Dehydrogenation processes via C–H activation within alkylphosphines
Mary
Grellier
*ab and
Sylviane
Sabo-Etienne
*ab
aCNRS, LCC (Laboratoire de Chimie de Coordination), 205 Route de Narbonne, F-31077 Toulouse, France. E-mail: mary.grellier@lcc-toulouse.fr; sylviane.sabo@lcc-toulouse.fr
bUniversité de Toulouse, UPS, INPT, F-31077 Toulouse, France
First published on 28th September 2011
Abstract
Phosphines are commonly used in organometallic chemistry and are present in a wide variety of catalytic systems. This feature article highlights the advances made in dehydrogenation processes occurring within alkylphosphines, with the aim of further developing catalytic processes involving C–H activation together with potential applications in the field of hydrogen storage.
 Mary Grellier | Mary Grellier received his PhD in 1996, from the University of Strasbourg, with Michel Pfeffer. He obtained a “Professeur Agrégé” position in Strasbourg in 1995. In 2000, he was promoted to “Maître de Conférences” at the University of Toulouse. In 2010, he received his HDR habilitation. His research at the LCC mainly concerns coordination and catalysis in organometallic chemistry. |
 Sylviane Sabo-Etienne | Sylviane Sabo-Etienne received her “Doctorat d'Etat” from the University of Toulouse. In 1985, she moved to the University of Brest, to work as “Chargée de Recherche CNRS”. After a year as a NSF-CNRS research associate with Maurice Brookhart (USA), she returned to Toulouse to work in collaboration with Bruno Chaudret on polyhydride chemistry. She was promoted “Directrice de Recherche” in 1997. In 2010, she received the RSC Frankland award, and the Glenn T Seaborg Memorial Lecturer award by the University of California, Berkeley. She is currently a group leader at the LCC and her research interests include organometallic chemistry and catalysis. |
A. Introduction
Homogeneous hydrogenation processes are often catalyzed by transition metal complexes featuring phosphine ligands in their coordination spheres. Significant progress has been made in preparing a wide variety of phosphines with varying electronic and steric properties and/or hapticity.1 Very often, the phosphine is regarded as a spectator ligand, but the problems of phosphine dissociation and consequent catalyst decomposition are recurrent. Depending on the class of phosphine and the conditions used, additional reactivity is observed and is of interest on its own right. This is indeed the case for alkylphosphines which can favour C–H activation.Chatt was a pioneer in this field through his demonstration of a C–H oxidative cleavage involving the bis(diphosphine) complex Ru(dmpe)2 and the hydrido species RuH(CH2PMe(CH2)2)PMe2)(dmpe) [(dmpe = Me2P(CH2)2PMe2)].2 It is remarkable that solid evidence for the origin of the hydride from the diphosphine was provided by NMR and IR labelling experiments. These early results were the origin of major developments in the field of C–H activation, an area that is now experiencing exponential growth with respect to catalytic applications.3
Taking into account the ability of an alkylphosphine to induce dehydrogenation processes, this article will focus on some recent advances linked to catalysis4 and to hydrogen storage.5 One of the hurdles in this area is the design of systems able to induce reversible dehydrogenation reactions. We have been working for several years on the chemistry of ruthenium polyhydrides and more particularly on the properties of dihydrogen complexes.6 Until recently, our preferred tool was the bis(dihydrogen) complex RuH2(H2)2(PCy3)2 (1) (Fig. 1).7 This compound, stabilized by two bulky and basic tricyclohexylphosphines (PCy3 = P(C6H11)3), was prepared in the early 80s by Bruno Chaudret et al. and first formulated as a hexahydride complex.8 The seminal paper of Kubas et al. in 1984 opened a new area in coordination chemistry and several polyhydrides were then reformulated as dihydrogen complexes.9 This was the case for the ruthenium bis(dihydrogen) complex 1 which proved to be a versatile starting material.10 For reasons directly linked to this article and detailed later on, we now focus on the corresponding complex RuH2(H2)2(PCyp3)2 (2) which incorporates tricyclopentylphosphines (PCyp3 = P(C5H9)3) (Fig. 1).11
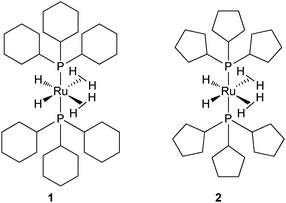 | ||
| Fig. 1 Two bis(dihydrogen) ruthenium complexes: PCy3vs. PCyp3. | ||
In this article we will first examine some key data relating to systems incorporating tricyclohexylphosphines and then will detail the most recent findings with the smaller cycloalkyl ring, i.e. tricyclopentylphosphine. These two phosphines display the several salient features that we want to focus on. In addition, we have selected some miscellaneous results relating to further trialkylphosphines that exhibit interesting features.
B. Tricyclohexylphosphine complexes
In 1978, Vrieze et al. reported partial dehydrogenation of PCy3 in iridium and rhodium systems.12 Addition of an excess of PCy3 to [MCl(coe)2]2 (M = Ir (3), Rh, coe = cyclooctene) in refluxing toluene led to a mixture including two isomers of four-coordinated metal(I) complexes MCl[PCy2(η2-C6H9)](PCy3) (4a–b) in which one phosphine had been partially dehydrogenated (Fig. 2). The double bond of the cyclohexenyl ring of the new ligand PCy2(η2-C6H9) was coordinated to the metal center as evidenced by NMR, IR and reactivity studies. Notably in the iridium case, oxidative addition was observed upon H2 bubbling, but without modifying the coordination mode of the PCy2(η2-C6H9) ligand. The authors postulated the intermediacy of a hydrido metalated product followed by β-hydrogen abstraction either through mononuclear or binuclear species.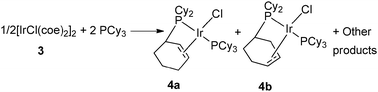 | ||
| Fig. 2 PCy2(η2-C6H9) ligands at iridium. | ||
Shortly after the publication of the first dihydrogen complex,9a the group of Kubas reported the X-ray structure of W(CO)3(PCy3)2 and evidenced an agostic interaction in one of the cyclohexyl rings.13 This agostic interaction was easily displaced by several ligands including dihydrogen. In 1991, Chaudret et al. reported two ruthenium systems where the PCy3 ligand was dehydrogenated. Protonation of Cp*RuH3(PCy3) (5) led to H2 evolution and formation of [Cp*Ru{PCy2(C6H9)}]+ (6) (Fig. 3).14 In the latter compound, the phosphine ligand was not only coordinated through the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond but also displayed a strong agostic interaction well characterized by NMR and X-ray data. Dehydrogenation of PCy3 was also observed upon reaction of the dihydride complex RuH2(OCOCF3)2(PCy3)2 with cyclooctene.15
C double bond but also displayed a strong agostic interaction well characterized by NMR and X-ray data. Dehydrogenation of PCy3 was also observed upon reaction of the dihydride complex RuH2(OCOCF3)2(PCy3)2 with cyclooctene.15
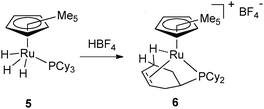 | ||
| Fig. 3 PCy2(η2-C6H9) ligand featuring an agostic interaction at ruthenium. | ||
The ethylene complex RuH[(η3-C6H8)PCy2](C2H4)(PCy3) (7) resulting from the reaction of 1 with ethylene turned out to be an excellent catalyst precursor for the selective dehydrogenation silylation of ethylene into vinylsilane. Remarkably in complex 7, the partially dehydrogenated cyclohexenyl ring is bonded to the metal in an allylic fashion.16 The dehydrogenation process was fully reversible under an atmosphere of dihydrogen,7 whereas silane addition led to a complex preserving the PCy2(η3-C6H8) ligand. This species, first proposed as resulting from silane oxidative addition, was formulated as a σ-silane complex RuH[(η3-C6H8)PCy2](σ-HSiEt3)(PCy3) after an in-depth analysis through multinuclear NMR, X-ray and DFT data on the analogous chlorosilane complex RuH[(η3-C6H8)PCy2](σ-HSiMe2Cl)(PCy3) (8) (Fig. 4).17 This is a unique example where two different JSi–H coupling constants were measured, illustrating the presence of a σ-Si–H bond and a SISHA interaction (secondary interaction between silicon and hydrogen atoms).18
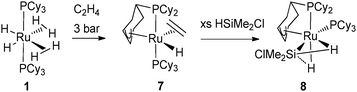 | ||
| Fig. 4 Dehydrogenation of a cyclohexyl ring featuring an allylic coordination at ruthenium. | ||
Further dehydrogenation of 1 was observed after reactions with functionalized olefins. Indeed, the addition of 5 equiv. of CH2CHtBu produced the compound RuH[(η3-C6H8)PCy2][(η2-C6H9)PCy2] (10) incorporating two partially dehydrogenated phosphines, one displaying an allylic bonding mode and the second one coordinated to the metal through a CC bond (Fig. 5).7 This compound could also be prepared in the absence of a hydrogen acceptor by heating 1 in the solid state above 90 °C.19
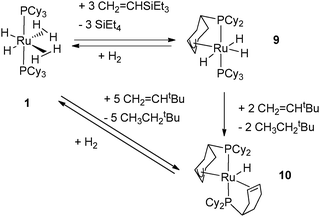 | ||
| Fig. 5 Dehydrogenation of cyclohexyl rings in two different modes. | ||
Allylic coordination of a cyclohexenyl ring was also observed in a ruthenium system by thermolysis of Ru(2-methylpropenyl)2(cod) (11) in the presence of Cy2P(CH2)nPCy2 with n = 3, 4 at 95 °C and 50 °C, respectively, leading to Ru{(η3-C6H8)CyP(CH2)nPCy2}(η3-C8H13) (12) (Fig. 6).20
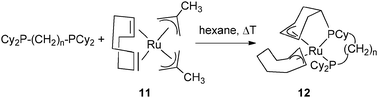 | ||
| Fig. 6 Dehydrogenation of a cyclohexyl ring in a chelating phosphine. | ||
Thermal dehydrogenation, without the need for a hydrogen acceptor, was also achieved when reacting a dioxane solution of [RuCl2(cymene)]2 (13) at 70 °C for 3 days with 1 equiv. of PCy3: partial dehydrogenation of the phosphine and cymene displacement led to the formation of the dinuclear chloro-bridged compound (cymene)Ru(μ-Cl)3RuCl{PCy2(η2-C6H9)} (14) (Fig. 7).21
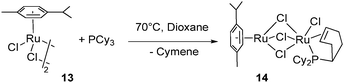 | ||
| Fig. 7 Thermal dehydrogenation of a cyclohexyl ring. | ||
The benzylidene complex Ru(CHPh)Cl[N(iPr2PS)2](PCy3) (15) reacted with NaOMe at 45 °C to produce the hydride RuH{N(iPr2PS)2}{PCy2(η2-C6H9)} (16) presumably through hydride migration to the alkylidene and partial dehydrogenation of the phosphine (Fig. 8).22 Closely related was the thermolysis of the benzylidene complex Ru(Tai)Cl(CHPh)(PCy3) (17) at 85 °C leading to the formation of Ru(Tai)Cl[PCy2(η2-C6H9)] (18) where Tai = HB(7-azaindolyl)3. Here also, the benzylidene acts as a hydrogen acceptor (Fig. 8).23
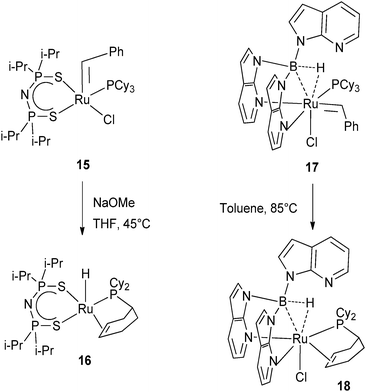 | ||
| Fig. 8 Benzylidene ligands acting as hydrogen acceptors. | ||
The unusual rearrangement affording the complex [CpRu{(η3-CH2C(CH2)3CCH2PCy2(η2-C6H9)]+ (21) resulted from the addition of 1,6-heptadiyne to [CpRu(NCCH3)2(PCy3)]+ (19) through an intermediate allyl carbene 20.24 In this system, the phosphonium ligand is coordinated to the metal centre through the CC double bond at the α position of the phosphorus, which is not bound to the ruthenium (Fig. 9).
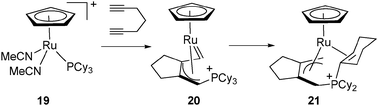 | ||
| Fig. 9 Dehydrogenation of a cyclohexyl ring in a phosphonium ligand. | ||
A series of carborane rhodium complexes incorporating PR3 ligands with R = iPr (23), Cyp (24), Cy (25) illustrate the different reactivities of these rather similar alkyl phosphines. In the case of PiPr3, alkene coordination was observed upon addition of CH2CHtBu to the dihydride complex 22, whereas partial dehydrogenation of the phosphine occurred in the case of the two cyclic substituents (Fig. 10).25
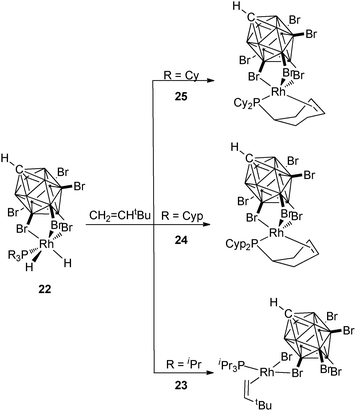
C. Tricyclopentylphosphine complexes
In comparison to the booming development of the chemistry of tricyclohexylphosphine complexes, Grubbs' catalyst being the most famous example,26 compounds incorporating tricyclopentylphosphine as a ligand received little attention until 2005. Possible explanations for this late arrival in interest could be (i) a higher commercial price; (ii) an easier oxidation; (iii) its liquid state (under normal conditions: rt and 1 atm) and (iv) little influence expected by reducing the size of the cycloalkyl ring, the two phosphines displaying similar basicity and steric hindrance. However there is a precedent for the use of tricyclopentyl rings in place of tricyclohexyl in overcoming solubility problems.27As mentioned above, our group has extensively developed the chemistry of the bis(dihydrogen) complex 1 by incorporating two tricyclohexylphosphines.6 During the course of reactivity and catalytic studies, we evidenced extremely facile reversible dehydrogenation pathways of either one or two cyclohexyl rings (Fig. 4 and 5).7,16,17,28 In some cases, this could be considered as an advantage: the phosphine can stabilize the metal center by acting as a bidentate ligand through P and η3- or η2-cycloalkenyl coordination. However, in catalysis, this might induce selectivity problems as a result of two competing cycles, one with a RuP2 fragment and one with a RuPP' fragment. We thus reasoned that dehydrogenation through C–H activation could be prevented by reducing the size of the cycloalkyl ring. The overall conformation adopted by cyclopentyl rings should be quite different from that defined by cyclohexyl rings. By analogy to the synthetic pathway used for the preparation of 1, we were able to isolate the bis(tricyclopentylphosphine) complex RuH2(H2)2(PCyp3)2 (2) (Fig. 1) which was fully characterized by neutron structure determination.11 A similar strategy was adopted by Weller's group for rhodium. The isoelectronic cationic rhodium complex [RhH2(η2-H2)2(PCyp3)2]+ (27) was prepared following the synthetic pathway employed for the analogous tricyclohexylphosphine complex.29
2 is less thermally stable than 1. Indeed, complex 2 can be kept for a few hours at room temperature under an argon atmosphere, instead of weeks for 1, before loss of dihydrogen and formation of a red dimeric compound formulated as Ru2H6(PCyp3)4. Its structure differs from that previously published for the analogous PCy3 dimeric complex.10a,30 A comparative study including a neutron structure determination will be published in due course.31
In contrast, the rhodium bis(dihydrogen) species 27 and its PCy3 analogue are only stable under a H2 atmosphere. Complexes 2 and 27 can be cleanly dehydrogenated in the presence of a hydrogen acceptor, ethylene or tert-butylethylene (tbe) to generate 26 (Fig. 11) and 28a–b (Fig. 12), respectively. In both cases, the dehydrogenation process is reversible. Complexes 26 and 28a–b not only result from dihydrogen evolution of the H2 and hydride ligands but also from partial dehydrogenation of the two phosphines leading to two cyclopentene rings coordinated to the metal center. Such a dehydrogenation process leads to the reduction of the metal center, Ru(II) to Ru(0) and Rh(III) to Rh(I), respectively. In contrast, the tricyclohexylphosphine ruthenium analogue leads to various Ru(II) complexes, when using C2H4 or tbe as a hydrogen acceptor, thus showing a huge difference in the final dehydrogenated product and highlighting the influence of the cycloalkyl substituents.7,16
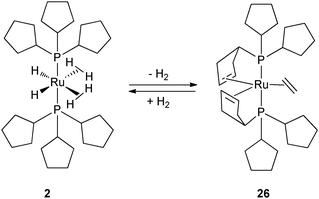 | ||
| Fig. 11 Reversible dehydrogenation of tricyclopentylphosphine ligands at the bis(dihydrogen) ruthenium complex 2. | ||
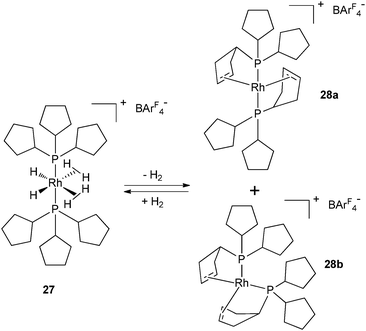 | ||
| Fig. 12 Reversible dehydrogenation of tricyclopentylphosphine ligands at the bis(dihydrogen) rhodium complex 27. | ||
The dehydrogenation process from 2 and 27 involves the loss of ten hydrogen atoms per metal, which represents 1.7% and 0.7% of the weight, respectively. These moderate values are far away from the DOE target,32 but give a unique opportunity to look at systems incorporating various hydrogen sources (hydrides, dihydrogen, C–H). It is remarkable that the dehydrogenation process is reversible in solution at low pressure of H2 (1–3 atm) and room temperature. In the context of hydrogen storage, the possibility to reversibly dehydrogenate auxiliary ligands is an attractive pathway to use them as a hydrogen source. In order to open this new route to further applications, it was important to demonstrate the viability of the process in the solid state. Weller's group has evidenced such a process on complexes 29 and 30 involving three types of hydrogen sources ((η2-H2), hydrides and C–H cycloalkyl protons) by 31P solid state NMR monitoring (Fig. 13).33 In solution, the formation of the rhodium(III) complex 31 was postulated on the basis of NMR data. The high field hydride chemical shift (−20.5 ppm) is in agreement with a weakly coordinating trans-ligand such as an agostic cycloalkyl or a solvent molecule. C–H bond activation followed by β-elimination was proposed for the last step. When 31 was placed under extended vacuum, 30 was recovered, in agreement with the intermediacy of 31 in the dehydrogenation process.
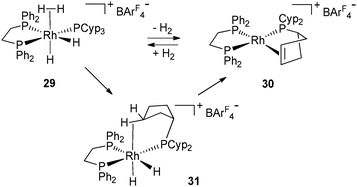 | ||
| Fig. 13 Dehydrogenation of PCyp3 both in solid and solution states. | ||
In the ruthenium system, it was also possible to trap intermediates. Indeed, upon short exposure to ethylene (2 min, 3 bar) 2 rapidly transfers 8 hydrogen atoms leading to a mixture of three dihydride complexes 32a–c. One of the three complexes has been isolated and identified by the X-ray structure as an unusual trans-dihydride complex 32a. On the basis of NMR studies, 32b–c have been proposed to be a mixture of isomers in equilibrium with 32a (Fig. 14).34
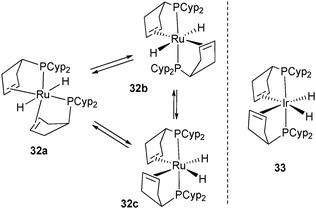 | ||
| Fig. 14 Intermediates in the reversible dehydrogenation of tricyclopentylphosphine ligands at ruthenium. | ||
The Ir complex 33 analogue to the ruthenium species 32c has been isolated by Weller et al. and fully characterized by X-ray diffraction (Fig. 14).29 It is possible that the cis geometry of the hydrides is required before reduction to Ru(0) (26) and Rh(I) (28a–b), respectively. This pre-organization might facilitate the reductive elimination of H2 and place the two alkene substituents in close proximity to their final positions. A bent coordination for the alkene fragments observed in several ruthenium complexes is in agreement with the occupancy of two of the three trigonal coordination available sites in 26 and 34 (Fig. 15), the carbonyl analogue of 26.35 This bent mode also observed in the rhodium(I) complex 28a was explained by DFT calculations. The bent bis(ethylene) model [Rh(C2H4)2(PH3)2]+ is more stable than the “rigorously” trans alkene isomer (+13.3 kJ mol−1) due to enhanced π-back donation from the Rh dz2 orbital to the alkene π* orbitals.29
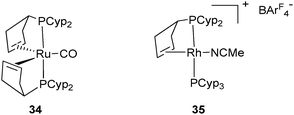 | ||
| Fig. 15 Ruthenium(0) and Rh(I) complexes. | ||
From the data described above, one can postulate that the presence of two PCyp3 in the coordination sphere of the metal always leads to the dehydrogenation of two cyclopentyl substituents. The trapping of intermediates from 27 was made possible by the use of acetonitrile, which enabled the isolation of the Rh(I) complex 35, in which only one cyclopentyl ring of one of the two phosphine ligands was dehydrogenated (Fig. 15). The preparation of mono-dehydrogenated phosphine complexes has also been conducted by our group. Reduction of electron density at ruthenium by coordination of one carbonyl ligand led to the formation of RuH2(η2-H2)(CO)(PCyp3)2 (36), which releases H2 in the presence of tbe as a hydrogen acceptor and affords complex 37 in which only one cyclopentyl ring has been dehydrogenated (Fig. 16). Further dehydrogenation was achieved by exposure of 37 to ethylene leading to reduction to a new ruthenium(0) complex 38. The system is reversible under a H2 atmosphere, and hydrogenation of complexes 38 and 37 to complex 36 was easily achieved (Fig. 16).35 The formation of 38 indicates that despite the presence of a hydrogen acceptor, dehydrogenation of the second phosphine ring can compete with coordination of another substrate.
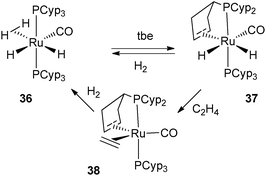 | ||
| Fig. 16 Reversible dehydrogenation of only one cyclopentyl ring. | ||
This competitive process has also been observed in the case of Rh complexes. At low temperatures, it is possible to isolate complex 40 resulting from the reaction of [Rh(nbd)Cl]2 (39) with NaBArF4 in the presence of 2 equivalents of PCyp3. However, 40 is not stable in solution and slowly transfers hydrogen from the cyclopentyl fragments to norbornadiene, thus affording 28a and 28b (Fig. 17 and 12).29
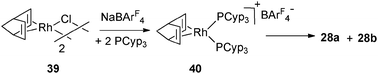 | ||
| Fig. 17 Intermediate 40 in the dehydrogenation process. | ||
The synthesis of 35 and 37 suggests that the dehydrogenation process should also be possible for a system bearing only one tricyclopentylphosphine. This is indeed the case at Rh and Ir, as illustrated in Fig. 18–20.36 Complex 42 is an interesting source of a “[(η2-cyclopentenyl)PCyp2M]+” fragment due to the weak coordination of the arene ring. It has been demonstrated by Hayashi et al. that hybrid phosphine–olefin type ligands may offer advantages over chelating phosphines or coordinated diene in rhodium catalysed 1,4-additions.3742 was tested and found to be efficient for such a catalysis.36b42 is also a good precursor for catalysed dehydrogenation processes of amineboranes. Recently, Weller et al. isolated complex 43 by incorporating a phospha-alkene ligand and two amineboranes κ1 and κ2 coordinated to the metal center (Fig. 19).38
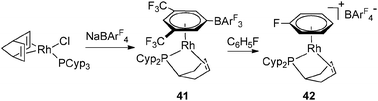
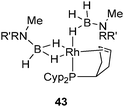 | ||
| Fig. 19 Stabilization of amineborane ligands at rhodium. | ||
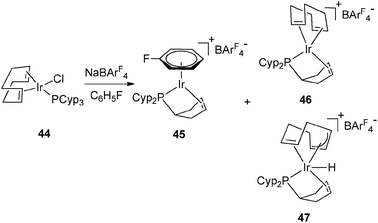 | ||
| Fig. 20 Dehydrogenation at iridium in arene solvents. | ||
The presence of a hydrogen acceptor as a ligand is helpful for hydrogen transfer from the cyclopentyl ring to the alkene. The choice of the hydrogen acceptor is important for selectivity. In the case of the Ir series, cyclooctadiene was preferred over nbd (Fig. 20). A mixture of different complexes was obtained even in arene solvents. When using C6H5F, the arene iridium complex (45) was formed together with [Ir(η2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :η2-C8H12){PCyp2(η2-C5H7)}]+ (46) and [IrH(η2:η3-C8H11){PCyp2(η2-C5H7)}]+ (47). Complex 45 could also be prepared in two steps from the mixture 46–47 (Fig. 20).39
:η2-C8H12){PCyp2(η2-C5H7)}]+ (46) and [IrH(η2:η3-C8H11){PCyp2(η2-C5H7)}]+ (47). Complex 45 could also be prepared in two steps from the mixture 46–47 (Fig. 20).39
The differences observed for C–H bond activation between cyclopentyl and cyclohexyl rings have been investigated using DFT calculations. The study made on simplified ligands (PH2Cyp versus PCyp3 and PH2Cy versus PCy3) showed thermodynamic and kinetic preferences for the first step of the dehydrogenation process. The thermodynamic balance is slightly favourable (ΔE = −0.5 kcal mol−1 and ΔG = −1 kcal mol−1) to activate the cyclopentyl agostic C–H bound to a Rh(III)-alkyl hydride and disfavored (ΔE = +4.8 kcal mol−1 and ΔG = +6.0 kcal mol−1) for the cyclohexyl fragment in the same environment. The activation barrier is also higher for the cyclohexyl system. Following this first activation step, the cyclohexyl fragment needs a conformational change from chair to twist-boat before ensuring β–H elimination. This conformation being obtained, all the different points calculated for the cyclohexylphosphine species (TS and intermediates) are on average +5 kcal mol−1 above the cyclopentyl analogues. This difference has been tested experimentally for a phosphine bearing cyclopentyl and cyclohexyl rings on the same phosphorous atom (PCy2Cyp). The activation of PCy2Cyp in complex 48, which produces 49 as a sole complex, leaves no doubt as to the facility of dehydrogenation of cyclopentyl versus cyclohexyl fragments with the corresponding formation of a phospha-η2-alkene ligand (Fig. 21).29
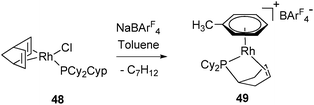 | ||
| Fig. 21 Preferred dehydrogenation at cyclopentyl vs. cyclohexyl rings. | ||
D. Triisopropylphosphine complexes
A series of osmium complexes incorporating dehydrogenated triisopropylphosphine ligands have been reported by Esteruelas et al.40 Dehydrogenation of one of the isopropyl substituents to a vinyl fragment is assisted by the presence of an alkyne acting as a hydrogen acceptor in the coordination sphere of the starting material 50 (Fig. 22). It is particularly interesting to note that upon reaction with H2, 51 is in equilibrium with the product of oxidative addition 52 in which the isopropenyl substituent is no longer coordinated to the metal, thus illustrating hemilabile character (Fig. 22).41 Moreover, the group of Esteruelas demonstrated that functionalization of the isopropenyl diisopropyl phosphine ligand is readily achieved into a variety of products such as α-allylphosphines via [2+2] cycloadditions, iminophosphines by insertion of the CN bond of benzonitriles into the C–H bond of the isopropenyl group, or dienylphosphines from reactions with alkyne or alkynols.40a Another feature of the reversible dehydrogenation process within PiPr3 was demonstrated by Glaser and Tilley in a Cp* osmium system with the preparation of the silylene complex 55 (Fig. 22).42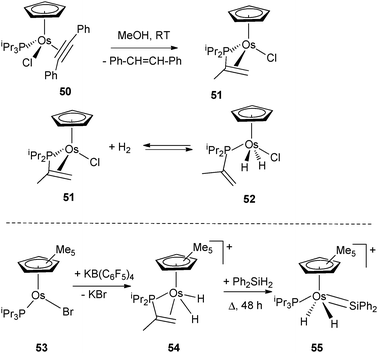 | ||
| Fig. 22 Dehydrogenation of triisopropylphosphine at osmium. | ||
The X-ray structure of the cationic tris(triisopropylphosphine) rhodium complex 56 evidences a β-agostic interaction of one of the isopropyl substituents, the complex thus achieving a 16 electron configuration (Fig. 23). Spontaneous dehydrogenation of the isopropyl substituent is observed with the formation of an equilibrium mixture of two complexes 57 and 58 as shown in Fig. 23. 57 results from dehydrogenation of one of the isopropyl groups into a vinyl fragment, whereas 58 is produced from the reaction of 56 with liberated H2. β–H transfer from the β–CH agostic bond is supported by DFT calculations.43
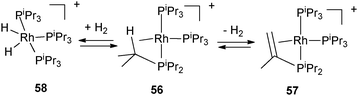 | ||
| Fig. 23 Dehydrogenation of triisopropylphosphine at rhodium. | ||
E. Miscellaneous systems
Similar dehydrogenation processes have also been reported in less common phosphine systems. As we concentrate on dehydrogenation pathways, we will refrain from discussion of phosphine–olefin ligand species, despite their interesting applications in asymmetric catalysis.37The group of Grützmacher synthesized unusual paramagnetic d9 metal complexes M(tropp)2 (M = Co, Rh, Ir) featuring tropylidene phosphine ligands (“tropp” ligand, Fig. 24).44 Of note is the characterization of the complexes by high-resolution EPR spectroscopy thanks to deuterium labelling of the phospha-alkenes. Grützmacher et al. have also conducted in-depth studies of the properties of chiral phosphine alkene systems. Hydrogenation of Ir(PhtroppPh)2+ (59) (PhtroppPh is a phenyl substituted tropp ligand) led to a cationic dihydride Ir(III) complex in which the phospha-alkene was hydrogenated through several intermediates identified by NMR as a function of H2 concentration. In the presence of an alkene acceptor, the initial complex 59 could be regenerated. This study shows that hydrogenation/dehydrogenation processes are not limited to simple ligands but can be extended to a large family of phosphines.45
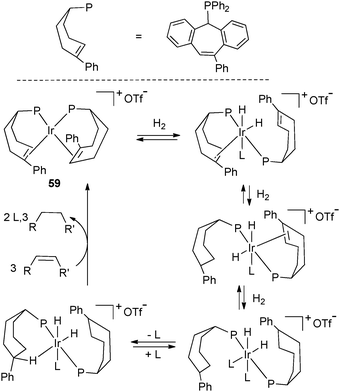 | ||
| Fig. 24 Iridium chemistry with tropylidene phosphine ligands. | ||
A few other unusual phosphines have been used to generate in one pot phospha-alkene ligands coordinated to a metal center in a κ–P, η2-CC mode. Bergman, Elman et al. have prepared a partially dehydrogenated cyclohexylphobane rhodium complex and used it as a catalyst precursor for the arylation of heterocycles via C–H bond activation.46 The synthesis of such a complex may prevent aryl halide hydrodehalogenation. Indeed, this undesired side reaction, observed in the case of PCy3 systems, was suspected to be induced by cycloalkyl dehydrogenation. In order to block this pathway, complex 61 was synthesized from the bis(phoban) complex 60. Complex 61 appeared to be a robust catalyst due to the chelating coordination mode of the phospha-alkene ligand (Fig. 25). To the best of our knowledge, reversible hydrogenation of the phospha-alkene ligand in complex 61, upon H2 addition, has not been reported.
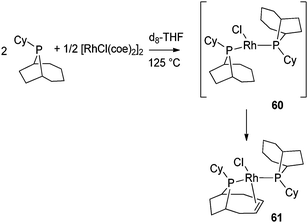 | ||
| Fig. 25 Cyclohexylphobane rhodium complexes. | ||
C–H activation occurs in a tBu pincer PNP rhodium complex leading to a hydride alkyl species which undergoes oxidative addition with arenes or H2 (Fig. 26).47 Reversible C–H activation of an iBu ligand in the rhodium complex [Rh(PtBuiBu2)2]+ is also observed.48 The use of phosphine incorporating a cyclopropyl group led to interesting activation pathways through C–C activation. Thus, addition of PtBu2CH2(C3H5) to [Rh(C8H14)2Cl]2 led to C–C bond cleavage of the cyclopropyl group. The resulting complex gives rise to a versatile reactivity: upon CO addition, reversible C–C coupling is demonstrated whereas selective C–C bond hydrogenation of the cyclopropane group can also be achieved in a few steps (Fig. 27).49 In relation to the important problem of hydrodesulfurization of thiophenes, PMe3 activation proved to play a key role in tungsten chemistry with a variety of bond activation processes starting from the hydride complex WH(η2-CH2PMe2)(PMe3)4.50
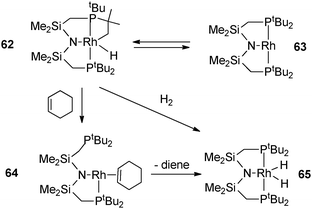 | ||
| Fig. 26 Dehydrogenation in a pincer complex. | ||
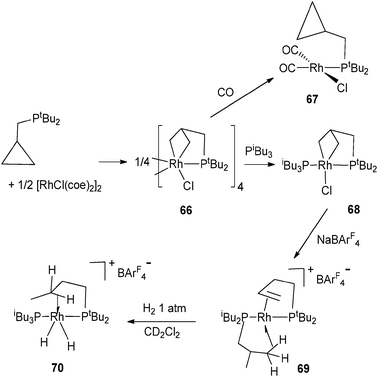 | ||
| Fig. 27 C–C activation at rhodium. | ||
Conclusion and outlook
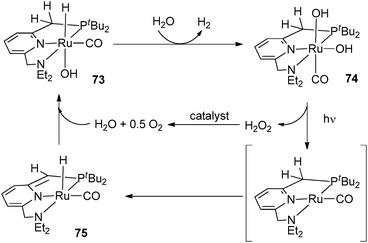
Through the selected examples reported here, one can see that subtle differences in phosphine substituents can induce dramatic changes in bond activation. Conformational factors are the key parameters to explain the differing behaviour of PCy3 and PCyp3 systems. We have thus launched a programme aimed at developing a new library of dihydrogen complexes by modifying the phosphine ligands. This will help us to strengthen the concept that catalyst precursors for hydrogen storage systems can be prepared from various sources of hydrogen atoms, i.e. hydride, dihydrogen ligands and from C–H activation within “non innocent” ligands such as phosphines. Ideally, we will search for reversible hydrogen uptake systems that do not require the presence of hydrogen acceptor molecules.Several systems incorporating arylphosphines can also induce C–H activation within their alkyl substituents to produce alkene fragments,51 but we have voluntarily restricted our discussion to alkylphosphines. Finally, we would like to point out a few related systems: (i) following their work on cyclopentylphosphine rhodium complexes, the group of Weller has demonstrated that it is possible to promote the dehydrogenation of cyclopentyl thioethers.52 (ii) The parallels often observed between phosphine and N-heterocyclic carbene ligands also hold true in this area. For example, the group of Whittlesey demonstrated the reversibility of the C–H bond cleavage as depicted in Fig. 28, a process that has direct implications in various catalytic applications.53 Related chemistry was also reported by the group of Aldridge for iridium systems incorporating Dipp substituted NHC ligands (Dipp = C6H3(iPr)2).54 (iii) Although not directly connected to the type of compounds we have selected, it is worth mentioning the achievements recently disclosed by Milstein et al. using PNP pincer ligands able to perform reversible deprotonation at a pyridinyl methylene group. Aromatization/dearomatization are key steps in various catalytic cycles (see Fig. 29 as an example).55
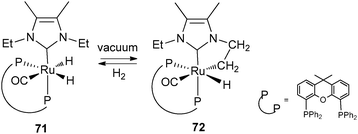 | ||
| Fig. 28 Reversible dehydrogenation at NHC ligands. | ||
 | ||
| Fig. 29 Aromatization/dearomatization steps in pincer ligands. | ||
We are confident that progress towards the design of innovative systems for an efficient reversible hydrogen uptake should appear in the near future, thanks in particular to an increased interplay between experimental and theoretical strategies.56
Acknowledgements
This work was supported by the CNRS and the ANR (Programme HyBoCat, ANR-09-BLAN-0184). The authors warmly thank their collaborators whose names are cited in the references for their contribution in this area.Notes and references
- (a) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS; (b) H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119–2176 CrossRef; (c) V. V. Grushin, Chem. Rev., 2004, 104, 1629–1662 CrossRef CAS; (d) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS; (e) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS; (f) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS; (g) J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832–8846 CrossRef CAS.
- J. Chatt and R. G. Hayter, J. Chem. Soc., 1961, 2605–2611 RSC.
- R. H. Crabtree, Chem. Rev., 2010, 110, 575 CrossRef CAS and the following articles of this special issue on “Selective Functionalization of C–H bonds”.
- R. Reguillo, M. Grellier, N. Vautravers, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2010, 132, 7854–7855 CrossRef CAS.
- (a) G. J. Kubas, Chem. Rev., 2007, 107, 4152–4205 CrossRef CAS; (b) F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613–2626 RSC; (c) A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 CrossRef CAS; (d) A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS; (e) G. Alcaraz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2010, 40, 7170–7179 CrossRef.
- G. Alcaraz, M. Grellier and S. Sabo-Etienne, Acc. Chem. Res., 2009, 42, 1640–1649 CrossRef CAS.
- A. F. Borowski, S. Sabo-Etienne, M. L. Christ, B. Donnadieu and B. Chaudret, Organometallics, 1996, 15, 1427–1434 CrossRef CAS.
- (a) B. Chaudret, G. Commenges and R. Poilblanc, J. Chem. Soc., Chem. Commun., 1982, 1388–1390 RSC; (b) B. Chaudret and R. Poilblanc, Organometallics, 1985, 4, 1722–1726 CrossRef CAS.
- (a) G. J. Kubas, R. R. Ryan, B. I. Swanson, P. J. Vergamini and H. J. Wasserman, J. Am. Chem. Soc., 1984, 106, 451–452 CrossRef CAS; (b) G. J. Kubas, Metal Dihydrogen and sigma-Bond Complexes, Kluwer Academic/Plenum Publishers, New York, 2001 Search PubMed.
- (a) T. Arliguie, B. Chaudret, R. H. Morris and A. Sella, Inorg. Chem., 1988, 27, 598–599 CrossRef CAS; (b) S. Sabo-Etienne and B. Chaudret, Coord. Chem. Rev., 1998, 178–180, 381–407 CrossRef CAS.
- M. Grellier, L. Vendier, B. Chaudret, A. Albinati, S. Rizzato, S. Mason and S. Sabo-Etienne, J. Am. Chem. Soc., 2005, 127, 17592–17593 CrossRef CAS.
- S. Hietkamp, D. J. Stufkens and K. Vrieze, J. Organomet. Chem., 1978, 152, 347–357 CrossRef CAS.
- H. J. Wasserman, G. J. Kubas and R. R. Ryan, J. Am. Chem. Soc., 1986, 108, 2294–2301 CrossRef CAS.
- T. Arliguie, B. Chaudret, F. A. Jalon, A. Otero, J. A. Lopez and F. J. Lahoz, Organometallics, 1991, 10, 1888–1896 CrossRef CAS.
- T. Arliguie, B. Chaudret, G. Chung and F. Dahan, Organometallics, 1991, 10, 2973–2977 CrossRef CAS.
- M. L. Christ, S. Sabo-Etienne and B. Chaudret, Organometallics, 1995, 14, 1082–1084 CrossRef CAS.
- (a) S. Lachaize, S. Sabo-Etienne, B. Donnadieu and B. Chaudret, Chem. Commun., 2003, 214–215 RSC; (b) S. Lachaize and S. Sabo-Etienne, unpublished results.
- (a) I. Atheaux, F. Delpech, B. Donnadieu, S. Sabo-Etienne, B. Chaudret, K. Hussein, J. C. Barthelat, T. Braun, S. B. Duckett and R. N. Perutz, Organometallics, 2002, 21, 5347 CrossRef CAS; (b) S. Lachaize and S. Sabo-Etienne, Eur. J. Inorg. Chem., 2006, 2115–2127 CrossRef CAS.
- B. Chaudret, P. Dagnac, D. Labroue and S. Sabo-Etienne, New J. Chem., 1996, 20, 1137–1141 CAS.
- C. Six, B. Gabor, H. Goerls, R. Mynott, P. Philipps and W. Leitner, Organometallics, 1999, 18, 3316–3326 CrossRef CAS.
- E. Solari, S. Gauthier, R. Scopelliti and K. Severin, Organometallics, 2009, 28, 4519–4526 CrossRef CAS.
- W.-M. Cheung, W.-H. Chiu, X.-Y. Yi, Q.-F. Zhang, I. D. Williams and W.-H. Leung, Organometallics, 2010, 29, 1981–1984 CrossRef CAS.
- N. Tsoureas, J. Nunn, T. Bevis, M. F. Haddow, A. Hamilton and G. R. Owen, Dalton Trans., 2011, 40, 951–958 RSC.
- K. Mauthner, K. M. Soldouzi, K. Mereiter, R. Schmid and K. Kirchner, Organometallics, 1999, 18, 4681–4683 CrossRef CAS.
- E. Molinos, S. K. Brayshaw, G. Kociok-Koehn and A. S. Weller, Dalton Trans., 2007, 4829–4844 RSC.
- R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS.
- R. L. Brainard, T. M. Miller and G. M. Whitesides, Organometallics, 1986, 5, 1481–1490 CrossRef CAS.
- F. Delpech, J. Mansas, H. Leuser, S. Sabo-Etienne and B. Chaudret, Organometallics, 2000, 19, 5750–5757 CrossRef CAS.
- T. M. Douglas, S. K. Brayshaw, R. Dallanegra, G. Kociok-Kohn, S. A. Macgregor, G. L. Moxham, A. S. Weller, T. Wondimagegn and P. Vadivelu, Chem.–Eur. J., 2008, 14, 1004–1022 CrossRef CAS.
- B. Chaudret, J. Devillers and R. Poilblanc, Organometallics, 1985, 4, 1727–1732 CrossRef CAS.
- M. Grellier, S. Sabo-Etienne, A. Albinati and S. Mason, to be published.
- T. K. A. Hoang and D. M. Antonelli, Adv. Mater., 2009, 21, 1787–1800 CrossRef CAS.
- T. M. Douglas and A. S. Weller, New J. Chem., 2008, 32, 966–969 RSC.
- M. Grellier, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2613–2615 CrossRef CAS.
- P. D. Bolton, M. Grellier, N. Vautravers, L. Vendier and S. Sabo-Etienne, Organometallics, 2008, 27, 5088–5093 CrossRef CAS.
- (a) T. M. Douglas, E. Molinos, S. K. Brayshaw and A. S. Weller, Organometallics, 2007, 26, 463–465 CrossRef CAS; (b) T. M. Douglas, J. Le Notre, S. K. Brayshaw, C. G. Frost and A. S. Weller, Chem. Commun., 2006, 3408–3410 RSC.
- R. Shintani, W.-L. Duan, T. Nagano, A. Okada and T. Hayashi, Angew. Chem., Int. Ed., 2005, 44, 4611–4614 CrossRef CAS.
- (a) R. Dallanegra, A. B. Chaplin and A. S. Weller, Angew. Chem., Int. Ed., 2009, 48, 6875–6878 CrossRef CAS; (b) R. Dallanegra, A. B. Chaplin, J. Tsim and A. S. Weller, Chem. Commun., 2010, 46, 3092–3094 RSC.
- S. Moret, R. Dallanegra, A. B. Chaplin, T. M. Douglas, R. M. Hiney and A. S. Weller, Inorg. Chim. Acta, 2010, 363, 574–580 CrossRef CAS.
- (a) M. A. Esteruelas and A. M. Lopez, Organometallics, 2005, 24, 3584–3613 CrossRef CAS; (b) R. Castro-Rodrigo, M. A. Esteruelas, A. M. López, S. Mozo and E. Oñate, Organometallics, 2010, 29, 4071–4079 CrossRef CAS; (c) M. A. Esteruelas, Y. A. Hernández, A. M. López, M. Oliván and E. Oñate, Organometallics, 2007, 26, 2193–2202 CrossRef CAS.
- M. Baya, M. L. Buil, M. A. Esteruelas and E. Oñate, Organometallics, 2004, 23, 1416–1423 CrossRef CAS.
- P. B. Glaser and T. D. Tilley, Organometallics, 2004, 23, 5799–5812 CrossRef CAS.
- A. B. Chaplin, A. I. Poblador-Bahamonde, H. A. Sparkes, J. A. K. Howard, S. A. Macgregor and A. S. Weller, Chem. Commun., 2009, 244–246 RSC.
- S. Deblon, L. Liesum, J. Harmer, H. Schönberg, A. Schweiger and H. Grützmacher, Chem.–Eur. J., 2002, 8, 601–611 CrossRef CAS.
- E. Piras, F. Läng, H. Rüegger, D. Stein, M. Wörle and H. Grützmacher, Chem.–Eur. J., 2006, 12, 5849–5858 CrossRef CAS.
- J. C. Lewis, A. M. Berman, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 2493–2500 CrossRef CAS.
- A. Y. Verat, M. Pink, H. Fan, J. Tomaszewski and K. G. Caulton, Organometallics, 2008, 27, 166–168 CrossRef CAS.
- L. J. Sewell, A. B. Chaplin, J. A. B. Abdalla and A. S. Weller, Dalton Trans., 2010, 39, 7437–7439 RSC.
- A. B. Chaplin and A. S. Weller, Organometallics, 2010, 29, 2332–2342 CrossRef CAS.
- A. Sattler and G. Parkin, J. Am. Chem. Soc., 2011, 133, 3748–3751 CrossRef CAS.
- See for example: (a) A. Baber, C. Fan, D. W. Norman, A. G. Orpen, P. G. Pringle and R. L. Wingad, Organometallics, 2008, 27, 5906–5910 CrossRef CAS; (b) W. Baratta, M. Ballico, A. Del Zotto, E. Zangrando and P. Rigo, Chem.–Eur. J., 2007, 13, 6701–6709 CrossRef CAS.
- R. Dallanegra, B. S. Pilgrim, A. B. Chaplin, T. J. Donohoe and A. S. Weller, Dalton Trans., 2011, 40, 6626–6628 RSC.
- A. E. W. Ledger, M. F. Mahon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 6941–6947 RSC.
- C. Y. Tang, W. Smith, D. Vidovic, A. L. Thompson, A. B. Chaplin and S. Aldridge, Organometallics, 2009, 28, 3059–3066 CrossRef CAS.
- (a) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS; (b) S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron and D. Milstein, Science, 2009, 324, 74–77 CrossRef CAS.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |