 Open Access Article
Open Access ArticleAmplifying vibrational circular dichroism by manipulation of the electronic manifold†
Sérgio R.
Domingos
a,
Matthijs R.
Panman
a,
Bert H.
Bakker
a,
Frantisek
Hartl
*b,
Wybren J.
Buma
*a and
Sander
Woutersen
*a
aVan 't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1089 XH, The Netherlands. E-mail: s.woutersen@uva.nl; w.j.buma@uva.nl
bDepartment of Chemistry, University of Reading, Whiteknights, Reading RG6 6AD, UK. E-mail: f.hartl@reading.ac.uk
First published on 14th October 2011
Abstract
Vibrational circular dichroism is a powerful technique to study the stereochemistry of chiral molecules, but often suffers from small signal intensities. Electrochemical modulation of the energies of the electronically excited state manifold is now demonstrated to lead to an order of magnitude enhancement of the differential absorption. Quantum-chemical calculations show that increased mixing between ground and excited states is at the origin of this amplification.
The development of tools to unambiguously assign the absolute configuration of naturally occurring and synthetic chiral molecules is of major importance for biochemical and pharmaceutical research. Vibrational circular dichroism (VCD), the infrared circular dichroism associated with vibrational transitions of chiral molecules, is a powerful spectroscopic method for this purpose since it makes it possible to determine the absolute configuration and conformation (or distribution of conformations) of a molecule without reference to any previous empirical rules. However, in many cases VCD measurements are difficult because of the small signal magnitudes (the differential absorption being typically 10−5 of the absorption itself). Such small intensities can be measured routinely nowadays, but at the expense of long measuring times. Strategies that would enhance VCD signals and thereby improve the ability of accurately differentiating between optical isomers by means of their vibrational signatures could therefore be of substantial importance.
It has been reported previously that molecular systems with low-lying electronically excited states can exhibit strongly enhanced VCD signals.1–3 The origin of this enhancement can be understood from the expression for the vibrational magnetic-dipole transition moment derived in the vibronic coupling theory for VCD.4,5 Here, we present a novel approach to amplify VCD signals by modulation of the energies of the excited-state manifold in a controlled way using spectroelectrochemistry.6–8
To investigate the effect of electrochemical reduction on the VCD response, we measured IR and VCD spectra of compounds (+)-1 and (−)-1 (Fig. 1) both in their neutral and radical anion forms (see ESI† for experimental details). Here, our main focus is on the comparison of the peak intensities in the VCD spectrum of the neutral and of the radical anion. A full conformational analysis of the IR and VCD spectra is given in the ESI.†
![Chemical structure of compounds (R)- and (S)-methyl 2-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)propanoate, (R)-1 and (S)-1 respectively.](/image/article/2012/CC/c1cc15369b/c1cc15369b-f1.gif) | ||
| Fig. 1 Chemical structure of compounds (R)- and (S)-methyl 2-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)propanoate, (R)-1 and (S)-1 respectively. | ||
The FTIR spectrum of neutral (−)-1 is shown in Fig. 2 (blue curve). In the 1500–1780 cm−1 frequency range four dominant vibrational modes (A1–4) can be identified and assigned by comparison with the literature9 (vide infra). The FTIR spectrum of the radical anion (red curve in Fig. 2) was obtained upon complete reduction of a 35 mM solution of (−)-1 in CD3CN (10−1 M TBAH) at 293 K. The conventional cyclic voltammogram for (−)-1 shows a completely reversible cathodic wave (see ESI†). The observed reduction potential (E1/2 = −1.3 V vs. Fc/Fc+) is in good agreement with previously reported values for similar systems.9 Full reversibility was also achieved with TL-CV on time scales of 2 mV s−1 (inset of Fig. 2). Intermediate spectra were recorded in small potential steps across the cathodic wave, and IR spectral changes were monitored at each step.
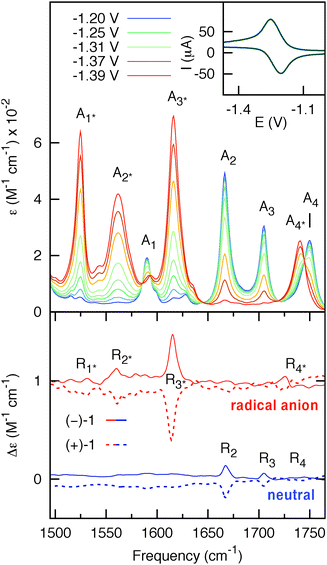 | ||
| Fig. 2 Upper panel: potential-dependent steady-state IR spectra of 35 mM (−)-1 (optical path = 200 μm). The blue and red curves represent the spectra of the initial (neutral) and final states (radical anion), respectively. Inset: thin-layer cyclic voltammogram of (−)-1 obtained using the IR OTTLE cell. Lower panel: VCD spectra of 7 mM (−)-1 (solid-line) and 7 mM (+)-1 (dashed-line) (optical path = 1.2 mm) for the neutral (blue) and radical anion (red) species. The spectra for the radical anion are vertically offset for clarity. | ||
Comparison of the IR spectra of neutral and anionic (−)-1 shows that the IR bands A2 and A3 of the neutral at 1704 cm−1 and 1667 cm−1, assigned to the symmetric and asymmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching modes of the naphthalimide moiety, are red-shifted in the radical anion to 1615 cm−1 (A2*) and 1562 cm−1 (A3*), respectively. The naphthalimide aromatic CC-stretching mode (A1) observed in neutral (S)-1 at 1589 cm−1 is red-shifted to 1525 cm−1 (A1*) in the radical anion. The methyl ester CO-stretching mode of the amino-acid moiety (A4) at 1750 cm−1 is red-shifted to 1740 cm−1 (A4*) in the radical anion. The VCD spectra of 7 mM solution of neutral and anionic (+)-1 and (−)-1 are displayed in the lower panel of Fig. 2. Spectra for both enantiomers have been obtained by averaging three consecutive sets of 20 min scans for the neutral, and three sets of 2 min scans from independent but consecutive CV cycles for the radical anion. For neutral (+)-1 and (−)-1, three features R2, R3 and R4 can be observed in the VCD spectra. These bands are red-shifted for the radical anion of (+)−1 and (−)-1 to the features R2*, R3* and R4*, respectively. Moreover, the spectra show another band R1* that is not visible in the spectrum of the neutral. Importantly, we find that the VCD signals are enhanced up to one order of magnitude compared to the neutral species. As will be argued in the following, this enhancement is due to a strong vibronically induced mixing of low-lying electronic excited states with the ground state in the electrochemically generated radical anion species. The peak intensities in VCD spectra are proportional to the rotational strength, given by:10
O stretching modes of the naphthalimide moiety, are red-shifted in the radical anion to 1615 cm−1 (A2*) and 1562 cm−1 (A3*), respectively. The naphthalimide aromatic CC-stretching mode (A1) observed in neutral (S)-1 at 1589 cm−1 is red-shifted to 1525 cm−1 (A1*) in the radical anion. The methyl ester CO-stretching mode of the amino-acid moiety (A4) at 1750 cm−1 is red-shifted to 1740 cm−1 (A4*) in the radical anion. The VCD spectra of 7 mM solution of neutral and anionic (+)-1 and (−)-1 are displayed in the lower panel of Fig. 2. Spectra for both enantiomers have been obtained by averaging three consecutive sets of 20 min scans for the neutral, and three sets of 2 min scans from independent but consecutive CV cycles for the radical anion. For neutral (+)-1 and (−)-1, three features R2, R3 and R4 can be observed in the VCD spectra. These bands are red-shifted for the radical anion of (+)−1 and (−)-1 to the features R2*, R3* and R4*, respectively. Moreover, the spectra show another band R1* that is not visible in the spectrum of the neutral. Importantly, we find that the VCD signals are enhanced up to one order of magnitude compared to the neutral species. As will be argued in the following, this enhancement is due to a strong vibronically induced mixing of low-lying electronic excited states with the ground state in the electrochemically generated radical anion species. The peak intensities in VCD spectra are proportional to the rotational strength, given by:10
| R = Im[〈Ψi|μel|Ψf〉〈Ψf|μmag|Ψi〉] | (1) |
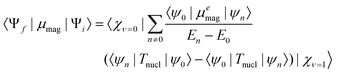 | (2) |
For large energy gaps between ground and electronically excited states, the coefficients in the perturbation expansion, 〈ψ0|μemag|ψn〉/(En − E0) (see eqn (2)), are small due to large energy denominators. As a consequence, the VCD signals are very small. This is the case for nearly all closed-shell organic molecular systems. In open-shell species, on the other hand, the first electronically excited states are generally at much lower energies. Eqn (2) shows that this leads to a strong increase in the magnetic transition-dipole moments, and thus in the VCD amplitudes. Indeed, the UV-Vis absorption spectrum of the radical anion displayed in Fig. 3 shows absorption bands at much longer wavelengths than the corresponding spectrum of the neutral system. We have confirmed the presence of these low-energy states and identified their electronic characteristics using time-dependent density functional theory (TD-DFT) calculations on the most stable conformer of the radical anion (see ESI†). The low-energy nature of these excited states induces mixing of their wave functions into the ground state, increasing the total number of adiabatic electronic states contributing to the vibrational eigenfunctions, and leading to strongly enhanced VCD signals, as confirmed by our experimental results.
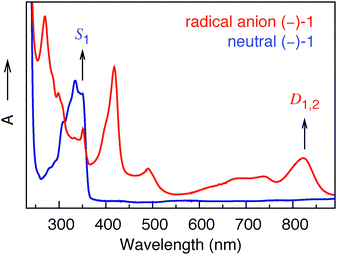 | ||
| Fig. 3 UV-Vis spectra of (−)-1 in the neutral (blue) and radical anion (red) forms. | ||
From the TD-DFT calculations we can assign two almost degenerate electronic states, D1 and D2, contributing to the absorption band at 822 nm in Fig. 3. Interestingly, we find that the electronic magnetic transition-dipole moment 〈ψ0|μemag|ψn〉 between the ground state and D2 is one order of magnitude larger than for any other of the first ten electronically excited states. Eqn (2) thus tells us that D2 will be a dominant contributor to the vibrational magnetic transition dipole moment, not only because of the relatively small energy gap (E2 − E0), but also because of the large value of the 〈ψ0|μemag|ψn〉 matrix element. It will therefore magnify the vibrational magnetic transition dipole moment, and thus the VCD intensity. The difference density plot between D2 and the ground state D0 shows that the electron density on the naphthalimide carbonyl groups is noticeably different in D2 and D0, while the electron density on the carbonyl group near the methyl ester is the same in the two states. The dominant role of D2 in the expansion of the vibrational magnetic transition moment (eqn (2)) makes one therefore expect that the VCD of the naphthalimide CO-stretching modes would be affected in particular. Our experimental results nicely confirm this expectation: we observe maximum VCD enhancement (one order of magnitude) for the symmetric naphthalimide CO-stretching mode. It is satisfying to note that our observations are internally consistent in other respects, as well. From a purely energetic point of view, D1 might also be expected to make a significant contribution to the vibrational magnetic transition dipole moment. Difference density plots between D1 and D0 show, however, that the electron density on the naphthalimide carbonyl groups is hardly different in D1 and D0 (see ESI†). A dominant contribution of D1 in eqn (2) would thus be at odds with an enhancement of the naphthalimide CO-stretching modes. Indeed, the influence of D1 is greatly reduced in the end on account of the small electronic magnetic transition dipole moment from the ground state (see ESI†).
In conclusion, we have observed a strong enhancement of VCD signals upon the electrochemical reduction of a closed-shell organic compound in solution: the conversion from neutral molecule to radical anion increases the VCD signals by up to one order of magnitude. Such intensity-enhanced VCD arising from the presence of electrochemically “created” low-lying electronic states should be a general phenomenon in organic compounds. As such, electrochemical VCD can be a valuable tool for amplifying signals in molecules that have small VCD amplitudes, or that can be studied only at low concentrations.
S. R. Domingos acknowledges financial support by the Portuguese FCT through Project SFRH/BD/48295/2008. The authors are grateful to Henk Luyten, Alexander Vdovin and Taasje Mahabiersing from the University of Amsterdam for technical support.
Notes and references
- C. Barnett, A. Drake, R. Kuroda, S. Mason and S. Savage, Chem. Phys. Lett., 1980, 70, 8–10 CrossRef CAS
.
- Y. He, X. Cao, L. A. Nafie and T. B. Freedman, J. Am. Chem. Soc., 2001, 123, 11320–11321 CrossRef CAS
- C. Johannessen and P. W. Thulstrup, Dalton Trans., 2007, 1028–1033 RSC
- L. A. Nafie and T. B. Freedman, J. Chem. Phys., 1983, 78, 7108 CrossRef CAS
- L. A. Nafie, J. Phys. Chem. A, 2004, 108, 7222–7231 CrossRef CAS
- W. E. Geiger, Organometallics, 2011, 30, 28–31 CrossRef CAS
- M. I. Bruce, N. N. Zaitseva, B. W. Skelton, A. H. White, M. A. Fox and P. J. Low, Dalton Trans., 2010, 1222–1234 RSC
- D. R. Laws, D. Chong, K. Nash, A. L. Rheingold and W. E. Geiger, J. Am. Chem. Soc., 2008, 130, 9859–9870 CrossRef CAS
- D. Jagesar, F. Hartl, W. Buma and A. Brouwer, Chem.–Eur. J., 2008, 14, 1935–1946 CrossRef CAS
-
D. J. Caldwell and H. Eyring, The Theory of Optical Activity, Wiley-Interscience, New York, 1971 Search PubMed
-
L. D. Landau and E. M. Lifschitz, Quantum Mechanics, Non-relativistic Theory, Butterworth-Heinemann, Oxford, 1999 Search PubMed
- P. J. Stephens, J. Phys. Chem., 1985, 89, 748–752 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, DFT and TD-DFT calculations. See DOI: 10.1039/c1cc15369b |
| This journal is © The Royal Society of Chemistry 2012 |