Chemoselective conversion of α-unbranched aldehydes to amides, esters, and carboxylic acids by NHC-catalysis†
Satoru
Kuwano
,
Shingo
Harada
,
Raphaël
Oriez
and
Ken-ichi
Yamada
*
Graduate School of Pharmaceutical Sciences, Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: yamak@pharm.kyoto-u.ac.jp; Fax: +81 75 753 4604; Tel: +81 75 753 4573
First published on 7th November 2011
Abstract
Depending on the N-heterocyclic carbene catalyst utilized, α-unbranched aldehydes selectively provided amides, esters, or carboxylic acids through oxidation by NCS. The α-unbranched aldehyde underwent these reactions chemoselectively in the presence of an aromatic or α-branched aldehyde.
N-heterocyclic carbenes (NHCs) are used as organocatalysts for various transformations.1 NHC-catalyzed esterification and amidation of α-oxidized aldehydes were demonstrated2 using α,β-epoxyaldehydes,2a,f α-haloaldehydes,2b,f,galkenal,2c,f,g and α-acyloxyaldehydes2k as substrates. Direct conversion of aldehydes to esters or amides was also achieved by NHC-catalysis,3 but only aromatic and unsaturated aldehydes were suitable for the reported direct amidation. Herein, we report a new method for direct conversion of α-unbranched aldehydes to amides, as well as esters and carboxylic acids, with NHC-catalysis. The first report of an NHC-dependent selectivity switch of nucleophiles is also described.
We unexpectedly found that diethylamide 3a was produced in 18% yield when hydrocinnamaldehyde (1a) and triethylamine were heated in refluxing toluene in the presence of benzoyl peroxide (BPO), N-hydroxyphthalimide (NHPI), and chiral NHC precursor 2a4 (Fig. 1 and Table 1, entry 1). Although benzaldehyde failed to undergo amide formation, the reaction proceeded even at room temperature, and produced 3a in 20% yield when triethylamine was replaced with diethylamine (entry 2). Without NHPI, the yield of 3a decreased to 7% (entry 3).
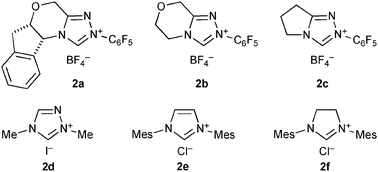 | ||
| Fig. 1 NHC precursors 2a–f. | ||
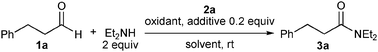
| Entry | Oxidant/equiv. | Additive | 2a/mol% | Time/h | 3a/%yield |
|---|---|---|---|---|---|
| a The solvent was toluene in entries 1–7 and CH2Cl2 in entries 8–14. b Under reflux with Et3N instead of Et2NH. c With 1.2 equiv. Et3N. | |||||
| 1b | (BzO)2/0.6 | NHPI | 20 | 19 | 18 |
| 2 | (BzO)2/0.6 | NHPI | 20 | 20 | 20 |
| 3 | (BzO)2/0.6 | — | 20 | 17 | 7 |
| 4 | (BzO)2/1 | NHPI | 20 | 19 | 29 |
| 5 | (BzO)2/1 | HOBt | 20 | 18 | 45 |
| 6 | (BzO)2/2 | HOBt | 20 | 20 | 55 |
| 7 | (3-ClC6H4CO2)2/2 | HOBt | 20 | 20 | 44 |
| 8 | NIS/1.3 | HOBt | 20 | 10 | 20 |
| 9 | NBS/1.3 | HOBt | 20 | 7 | 69 |
| 10 | NCS/1.3 | HOBt | 20 | 6 | 76 |
| 11c | NCS/1.3 | HOBt | 20 | 6 | 96 |
| 12c | NCS/1.3 | HOBt | 10 | 12.5 | 92 |
| 13c | NCS/1.3 | HOBt | 5 | 12.5 | 88 |
| 14c | NCS/1.3 | HOBt | 2 | 12.5 | 79 |
These results led us to speculate that the pathway to amide 3a was as follows (Scheme 1). First, aldehyde 1a and diethylamine formed enamine, which was then oxidized by BPO to give α-benzoyloxy aldehyde 4.5Diethylamine may have been produced by the reaction of triethylamine with BPO (entry 1).6 The NHC underwent addition with 4 to form Breslow intermediate 5. Liberation of benzoate followed by tautomerization gave acyltriazolium 6,2k which was then converted into activated ester 7 by NHPI, and the diethylamine underwent acylation to produce amide 3a. The failed reaction with non-enolizable benzaldehyde is also explained by this enamine-pathway.
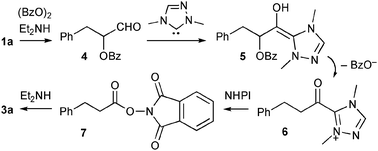 | ||
| Scheme 1 Working hypothesis. | ||
Based on this hypothesized pathway, the reaction conditions were optimized. First, the reaction was performed with a stoichiometric amount of BPO, and 3a was obtained in 29% yield after 19 h (entry 4). The use of 1-hydroxybenzotriazole (HOBt) instead of NHPI made the reaction cleaner, and gave 3a in 45% yield (entry 5). Although other NHC precursors 2b–f were tested, less satisfactory results were obtained.
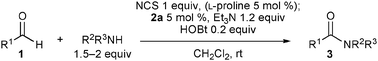
Then, the reaction was performed with an increased amount of BPO (2 equiv.), and the yield of 3a slightly improved to 55% (entry 6). No improvement was observed when m-chlorobenzoyl peroxide was used in place of BPO, and 3a was obtained in 44% yield (entry 7). Then, NCS was tested as the oxidant, replacing BPOs. The reaction of 1a with NCS (1.3 equiv.) in dichloromethane produced 3a in 76% yield (entry 10), though NIS and NBS gave less satisfactory results (entries 8 and 9). Finally, when the reaction was conducted with triethylamine (1.2 equiv.) to neutralize the hydrogen chloride liberated during the reaction, 3a was obtained in excellent yield after 6 h (entry 11). When the reaction was quenched after 30 min, 3a was produced in 32% yield and α-chlorohydrocinnamaldehyde was mainly obtained in 60% yield. The chloroaldehyde and diethylamine were then converted into 3a in 94% yield after 6 h in the presence of 2a and triethylamine in dichloromethane at room temperature. These results indicate that the reaction proceeds mainly through the α-chlorination of aldehyde followed by NHC-catalyzed acylation of nucleophiles, and not through oxidation of a Breslow intermediate. Thus, the reaction was best performed by pre-mixing 1a and NCS in the presence of diethylamine before the addition of 2a, triethylamine, and HOBt, and catalyst loading of 2a was reducible (2–10 mol%) with only a slight decrease in the product yield (entries 12–14).
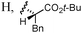
Other aldehydes and amines were applied to the reaction (Table 2). Linear aliphatic aldehyde 1b was a good substrate, and amide 3b was obtained in 91% yield (entry 2). A siloxy group was compatible with this transformation, and the reaction with aldehyde 1c provided 3c in 87% yield using 10 mol% 2a (entry 3), though the yield was decreased to 56% with 5 mol% 2a. α-Branched aldehyde was not suitable; the reaction of cyclohexanecarbaldehyde gave the corresponding diethylamide in only 25% yield. The reaction of 1a and dibenzylamine gave N,N-dibenzyl amide 3d in 49% yield along with N-benzyl amide 3e in 11% yield. The production of 3e indicates that dibenzylamine was debenzylated by the action of NCS. To avoid the reaction of the amine and NCS, an α-chlorination step was performed using L-proline as a catalyst; a solution of 2a, triethylamine, HOBt, and dibenzylamine was added to a solution of 1a, NCS, and L-proline (5 mol%) in dichloromethane pre-mixed for 9 h, and 3d was obtained in 71% yield (entry 4). The use of L-proline was effective for the reactions of other amines. In the reaction of benzylamine, however, slow addition of the amine over 3 h was important to obtain 3e in 72% yield (entry 5), and adding the amine in one portion decreased the yield to 39%. Formation of Weinreb amide efficiently proceeded, and the reaction of 1a with methoxyamine hydrochloride salt provided 3f in 81% yield (entry 6). An amino acid was also a good reaction partner; the reaction of 1a and phenylalanine tert-butyl ester hydrochloride salt produced N-acyl amino acid 3g without racemization (entry 7). The reaction rates of the amino acid enantiomers, however, were not significantly different, suggesting that amidation proceeded via an achiral intermediate such as 11 as shown in Scheme 2.
| Entry | 1/R1 | Amine b R2, R3 | Time/h | 3/%yield |
|---|---|---|---|---|
| a Entries 1–3 were conducted without L-proline, while entries 4–7 were conducted with L-proline. b Used 2 equiv. in entries 1–3 and 1.5 equiv. in entries 4–7. c From Table 1, entry 13 for comparison. d With 10 mol% 2a. e BnNH2 was added over 3 h. f HCl salt of R2R3NH was added instead of free amine. g Without racemization. | ||||
| 1c | 1a/Ph(CH2)2 | Et, Et | 12.5 | 3a/88 |
| 2 | 1b/Me(CH2)5 | Et, Et | 12 | 3b/91 |
| 3d | 1c/TBSO(CH2)3 | Et, Et | 9 | 3c/87 |
| 4 | 1a/Ph(CH2)2 | Bn, Bn | 20 | 3d/71 |
| 5e | 1a/Ph(CH2)2 | H, Bn | 23 | 3e/72 |
| 6f | 1a/Ph(CH2)2 | H, OMe | 23 | 3f/81 |
| 7f | 1a/Ph(CH2)2 |
|
19 | 3g/76g |
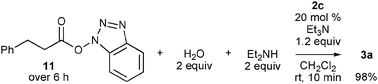 | ||
| Scheme 2 Reaction of benzotriazolyl ester 11 with diethylamine in the presence of water and 2c-derived NHC. | ||
Investigation for the best NHC catalyst under the conditions using NCS revealed that 2a was the best among 2a–f, and also led to an interesting NHC-dependent selectivity switching of the nucleophilic partner. When the reaction was conducted in the presence of triazolium 2b, instead of 2a, along with amide 3a in 67% yield, carboxylic acid 8a was obtained in 14% yield (Table 3, entry 2). With triazolium 2c, 8a and 3a were obtained in similar amounts (31–38% and 23–37%, respectively) (entry 3), while complex mixtures were produced using 2d–f. The varying yields of 3a and 8a in the reaction with 2c indicate that the formation of the carboxylic acid is due to a reaction of intermediate 6 or 11 with exogenous water. Indeed, additional water (2 equiv.) increased the yield of the carboxylic acid, and we obtained 8a in 83% yield and 3a in 10% yield (entry 5). In contrast, the reaction with NHC derived from 2a preferentially produced amide 3a even in the presence of water (entry 4).
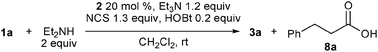
In this reaction, amides were likely formed via activated esters 11, because acylazoliums, such as 6, react predominantly with water and alcohols over amines,7 and indeed, the yield of amide 3a was poor without NHPI and HOBt (Table 1, entry 3). Recently, activation of O-nucleophiles by hydrogen bonding with NHC was proposed to explain the O-preference of acylazoliums;3e,8 thus, a competitive reaction of water and diethylamine with benzotriazolyl ester 11 was conducted in the presence of 20 mol% 2c (Scheme 2). Although 11 was slowly added over 6 h, no activation of water over amine was observed, and amide 3a was quantitatively produced. This result also indicates that carboxylic acid 8a was directly produced by the reaction of acyltriazolium 6 with water.
As expected from the pKa values (HOBt 4.6,9water 15.7), DFT calculations suggested higher stability of an NHC–HOBt hydrogen-bond complex, in which the O–H bond of HOBt was more elongated and thus activated, than an NHC–water complex.10 The reaction of HOBt was, however, faster than that of water with more bulky 2a-derived NHC (entry 4), and became slower with less bulky 2c-derived NHC (entry 5). In contrast to entry 5, using 5 mol% 2c, 3a was produced in 43% yield with 8a in 53% yield. These results are contradictory to the hydrogen-bond activation model, and seem to suggest that a hydrogen bond with NHC is not an important factor of the chemoselectivity of acylazoliums, at least in this reaction, although acyltriazolium 6 reacts with HOBt or water, depending on the choice of the NHC catalyst.
Thus, the reaction of O-nucleophiles was best performed with 1.1 equiv. of diethylamine in the absence of HOBt. In the presence of 5 mol% 2c, carboxylic acid 8a was obtained in 85% yield without production of amide 3a (Scheme 3). Alcohols such as benzyl and allyl alcohols were also good nucleophiles, and aldehyde 1a was converted into the corresponding esters 9 and 10, respectively, in good yields.
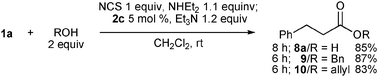 | ||
| Scheme 3 Reaction of 1a with O-nucleophiles. | ||
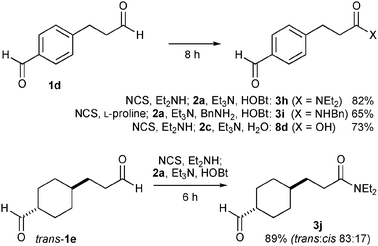
Taking advantage of this reaction, chemoselective conversion of dialdehydes 1d and 1e was demonstrated (Scheme 4). With 1d having both aliphatic and aromatic formyl groups, the reaction with diethylamine and water gave amide 3h in 82% yield and carboxylic acid 8d in 73% yield. The reaction of benzylamine also proceeded in a chemoselective manner using proline as a co-catalyst to give α-unbranched amide 3i in 65% yield and no amidation of the aromatic aldehyde moiety was observed. The reaction of diethylamine and 1e having both α-branched and α-unbranched aldehyde moieties proceeded selectively at the α-unbranched moiety to provide mono-amide 3j in 89% yield. Partial isomerization (trans only to trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis 83:17) was observed at the α-position of the branched aldehyde moiety in the reaction of 1e, suggesting reversible enamine formation of the α-branched aldehyde moiety. Therefore, the selectivity is likely due to the slower chlorination of the more hindered enamine.
:cis 83:17) was observed at the α-position of the branched aldehyde moiety in the reaction of 1e, suggesting reversible enamine formation of the α-branched aldehyde moiety. Therefore, the selectivity is likely due to the slower chlorination of the more hindered enamine.
 | ||
| Scheme 4 Chemoselective conversion of α-unbranched aldehydes to amides and carboxylic acids. | ||
In summary, we developed a new one-pot transformation of α-unbranched aldehydes to amides, esters, and carboxylic acids with NHC-catalysis. It is advantageous that selective conversion of α-unbranched aldehydes is possible and isolation of unstable α-chloroaldehyde intermediates is unnecessary. The observed NHC-dependent nucleophile-selectivity shows that chemoselectivity can be controlled by the selection of the NHC-catalyst.
We thank financial support by a Grant-in-Aid for Young Scientist (B) from JSPS, Targeted Protein Research Program from JST, and Uehara Memorial Foundation.
Notes and references
- Reviews: (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS; (b) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS; (c) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691 RSC.
- (a) K. Y.-K. Chow and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 8126 CrossRef CAS; (b) N. T. Reynolds, J. Read de Alaniz and T. Rovis, J. Am. Chem. Soc., 2004, 126, 9518 CrossRef CAS; (c) A. Chan and K. A. Scheidt, Org. Lett., 2005, 7, 905 CrossRef CAS; (d) K. Zeitler, Org. Lett., 2006, 8, 637 CrossRef CAS; (e) B. Alcaide, P. Almendros, G. Cabrero and M. P. Ruiz, Chem. Commun., 2007, 4788 RSC; (f) H. U. Vora and T. Rovis, J. Am. Chem. Soc., 2007, 129, 13796 CrossRef CAS; (g) J. W. Bode and S. S. Sohn, J. Am. Chem. Soc., 2007, 129, 13798 CrossRef CAS; (h) G.-Q. Li, L.-X. Dai and S.-L. You, Org. Lett., 2009, 11, 1623 CrossRef CAS; (i) Y. Kawanaka, E. M. Phillips and K. A. Scheidt, J. Am. Chem. Soc., 2009, 131, 18028 CrossRef CAS; (j) K. Thai, L. Wang, T. Dudding, F. Bilodeau and M. Gravel, Org. Lett., 2010, 12, 5708 CrossRef CAS; (k) K. B. Ling and A. D. Smith, Chem. Commun., 2011, 47, 373 RSC; (l) J. Qi, X. Xie, J. He, L. Zhang, D. Ma and X. She, Org. Biomol. Chem., 2011, 9, 5948 RSC.
- (a) B. E. Maki, A. Chan, E. M. Phillips and K. A. Scheidt, Org. Lett., 2007, 9, 371 CrossRef CAS; (b) B. E. Maki and K. A. Scheidt, Org. Lett., 2008, 10, 4331 CrossRef CAS; (c) B. E. Maki, A. Chan, E. M. Phillips and K. A. Scheidt, Tetrahedron, 2009, 65, 3102 CrossRef CAS; (d) M. Yoshida, Y. Katagiri, W.-B. Zhu and K. Shishido, Org. Biomol. Chem., 2009, 7, 4062 RSC; (e) S. De Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190 CrossRef CAS; (f) S. De Sarkar and A. Studer, Org. Lett., 2010, 12, 1992 CrossRef CAS.
- M. S. Kerr, J. R. de Alaniz and T. Rovis, J. Org. Chem., 2005, 70, 5725 CrossRef CAS.
- (a) T. Kano, H. Mii and K. Maruoka, J. Am. Chem. Soc., 2009, 131, 3450 CrossRef CAS; (b) M. J. P. Vaismaa, S. C. Yau and N. C. O. Tomkinson, Tetrahedron Lett., 2009, 50, 3625 CrossRef CAS.
- K. Gambar'jan, Zh. Obshch. Khim., 1933, 3, 222.
- (a) T. C. Bruice and N. G. Kundu, J. Am. Chem. Soc., 1966, 88, 4098 CrossRef; (b) T. C. Owen and A. Richards, J. Am. Chem. Soc., 1987, 109, 2520 CrossRef CAS; (c) R. J. Mahatthananchai, P. Zheng and J. W. Bode, Angew. Chem., Int. Ed., 2011, 50, 1673 CrossRef CAS.
- M. Movassaghi and M. A. Schmidt, Org. Lett., 2005, 7, 2453 CrossRef CAS.
- I. Koppel, J. Koppel, I. Leito, V. Pihl, L. Grehn and U. Ragnarsson, J. Chem. Res., Miniprint, 1993, 11, 3008 Search PubMed.
- A complex of 1,3,4-trimethyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene with 1-hydroxy-1H-1,2,3-triazole was more stable by 8 kcal mol−1 and elongation of its H–O bond was larger by 0.07 Å than a complex with H2O at the B3LYP/6-311+G** level of theory with counterpoise corrections, which would be sufficient for qualitative discussion. For the H-bond activation by NHC, see ref. 3e and 8.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data and NMR charts of products, HPLC traces of 3g, and results of DFT calculations. See DOI: 10.1039/c1cc15539c |
| This journal is © The Royal Society of Chemistry 2012 |