehyde and aniline derivatives using FeCl3 and PHMS was reported: S. Enthaler, ChemCatChem, 2010, 2, 1411–1415 Search PubMed.
- (a) X.-F. Wu and C. Darcel, Eur. J. Org. Chem., 2009, 1144–1147 CrossRef CAS; (b) X.-F. Wu, D. Bezier and C. Darcel, Adv. Synth. Catal., 2009, 351, 367–370 CrossRef CAS; (c) X.-F. Wu, C. Vovard-Le Bray, L. Bechki and C. Darcel, Tetrahedron, 2009, 65, 7380–7384 CrossRef CAS; (d) X.-F. Wu and C. Darcel, Eur. J. Org. Chem., 2009, 4753–4765 CrossRef CAS.
- L. C. Misal Castro, D. Bézier, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2011, 353, 1279–1284 CrossRef.
- (a) F. Jiang, D. Bézier, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2011, 353, 239–244 CrossRef CAS; (b) D. Bézier, G. T. Venkanna, J.-B. Sortais and C. Darcel, ChemCatChem DOI:10.1002/cctc.201100202; (c) D. Bézier, F. Jiang, J.-B. Sortais and C. Darcel, Eur. J. Inorg. Chem., 2000 DOI:10.1002/ejic.201100762.
- (a) H. Nakazawa, K. Kamata and M. Itazaki, Chem. Commun., 2005, 4004–4006 RSC; (b) H. Nakazawa, M. Itazaki, K. Kamata and K. Ueda, Chem.–Asian J., 2007, 2, 882–888 CrossRef CAS; (c) H. Nakazawa, T. Kawasaki, K. Miyoshi, C. H. Suresh and N. Koga, Organometallics, 2004, 23, 117–126 CrossRef CAS.
- The conversion was 36% and only starting material and product were detected in the crude 1H NMR spectrum.
Footnotes |
| † Dedicated to Dr Christian Bruneau on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cc14403k |
| This journal is © The Royal Society of Chemistry 2012 |