Recent efforts directed to the development of more sustainable asymmetric organocatalysis
José G.
Hernández
and
Eusebio
Juaristi
Departamento de Química, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional (CINVESTAV-IPN), Apartado Postal 14–740, 07000 México, D.F., Mexico. E-mail: gregoriojose_@relaq.mx; juaristi@relaq.mx; Fax: +52 55 5747 3389; Tel: +52 55 5747 3722
First published on 19th April 2012
Abstract
In line with the principles of “green” chemistry, organocatalysis seeks to reduce energy consumption and to optimize the use of the available resources, aiming to become a sustainable strategy in chemical transformations. Nevertheless, during the last decade diverse experimental protocols have made organocatalysis an even “greener” alternative by the use of friendlier reaction conditions, or via the application of solvent-free methodologies, or through the design and synthesis of more selective catalysts, or via the development of multicomponent one-pot organocatalytic reactions, or by the recycling and reuse of organocatalysts, or by means of the application of more energy-efficient activation techniques, among other approaches. In this feature article we review some of the remarkable advancements that have made it possible to develop even more sustainable asymmetric organocatalyzed methodologies.
 José G. Hernández | José Gregorio Hernandez obtained his Bachelor in Science degree in 2007 from Universidad Industrial de Santander in Colombia under the direction of Prof. Vladimir V. Kouznetsov. In 2008 he joined Prof. Eusebio Juaristi's group at CINVESTAV-IPN in Mexico, where he is currently completing his PhD studies. His research interests include organic synthesis, green chemistry and asymmetric organocatalysis. |
 Eusebio Juaristi | Eusebio Juaristi was born in Querétaro (México) and studied Chemistry at Instituto Tecnológico y de Estudios Superiores de Monterrey (B.Sc.) and at the University of North Carolina in Chapel Hill (PhD). Following a postdoctoral stay in the University of California-Berkeley, he returned to Mexico in 1979 as a Professor of Chemistry at CINVESTAV-IPN. He has been a Visiting Professor at the ETH-Zurich and UC-Berkeley. In 1998 he received Mexico's Presidential Medal of Science and in 2006 he became a member of El Colegio Nacional, highest academic distinction in Mexico. |
Introduction
Since its renaissance at the beginning of the last decade,1 organocatalysis has proved to be an attractive strategy to carry out asymmetric processes in ways that are competitive with those involving catalysis with metals, enzymes or chiral metal complexes. From the beginning, it was evident that organocatalytic processes fulfill some of the guidelines of “green” chemistry. For example, in the asymmetric aldol reaction organocatalyzed by L-proline reported by List and Barbas et al.1a in addition to the remarkable novelty of constituting “the first example of a nonmetallic small-molecule catalyst for direct intermolecular asymmetric aldol reactions”,1a this development can also be classified as a “green” process, since the direct asymmetric aldol reaction takes place in the absence of preformed metal enolates, simultaneously enhancing the atom economy of the process. Indeed, since organocatalysis avoids the use of metals, this renders the organocatalytic process less hazardous. Moreover, by comparison with precious or transition metals, organic compounds such as L-proline are usually (but not always) more stable, less expensive, less toxic, and more easily applied to a broader number of substrates. Finally, by using less than stoichiometric amounts of L-proline as the chiral activator that afforded the aldol products with good yields and remarkable enantioselectivities, List and co-workers showed that L-proline is indeed an efficient and selective catalyst that satisfies some of the essential attributes associated with “green” chemistry (Scheme 1).2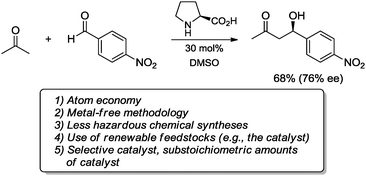 | ||
| Scheme 1 Some characteristics fulfilling the “green” chemistry principles in the asymmetric aldol reaction catalyzed by L-proline.1a | ||
Nevertheless, during the last ten years significant efforts have been made by several researchers in order to render organocatalysis even more sustainable. Particular improvements involve the use of friendlier reaction conditions, the development of even more regio-, diastereo-, and enantioselective organocatalysts, the design of more energy-efficient activation techniques, the application of novel multicomponent “one-pot” protocols, the implementation of simpler work-up and purification procedures, the application of continuous flow processes, and the development of supported and recyclable catalysts. All these advancements contribute to the making of more sustainable organocatalytic processes.
With the purpose of reviewing recent advancements in the field of asymmetric organocatalysis from the point of view of “green” chemistry, we present and evaluate in this feature article some novel and ingenious approaches directed towards the development of “greener” organocatalytic processes. The examples reported here arise from a selection of significant contributions from the recent literature, which in our opinion demonstrate how progress in an emerging new area can be achieved in a sustainable manner.
Friendlier reaction media. Use of water
One of the most important features in the seminal work of List, Lerner and Barbas1a is the fact that the reaction does not require inert conditions to proceed efficiently. This characteristic offers enormous advantages, especially during scale-up procedures in the laboratory and for industrial-scale applications in chemical plants.In this regard, the attractiveness of water as solvent in organic reactions was first highlighted in the 1980s with the reports where Breslow describes a strong rate acceleration in Diels–Alder reactions that are carried out in water relative to organic solvents.3,4 Indeed, in addition to the unique physical properties displayed by water, aqueous reactions have been catalogued as privileged “green” processes, because they present desirable advantages from the point of view of environmental concerns, safety, and low cost.5
Nevertheless, the real “greenness” of aqueous-based organocatalytic reactions has been questioned.6 Indeed, for reactions performed in aqueous media, there is a significant possibility that the water being used as solvent will get contaminated by the organic compounds involved. Furthermore, Blackmond et al.6 argue that the disposal of discarded water by discharge into the normal drainage can end up being more polluting than a reaction carried out in traditional organic solvents with the proper after-treatment work-up.
Returning to the evaluation of the potential benefits attained from the use of water in chemical reactions, the first report of the use of water in an organocatalyzed aldol reaction comes from Barbas et al.7 In this work, it was found that the reaction between acetone and 4-nitrobenzaldehyde proceeds well in the presence of a small amount (less than 4 vol%) of water. By contrast, with increased amounts of water the enantioselectivity of the process decreased.
Following the work of Barbas and co-workers published in 2001,7 many reports have appeared describing the use of water, either as solvent, as co-solvent, or as additive in organocatalytic reactions.8 In an illustrative example, Barbas et al.9 found that hydrophobic diamine 1 (10 mol%) constitutes an efficient bifunctional catalyst in the aldol reaction of cyclic ketones with aromatic aldehydes in solvent water and in the presence of one equivalent of TFA, affording the aldol products with high diastereo- and enantioselectivity (Scheme 2).
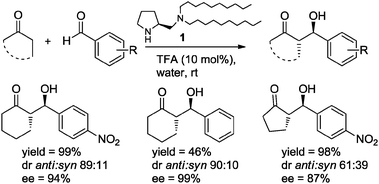 | ||
| Scheme 2 Aldol reactions catalyzed by diamine 1–TFA in water.9 | ||
Simultaneously and independently, Hayashi and co-workers10 reported the use of TBDPS-protected hydroxyproline 2 (10 mol%) as organocatalyst in the aldol reaction of cyclic ketones with aliphatic and aromatic aldehydes in water, obtaining the anti-aldol adducts with up to 99% ee (Scheme 3).
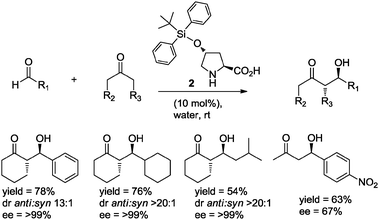 | ||
| Scheme 3 The asymmetric aldol reaction in water catalyzed by 4-tert-butyldiphenylsiloxyproline 2. | ||
In addition to the work mentioned above, a great number of studies in aqueous media or in brine solution have been reported in the last decade.11Fig. 1 shows some of the most prominent organocatalysts employed in aldol, Michael, Mannich, and cycloaddition reactions, which have proven not only to tolerate aqueous media, but that actually benefit from the presence of water in the reaction medium.
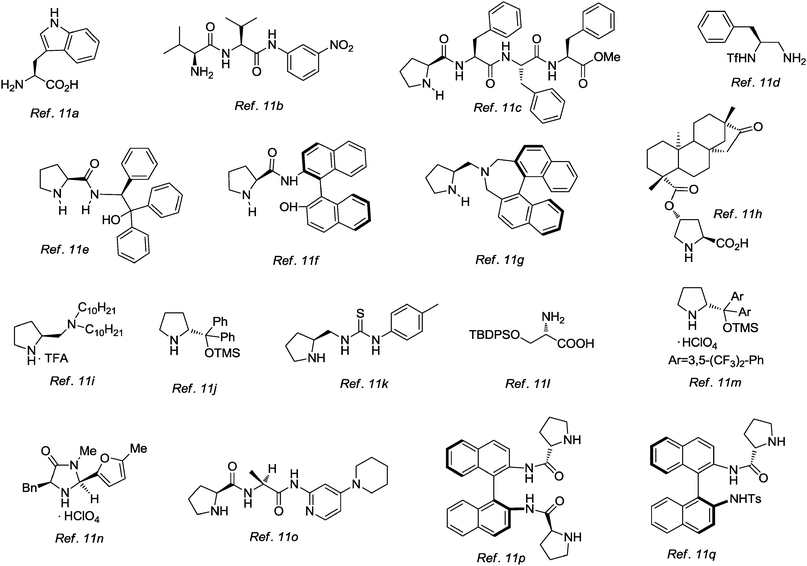 | ||
| Fig. 1 Examples of some water-compatible organocatalysts. | ||
In spite of the well-established effect of water as an enhancer of reactivity and selectivity in many organocatalytic processes, the role of water is not fully understood. Nevertheless, the exceptional physical properties of water, such as its high polarity, high surface tension, the ability to induce the hydrophobic effect, and especially water's capability to participate in hydrogen bonding, are frequently associated with the observed improvements.12
In this context,11h,x Tao and co-workers recently reported examples of aldol and Mannich reactions catalyzed by several amphiphilic organocatalysts derived from isosteviol and proline. In the presence of water the reactions proceeded efficiently affording the expected products in good to excellent yields and high stereoselectivity. The authors propose that water plays an important role in the transition state of the addition reaction through the formation of a hydrophobic chiral pocket induced by the presence of the lipophilic isosteviol unit. Additionally, the free hydroxyl groups of water in the interfacial surface could be involved in hydrogen bond formation, which leads to the observed improvement of the activity and stereoselectivity as a consequence of the more rigid transition state (Fig. 2).
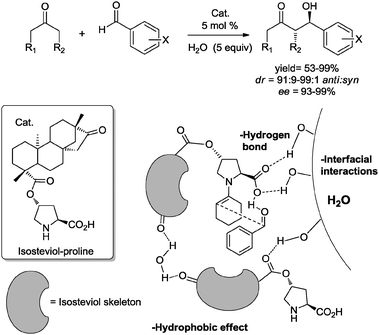 | ||
| Fig. 2 Proposed transition state for aldol reaction in the presence of water.11h | ||
In conclusion, from the “green” chemistry perspective, the use of water as a reaction medium offers a number of benefits: (1) it obviously avoids the use of toxic, volatile and/or corrosive solvents, (2) it usually minimizes the amount of catalyst that is required, (3) it frequently induces a significant acceleration of the reaction rate, and (4) it facilitates the isolation and purification of the products. Nevertheless, a frequent criticism directed to reactions using water consists in the lack of a general or practical procedure for the treatment of the aqueous phase at the end of the reaction to prevent the contamination of clean water sources.13
Ionic liquids
A different type of environmentally attractive reaction medium that is used in organocatalytic reactions is ionic liquids (IL), which, in contrast with the recent development of organocatalytic processes, have been known for nearly a century.14 ILs are liquid organic salts that essentially present no vapor pressure, and hence are not anticipated to emit potentially toxic volatile organic compounds. This property has attracted much attention owing to the possibility of using ILs as reaction media for “green” synthetic procedures. In addition, in the majority of the applications of ILs in catalysis emphasis has centered on the feasibility of recycling the solvent (the ILs). Simultaneously, separation of the ILs allows for the efficient recovery of the catalyst, which usually maintains its activity for several cycles. This was precisely one of observations reported independently in 2002 by Toma15a and Loh15b during their pioneering studies of organocatalyzed aldol reactions using ILs as solvent. Both research groups studied the asymmetric aldol reaction between acetone and aromatic aldehydes using (S)-proline as catalyst (1–30 mol%). The best IL turned out to be [bmim][PF6], which afforded the expected aldol products in good chemical yields and with moderate to good enantioselectivities (Scheme 4). The chiral immobilized organocatalysts were reused two to four times, with only a minor decrease in yield and enantioselectivity (for R = Ph,15b first cycle: yield 58% and ee 71%; fourth cycle: yield 52% and ee 67%).15![Proline-catalyzed aldol reaction in [bmim][PF6].15](/image/article/2012/CC/c2cc30951c/c2cc30951c-s4.gif) | ||
| Scheme 4 Proline-catalyzed aldol reaction in [bmim][PF6].15 | ||
Since the disclosure of the two previous experiments, the popularity of ionic liquids has increased notoriously.16 In particular, a number of novel organocatalysts have been evaluated using ionic liquids as reaction media, for example in the aldol reaction,17 as well as in other important reactions such as Michael additions,18 α-amination19 and α-aminoxylation,20 Mannich reaction,21 Diels–Alder reaction,22 among others.23
A common problem encountered with immobilized organocatalysts when using ionic liquids or modified-ionic liquids is the substantial loss of catalyst during the stages of recovery and recycling, making it difficult to maintain high reaction yields and stereoselectivities after 3 or 4 cycles. A convenient way to minimize this drawback is to modify the organocatalyst's structure by incorporation of an imidazolium moiety, which reduces losses of the recovered catalysts. For instance, in 2006 Miao and Chan described the synthesis of novel organocatalyst 3 derived from (2S,4R)-4-hydroxyproline, which was evaluated in the direct aldol reaction between various ketones and several aldehydes.24 By comparison with reaction in conventional solvents such as acetone and dimethylsulfoxide, the ionic liquid-supported catalyst 3 (30 mol%) afforded the aldol products in good to excellent yields (40–92%) and with good enantioselectivities (60–87% ee). Furthermore, organocatalyst 3 showed superior catalytic efficiency than (S)-proline, and could be recovered readily and reused up to 4 cycles24 (Scheme 5).
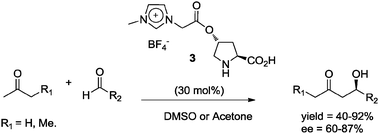 | ||
| Scheme 5 Aldol reaction catalyzed by ionic liquid-supported catalyst 3. | ||
Shortly thereafter, two reports in the literature described the use of structurally related catalysts in aldol reactions with IL as solvent.25 For instance, catalyst 4 (5 mol%) was used in the aldol reaction of cyclohexanone or hydroxyacetone with aromatic aldehydes in [bmim][Tf2N]. The expected products were obtained with good yields and stereoselectivities (Scheme 6a).25a
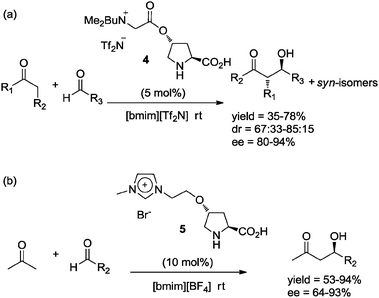 | ||
| Scheme 6 Aldol reaction catalyzed by 4 and 5 in ionic liquid media. | ||
On the other hand, ionic liquid-supported proline 5 (10 mol%) was also evaluated in the aldol reaction of acetone and several aldehydes in [bmim][BF4]. The reaction proceeded efficiently and the products were generated with good enantioselectivities (Scheme 6b).25bFig. 3 shows salient examples of organocatalysts that have proved to be efficient in ionic liquid or as ionic liquid-supported catalysts.
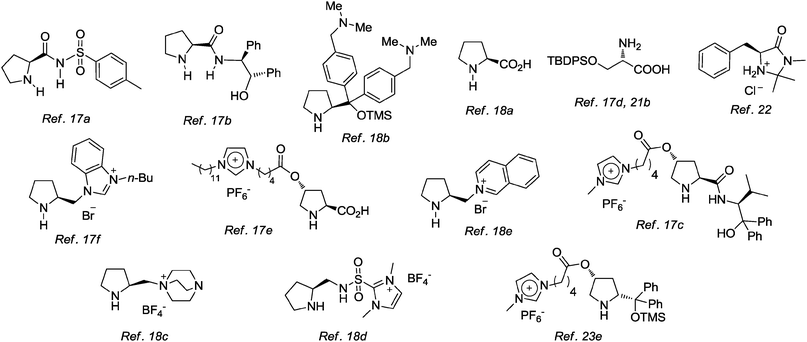 | ||
| Fig. 3 Representative examples of organocatalysts used in ILs and ionic liquid-supported catalysts. | ||
Relative to organocatalytic reactions performed in traditional organic solvents, the main benefits derived from the use of ILs as the reaction medium are (1) the possibility of exploring many diverse, potentially efficient combinations of functionalized ionic liquids, (2) the convenient recovery and efficient reuse of the catalyst, and (3), also in keeping with the tenets of “green” chemistry, the easy reutilization of the reaction medium.
However, it must be mentioned that moving from an organic solvent to an IL not always leads to a significant improvement in the selectivity of the catalyst or in the associated reaction rate. Furthermore, there are still many issues and unanswered questions regarding the real benefits of using ILs as reaction media. Some of the main concerns refer to their relatively high cost, potential human and environmental toxicity, poor biodegradability, high viscosity, and the need for laborious purification methodologies.26
Organic carbonate solvents
Linear and cyclic organic carbonates (Fig. 4) are a class of alternative solvents that have been extensively used during recent years.27 Because of their apparent easy availability and low cost, low toxicity and biodegradability, organic carbonates have also been considered as “green” solvents in organocatalytic reactions.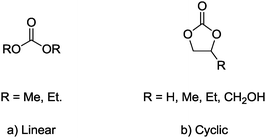 | ||
| Fig. 4 Common organic carbonates as potential “green” solvents. | ||
In 2009 North et al.28 evaluated ethylene and propylene carbonate (6 and 7, respectively, Scheme 7) as sustainable solvents in the asymmetric aldol reaction catalyzed by (S)-proline (10 mol%). The reaction of cyclic and acyclic ketones with aromatic aldehydes led to the formation of the desired aldol products in good chemical yields and good to excellent stereoselectivities (Scheme 7).28
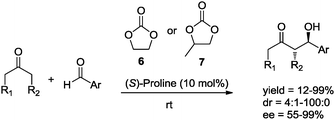 | ||
| Scheme 7 Asymmetric aldol reaction in cyclic carbonate solvents. | ||
In related work, North and co-workers studied the chiral solvent effect of propylene carbonate (R)-7 in organocatalyzed aldol reactions. These researchers found that using the appropriate combination of carbonate solvent and the “matching” proline enantiomer, the stereoselectivity of aldol adducts can be improved considerably (Scheme 8).29
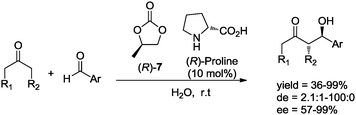 | ||
| Scheme 8 Asymmetric aldol reaction in chiral propylene carbonate. | ||
Recently, North et al. extended the application of cyclic carbonate solvents while studying the α-hydrazination of aldehydes and ketones by diazodicarboxylates organocatalyzed by (S)-proline (5 mol%).30
Regarding the actual “greenness” of organic carbonate solvents, the principal drawback is related to their preparation. Traditionally, the synthetic routes followed in the industrial-scale preparation of linear carbonates include the use of hazardous phosgene. On the other hand, the preparation of cyclic carbonates, despite involving 100% atom economical reactions, requires as starting material propylene oxide, which is highly toxic.
Nevertheless, new approaches directed to the preparation of organic carbonates using “greener” methodologies have been reported recently.27,31a–d
Solvent-free organocatalyzed reactions
As it can be appreciated in the sections discussed above, considerable improvement in the “greenness” of reaction media being used in organocatalyzed reactions has been achieved.31e Initially, the reactions of interest were typically performed under mild temperature conditions in organic solvents such as DMSO, DMF, and chlorinated solvents. Subsequently, alternative reaction media were explored, e.g. water, ionic liquids, or cyclic organic carbonates. However, as we have seen, all of these solvents have some associated disadvantages related to cost, production, handling, or toxicity, among others.A strategy explored in some organocatalyzed processes has been the use of a large excess of a reagent which acts as reaction medium, avoiding the use of auxiliary solvents. For example, in aldol and Michael reactions it is possible to employ an excess of the nucleophilic component to perform the reactions under solvent-free conditions.32 In this sense, Gong et al.,32a Zhao et al.32c and Xiao et al.32e evaluated the aldol reaction of aromatic aldehydes using an excess of acetone (27–45 equiv.) under “solvent-free” conditions. For example, (S)-proline derivatives 8 and 9 catalyzed the desired reaction affording the aldol adducts in high yields and with excellent enantioselectivities. In the case of derivative 10, its organocatalytic activity was evaluated in several traditional solvents (e.g., CHCl3, CH2Cl2, THF, DMF, water); nevertheless, highest enantioinduction was obtained when using acetone both as reagent and solvent (>45 equiv.) (Scheme 9).32a,c,e
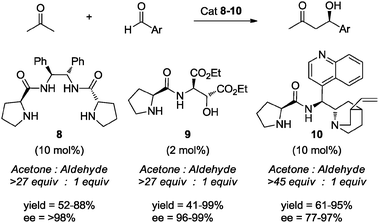 | ||
| Scheme 9 Organocatalyzed asymmetric aldol reactions under solvent-free conditions using excess of acetone. | ||
Nevertheless, the use of a large excess of one of the reagents is not a true “green” solution. A better option has been the use of approximately equimolar amounts of reactants (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–1:10 equiv.), in more real solvent-free green methodologies.33 For instance, in 2007 Oriyama and Ishino33a reported a highly enantioselective asymmetric conjugate addition of thiols to α,β-unsaturated aldehydes catalyzed by organocatalyst 11 (10 mol%) under solvent-free conditions, using only a slight excess of aldehyde (1.5 equiv.). The reaction proceeded to give the corresponding chiral sulfides with excellent enantioselectivity (up to 99% ee) (Scheme 10).
:1–1:10 equiv.), in more real solvent-free green methodologies.33 For instance, in 2007 Oriyama and Ishino33a reported a highly enantioselective asymmetric conjugate addition of thiols to α,β-unsaturated aldehydes catalyzed by organocatalyst 11 (10 mol%) under solvent-free conditions, using only a slight excess of aldehyde (1.5 equiv.). The reaction proceeded to give the corresponding chiral sulfides with excellent enantioselectivity (up to 99% ee) (Scheme 10).
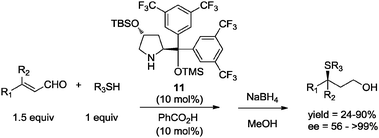 | ||
| Scheme 10 Asymmetric conjugate addition of thiols under solvent-free conditions.33a | ||
By the same token, prolinol sulfinyl ester 12 has been employed as organocatalyst in the absence of solvent. In particular, Zeng and Zhong33e found that when using only 3 equivalents of cyclohexanone as substrate, the Michael addition to nitroolefins catalyzed by 12 (10 mol%) proceeded in excellent yields and with high enantio- and diastereoselectivities, under solvent-free conditions at room temperature (Scheme 11).
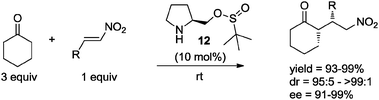 | ||
| Scheme 11 Michael addition of cyclohexanone to β-nitrostyrenes catalyzed by 12 under neat conditions.33e | ||
Recently, Li and co-workers reported the asymmetric Michael addition of diethyl malonate to nitroalkenes under solvent-free conditions using thiourea 13 as catalyst (10 mol%). The reaction was performed using stoichiometric amounts of reagents, in the complete absence of solvent, affording the corresponding Michael adducts in good to excellent yields and enantioselectivities. The versatility of the methodology was demonstrated in the key step of the synthesis of pregabalin, an amino acid derivative of γ-amino butyric acid, which has demonstrated to be an anticonvulsant drug used for neuropathic pain. The reaction allowed the formation of pregabalin hydrochloride 14 with high enantioselectivity (98% ee) (Scheme 12).33h
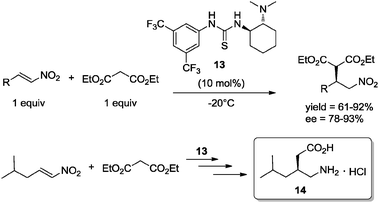 | ||
| Scheme 12 Enantioselective Michael addition under solvent-free conditions. Application of chiral organocatalyst 13 in the enantioselective synthesis of pregabalin.33h | ||
The organocatalyzed aldol reaction has also been performed under solvent-free conditions. In 2009, Worch and Bolm applied the prolyl-substituted sulfonimidamides (S,S)- and (S,R)-15 as catalysts (10 mol%) in solvent-free asymmetric aldol reactions.33d Using 5 equivalents of cyclohexanone, the reaction proceeded to afford the aldol products in moderate to very good yields and with good to excellent stereoselectivities (Scheme 13). Also in 2009 Nájera and co-workers prepared and evaluated a series of prolinamides and prolinethioamides as catalysts in the direct asymmetric aldol reaction between various ketones and aromatic aldehydes under solvent-free reaction conditions.33f In particular, prolinethioamide 16 (5 mol%) proved to be an excellent catalyst when using only 2 equivalents of cyclohexanone as substrate under traditional magnetic stirring. The reaction took place efficiently, and the major anti-aldol products were obtained with high enantioselectivities (Scheme 13). Very recently, Lygo and co-workers reported the synthesis and application of the chiral primary tertiary diamine salt 17, as an organocatalyst (5 mol%) in the enantioselective aldol reaction between cyclohexanone and a series of aromatic aldehydes. After evaluation of several different solvents, the reaction proved to be more efficient under solvent-free conditions, using 3 equivalents of ketone (Scheme 13).33i
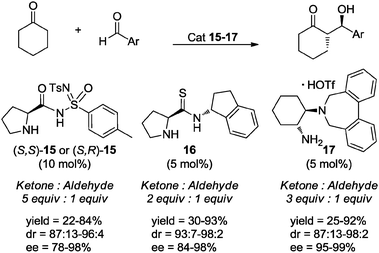 | ||
| Scheme 13 Examples of organocatalyzed asymmetric aldol reactions in the absence of solvent. | ||
An interesting research work was recently reported by Concellón, del Amo, and co-workers. These researchers studied the direct aldol reaction organocatalyzed by (S)-proline but using TBD (triazabicyclo[4.4.0]dec-5-ene)-derived guanidinium salts as co-catalysts. The results revealed that the use of (S)-proline and achiral guanidinium salt 18 led to the formation of the anticipated aldol products in good to very good yields, and with very high diastereo- and enantioselectivities under solvent-free conditions and with no stirring (Scheme 14).33j,34
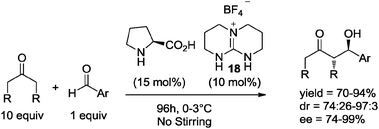 | ||
| Scheme 14 Asymmetric aldol reaction under solvent-free conditions, in the absence of stirring. | ||
The historically most significant organocatalyzed transformation is the intramolecular aldol reaction (Robinson-type annulation),1i–m which has proved to be a most useful strategy in the asymmetric preparation of the Wieland–Miescher ketone 19 (WMK), a well-recognized synthon in the synthesis of natural products.35 A solvent-free Robinson annulation procedure in the synthesis of WMK 19 was reported by Rajagopalan and Swaminathan in 1996.36 Using L-proline as catalyst and under neat conditions the reaction afforded the ketone 19 with low enantioinduction. More recently, Nájera, Bonjoch and co-workers have reported solvent-free, highly stereoselective syntheses of WMK 19 and Wieland–Miescher ketone analogues.11q,33f,34,37a,b For example, using the binam-prolinamide 20 as catalyst, the reaction proceeded efficiently to give the corresponding bicyclic diketones in high isolated yields and with excellent enantioselectivities (Scheme 15).
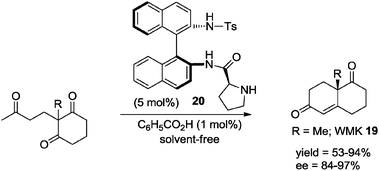 | ||
| Scheme 15 Solvent-free intramolecular aldol reaction.37a | ||
In a very recent report, Luo et al.37c described the asymmetric synthesis of WMK 19 as well as several analogues, through a solvent-free intramolecular aldol reaction catalyzed by chiral primary amine 21 (10 mol%). The protocol was also applied in the preparation of Hajos–Parrish ketone, affording in general the desired cyclic diketones in high yields and enantioselectivities (Scheme 16).
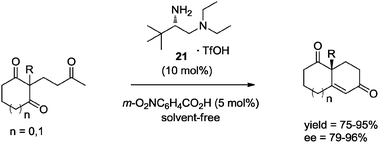 | ||
| Scheme 16 Primary amine catalyzed intramolecular aldol reaction in the absence of solvent. | ||
The implementation of the solvent-free philosophy in organocatalyzed reactions represents a genuine advancement towards the goal to make the organocatalytic process “greener”. Indeed, reactions performed under highly concentrated conditions or in the total absence of solvent present relevant advantages such as environmental safety, cost savings, and decreased reaction times. The attributes associated with solvent-free strategies help make the overall organocatalyzed process more sustainable. Nevertheless, it is important to understand that although the reactions may take place efficiently, the work-up, the separation of the organocatalyst from the product mixture and the subsequent purification steps generally involve the use of chromatographic techniques which require large amounts of volatile solvents.
Alternative reaction techniques: High-Speed Ball Milling
Following the previous discussion of solvent-free organocatalyzed methodologies, it is important to state that High-Speed Ball Milling (HSBM) is a sustainable mechanochemical technique that is being increasingly used in synthetic organic chemistry to promote several synthetically useful reactions, under solvent-free conditions.38,39 However, it is important to clarify that mechanochemical activation does not necessarily imply a solvent-free process, because mechanochemistry can occasionally be performed in the presence of solvents which facilitate the stirring, grinding or milling process. The advantages of the HSBM technique have also been applied in the area of organocatalysis. Pioneers in this field, Bolm et al., reported that the asymmetric aldol reaction under solvent-free conditions and catalyzed by (S)-proline (10 mol%) in a ball mill, employing a 1.1:1 ketone/aldehyde ratio, proceeded efficiently to give selectively the anti-aldol diastereomeric products in high yields and with up to 99% enantiomeric excess (Scheme 17).40 In this study, Bolm and co-workers demonstrated that the efficiency of the process using High-Speed Ball Milling (HSBM) was superior relative to traditional magnetic stirring: reaction times were shorter, and chemical yields and stereoselectivities were higher.
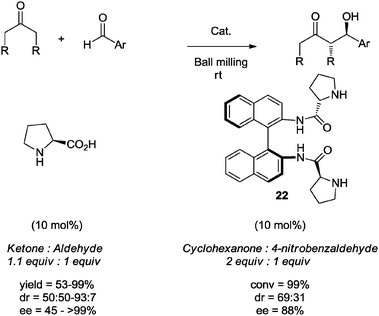 | ||
| Scheme 17 Enantioselective aldol reactions under solvent-free conditions, using High-Speed Ball Milling (HSBM). | ||
In this regard, Nájera and co-workers have reported the use of (Sa)-binam-L-prolinamides as organocatalysts in solvent-free aldol reactions. In particular, the prolinamide 22 (10 mol%) proved to work well as an organocatalyst in the reaction between cyclohexanone and 4-nitrobenzaldehyde using a planetary ball mill. The reaction was carried out using only 2 equivalents of ketone at room temperature. The anti-aldol adduct was obtained in high yield and with good diastereo- and enantioselectivity (Scheme 17).33c,41
Recently, Hernández and Juaristi42 carried out the asymmetric aldol reaction between representative ketones and various aromatic aldehydes, catalyzed by the methyl ester of (S)-proline-(S)-phenylalanine, (S,S)-23 (7 mol%). The reactions were performed under solvent-free conditions in a ball mill, using essentially stoichiometric amounts of the starting ketone and aldehyde (1.1:1).42 In general, the reaction proceeds efficiently affording the anti aldol products with good diastereo- and enantioselectivities. Furthermore, it could be demonstrated that dipeptide (S,S)-23 is a more efficient organocatalyst in the asymmetric aldol reaction under solvent-free conditions, relative to the same reaction in solution. In addition, it was found that reaction times were shortened relative to the same reaction employing 10 mol% of amino acid (S)-proline, also in a ball mill under solvent-free conditions (Scheme 18).
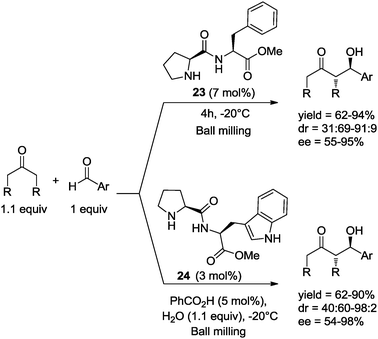 | ||
| Scheme 18 Asymmetric aldol reaction catalyzed by dipeptides 23 and 24 under ball-milling conditions. | ||
In more recent work, Hernández and Juaristi reported the asymmetric aldol reaction between ketones and various aromatic aldehydes under solvent-free conditions in a ball mill, with organocatalysis by several (S)-proline-containing dipeptides. It was observed that in the presence of an equimolar amount of water and a catalytic amount of benzoic acid, dipeptide (S,S)-proline-tryptophan-CO2Me 24 (3 mol%) was a particularly efficient catalyst affording the anti aldol products in good yields with high diastereo- and enantioselectivities. The better organocatalytic activity observed for 24 was explained as a result of the large aromatic area present in the lipophilic residue of tryptophan, which under solvent-free conditions and in the presence of an equimolar amount of water may promote the creation of a hydrophobic environment that could enhance non-covalent π–π interactions between the aromatic rings of the catalyst and the aldehydes, creating a more rigid transition state that induces higher stereoselectivity (Scheme 18).43
In related work, Hernández and Juaristi prepared and evaluated a series of (S,S)-proline-containing thiodipeptides, 25–27, as potential organocatalysts in aldol reactions under solvent-free reaction conditions using the HSBM technique. The thiodipeptide 25 (7 mol%) proved to be the most efficient catalyst in the reaction between cyclic ketones and aromatic aldehydes, affording the anti aldol products in good yields with high diastereo- and enantioselectivity. It is worth mentioning that the activity of catalysts 25–27 was also evaluated in the challenging aldol reaction between isatin derivatives and acetone. In that case the thiodipeptide 27 showed the major catalytic activity generating the hydroxy oxyindole derivatives in moderate to good yields and regular enantioselectivities (Scheme 19).44
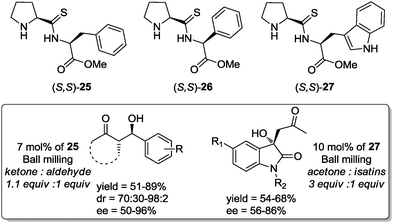 | ||
| Scheme 19 Solvent-free aldol reaction catalyzed by thiodipeptides 25–27 under ball-milling conditions.44 | ||
Recently, Xu et al. reported the Michael addition reaction of 1,3-dicarbonyl compounds to nitroolefins under solvent-free conditions in a ball mill. The reaction using catalyst 28 (0.5 mol%) afforded the corresponding adducts in very short milling times with high yields and stereoselectivities (Scheme 20).45
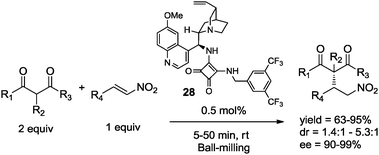 | ||
| Scheme 20 Organocatalyzed Michael addition reaction under ball-milling conditions.45 | ||
Very recently, Lamaty and co-workers reported the enantioselective synthesis of amino esters under ball-milling conditions, using a chiral ammonium salt derived from cinchonidine 29 (10 mol%). The reaction of the Schiff base of glycine tert-butyl ester with several electrophiles under basic conditions afforded the desired alkylated products in excellent yields and with moderate enantioselectivities (Scheme 21).46
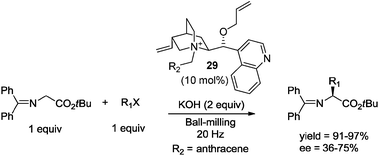 | ||
| Scheme 21 Enantioselective alkylation reaction under ball-milling conditions.46 | ||
In summary, the High-Speed Ball Milling technique has proved in the last few years to be a useful option to promote solvent-free organocatalyzed reactions. Relative to solution reactions, under HSBM conditions reaction yields tend to be higher, milling times (reaction times) are usually shorter, and selectivities better. The latter is probably because in the absence of solvent no solvation effects exist and non-covalent interactions (e.g. hydrogen bonding, hydrophobic effect, π–π stacking) are enhanced. Another highlight of HSBM is that relative to other activation methods such as microwaves and ultrasound, the ball milling technique is more energy efficient, which is directly related to the “greenness” of the process.38c On the other hand, the principal drawback of HSBM is associated with the difficulty associated with the accurate measurement and control of the temperature and pressure inside the grinding jars.
Microwaves, ultrasound and high pressure
Besides HSBM, other special techniques which have been classified as “green” alternatives in organic synthesis have also been applied to organocatalytic reactions. In particular, microwave irradiation, ultrasound or the use of high pressure have proved to be most promising. Indeed, organocatalytic processes performed using the above-mentioned non-traditional activation methodologies have been authoritatively reviewed.16a,38a Nevertheless, it is worth commenting that in connection with one of the 12 principles of “green” chemistry (‘the design for energy efficiency’), “green” synthetic methods should be preferably conducted at ambient temperature and atmospheric pressure, with the purpose to minimize the amount of energy required to increase the temperature or the pressure in the reactor. However, in the case of MW irradiation one has to recall that the observed enhancement of the reaction rate is in part associated with the rapid heating caused by MW irradiation relative to the same reaction using conventional heating methods.47On the other hand, the use of supercritical fluids as a reaction medium or the use of high pressure methodologies have the inconvenience of requiring extreme conditions (thus, high energy costs), and are accompanied by the requirement of special high-pressure apparatus to achieve the appropriate reaction conditions. Nevertheless, Hayashi and co-workers have reported an easy procedure to carry out organocatalytic reactions under high pressure. These researchers developed a convenient method for inducing high pressure by water-freezing in a sealed autoclave, taking advantage of the increased volume of liquid water when it freezes.48 This methodology has been used successfully in the asymmetric Baylis–Hillman reaction as well as in stereoselective aldol and Mannich reactions. The most evident beneficial effect of high pressure was observed in the Mannich reaction between various aldehydes, p-anisidine and acetone, organocatalyzed by (S)-proline (30 mol%) at −20 °C and 200 MPa. In particular, using p-bromobenzaldehyde the corresponding product was formed with better yield and with similar enantioinduction when compared with the same reaction at low pressure (Scheme 22).48
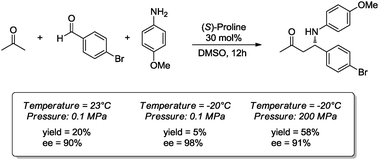 | ||
| Scheme 22 Mannich reaction under ambient pressure and high pressure induced by water-freezing. | ||
It must be said that in addition to energy efficiency considerations, organocatalytic reactions employing MW, ultrasound or high pressure are generally performed in organic solvents such as DMF, DMSO, THF, CH3Cl or CH2Cl2, which are considered undesired reaction media in the “green” chemistry field. A real improvement in the “greenness” of organocatalysis using these special techniques could be obtained by performing the MW, ultrasound or high pressure reactions in the absence of solvent. In this context, in 2010 Procopio and co-workers reported the microwave-assisted organocatalytic conjugate addition of diethyl malonate to several enones, under various conditions. Surprisingly, the reaction catalyzed by 15 mol% of (S)-proline proved to work well under solvent-free conditions, using only a slight excess of diethyl malonate (1.2 equiv.).49 The Michael adducts were obtained in short reaction times with good yields and moderate to excellent enantioselectivities (40–99% ee) (Fig. 5).
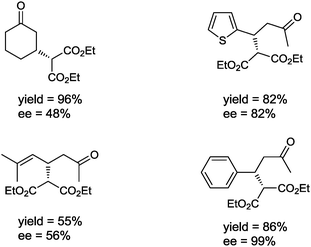 | ||
| Fig. 5 Michael adducts obtained under solvent-free conditions using MW irradiation. Reaction conditions: enone (5.0 mmol), diethyl malonate (6.0 mmol), piperidine (6.0 mmol), (S)-proline (15 mol%), 1.5–2.0 h of reaction time at 55 °C under MW irradiation. | ||
Catalytic efficiency and catalyst loading
Following the discussion above, regarding the impact of reaction media and alternative activation methodologies on “green” chemistry, it is essential to examine a central aspect of organocatalysis: the structural and functional characteristics of organocatalysts. Indeed, recently organocatalysts have been the target of criticism derived from consideration of the (sometimes) large amounts required in order to compare favorably with results from metal-catalyzed processes. In this regard, at the beginning of the last decade typical organocatalytic reactions used around 20 to 30 mol% of the catalyst. Nowadays, the general aim is to use lower amounts, preferably less than 5–10 mol%.50Indeed, the central principle of green chemistry is the high selectivity and efficiency of the catalytic materials. Of course, the use of highly selective catalytic reagents is superior relative to the employment of stoichiometric reagents. In this sense, it is common to find in organocatalytic reports that special attention is given to the specific effect of the amount of catalyst used. Generally, evaluation of the use of different amounts of organocatalyst shows that when using lower amounts reaction times become longer. Nevertheless, it is important to note that in many cases the stereoselectivity of the process remains remarkably high even when the amount of catalyst is diminished.
This at first sight unexpected effect was recently studied by Gröger, Berkessel and co-workers, who performed the aldol reaction between acetone and 3-chlorobenzaldehyde organocatalyzed by (R,R)-30. Surprisingly, these researchers found that a reduction in catalyst loading led to a dramatic improvement of the enantioselectivity for the formation of aldol product 31 (Scheme 23).51
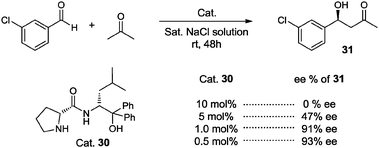 | ||
| Scheme 23 Effect of the amount of catalyst on the direct aldol reaction of 3-chlorobenzaldehyde with acetone catalyzed by (R,R)-30. | ||
The authors explained this result in terms of a change from kinetic control to thermodynamic control when the quantity of the catalyst is increased.51 This observation offers a new “green” alternative for making organocatalysts even more efficient: the traditional screening of the amount of catalyst in organocatalytic processes has to be carried out more carefully, since it could reveal that the ideal quantity of the catalyst is lower than anticipated, in fact diminishing costs by simply reducing the catalyst loading, and avoiding the use of excess catalyst and the addition of additives in the reaction media.
Indeed, several recent reports in the literature describe reactions that proceed more efficiently with rather low amounts of organocatalysts. Salient organocatalysts used in low quantity in aldol reactions, Michael reactions, Mannich reactions are presented in Fig. 6.52
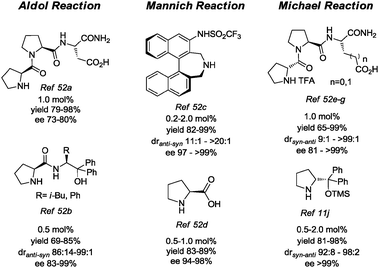 | ||
| Fig. 6 Structure of representative organocatalysts used in low loading. | ||
Regarding the recovery of organocatalysts, it is worth mentioning that some unsupported catalysts have been recovered by extractive acid–base work-up, simple filtration or centrifugation and reused with only minor decrease in its organocatalytic activity.11p,33f,34,37a,53
Nevertheless, a convenient strategy to render organocatalysts “greener” consists in their immobilization on a support with the purpose of facilitating catalyst recovery and reuse by simple work-up protocols.54 Additionally, in some cases immobilization processes allow the use of low amounts of the catalyst, because the active molecules are better distributed in the supporting material relative to a non-supported catalyst. The immobilization approach has been applied to simple organocatalysts such as proline, although the major benefits have been reported with support of more complex organocatalysts.
Recent strategies employ immobilized organic catalysts in friendly reaction media such as ionic liquids, water,8b or solvent-free methodologies. For example, Wennemers and co-workers55 reported the immobilization of a potent tripeptide H-L-Pro-L-Pro-L-Asp-NH2 organocatalyst on different resins. The anchored catalysts were evaluated in the aldol reaction between acetone and various aldehydes. Best results were obtained using TentaGel (polyethylene glycol–polystyrene) and PEGA (polyethylene glycol–polyacrylamide) as solid support. In particular, under solvent-free conditions and using an excess of acetone, solid-supported tripeptide 32 exhibited comparable or better reactivity and selectivity relative to a non-supported tripeptide (Scheme 24).55 Catalyst 32 could be recycled and reused up to 8 cycles, preserving the stereoselectivity of the aldol process, although the chemical yield diminished after 3 runs.
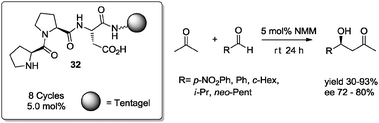 | ||
| Scheme 24 Asymmetric aldol reaction under solvent-free conditions catalyzed by supported tripeptide 32. | ||
Recently, Gao, Lu and Li used the chiral diamine-polyoxometalate (POM) acid combined organocatalyst 33 in the asymmetric direct aldol reaction of long-chain aliphatic ketones with various aromatic aldehydes, in the absence of solvent and using an excess of ketone. Catalyst 33 (2.5 mol%) activated the formation of the corresponding aldol adducts in good to high yields (54–95%) and with good enantioselectivities (80–91% ee) (Scheme 25).56 Chiral diamine-POM 33 could be easily recovered by simple filtration and recyclability was verified in 5 consecutive cycles showing only minor diminution in yield and enantioselectivity.
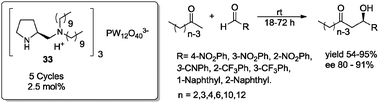 | ||
| Scheme 25 Solvent-free aldol reaction promoted by catalyst 33.56 | ||
In this context, in 2008 Yan and Wang prepared several Merrifield resin-supported dipeptides and tripeptides, and evaluated them as organocatalysts in the asymmetric direct aldol reaction. The best catalyst proved to be dipeptide H-L-Pro-L-Ala-O-resin 34 (10 mol%), whose efficiency was then optimized via screening of different reaction media, observing best results under neat conditions. For example, the reaction of cyclic and acyclic ketones with aromatic aldehydes afforded the corresponding aldol products with good to excellent yields (52–98%), good diastereoselectivities (anti:syn = 20:80–90:10) and enantioselectivities (75–95% ee) (Fig. 7).57 Additionally, catalyst 34 was reused up to 7 cycles without loss of activity or stereoselectivity.
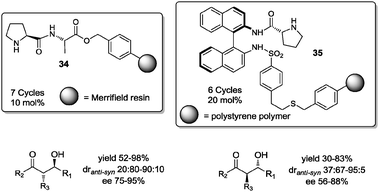 | ||
| Fig. 7 Representative supported organocatalysts used under solvent-free conditions. | ||
In related work, Nájera et al.58 described the preparation of polystyrene-supported binam-prolinamide 35, and its evaluation as an organocatalyst in asymmetric aldol reactions. Immobilized prolinamide 35 proved to be an efficient catalyst under solvent-free conditions (8 equiv. of ketone) or aqueous conditions. The reaction afforded the aldol adducts in high yields and stereoselectivities (Fig. 7). Furthermore, anchored prolinamide 35 showed recyclability in the reaction between cyclohexanone and 4-nitrobenzaldehyde, up to 6 cycles with only a slight decrease of yield and without loss of stereoselectivity in the aldol reaction.58
To close our discussion about “greener” organocatalysts, it is important to mention their potential role in asymmetric one-pot reactions, which is one of the most effective strategies for the “green” synthesis of complex molecular structures by carrying out sequentially multiple chemical steps in a single reaction vessel.59 The field of organocatalytic one-pot reactions is in fact an emblematic example of “green” chemistry, because in addition to all the advantages associated with organocatalysts, one-pot processes avoid the time and cost-consuming steps of protection and deprotection, as well as the corresponding work-up procedures. Furthermore, organocatalytic one-pot reactions minimize or avoid the generation of waste associated with the purification of intermediates. Finally, organocatalytic one-pot reactions frequently exhibit high yields and stereoselectivities. The above-mentioned attributes have made multicomponent one-pot protocols a priority option in natural product synthesis.60
Fig. 8 presents some of the most representative organocatalysts used in one-pot reactions, such as the Jørgensen–Hayashi catalyst, MacMillan imidazolidinones, cinchona derivatives, thioureas, and bifunctional amino-thiourea catalysts.61–66
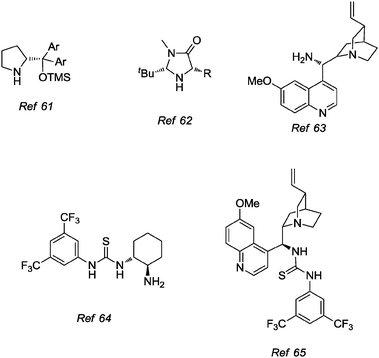 | ||
| Fig. 8 Examples of salient catalysts used in organocatalytic one-pot reactions. | ||
Unfortunately, space limitations prevent discussion in this feature article of the “green” characteristics of organocatalyzed cycloaddition, epoxidation, multicomponent, cascade, and other interesting reactions. Nevertheless, the analysis made here with aldol, Michael, Mannich or alkylation reactions is also valid and easily extendable to other organocatalyzed processes.
Concluding remarks
There is no doubt that “green” chemistry and organocatalysis represent remarkable developments in the chemistry of the XXI century. In spite of the fact that these two areas are still in their “infancy” (for example, compared with enzymatic catalysis, organocatalysis usually exhibits significantly lower reaction rates, enantioselectivities and turn-over efficiency), they already have contributed very significantly to the progress of science. “Green” chemistry has expanded and it now encompasses wide areas of the chemical enterprise. Of particular interest are developments with potential impact on industry and laboratory research as a means to continue chemical development in a more sustainable manner.67 Indeed, R. Noyori has stated that the future of chemists and actually the survival of humankind depend on our ability to manufacture useful compounds in an economical, energy-efficient, resource-preserving, and environmentally benign way.68 Thus, progress in the area of organocatalysis will accelerate chemical development in a more sustainable manner.All “green” contributions in the field of organocatalysis are very valuable because in addition to the preparation of the desired products, chemists implicitly have the social compromise to be more creative in the design of friendlier methodologies that prevent or at least reduce deleterious environmental impact.
During the last ten years, the progress of organocatalysis has been accompanied, in a synergetic combination, by “green” approaches that have made organocatalytic processes even “greener”. The above considerations motivated us to review and highlight here some salient approaches that from our perspective show clearly how the expansion of a new area can be made in a sustainable way.
Having discussed in the above sections the advantages and drawbacks associated with some “green” alternatives in organocatalysis, it is worth mentioning that a “green” reaction does not imply a “green” global process. Indeed, “green” processes not only have to evaluate the type of reaction media, the recyclability of the catalyst, or the activation strategy: a true “green” process must take into account all aspects involved, from the design of the chemical route to the final after-treatment of undesirable waste. Nevertheless, all the approaches described here, as well as others such as the use of fluorinated solvents, continuous flow, and microreactor techniques, are already contributing to the important goal to make organocatalytic processes more sustainable.
Acknowledgements
The authors are indebted to Conacyt, Mexico, for financial support via grant No. 130826. J. G. H. thanks Conacyt, Mexico, for a doctoral fellowship (No. 22211).Notes and references
- (a) B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS; (b) K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS; (c) W. Notz and B. List, J. Am. Chem. Soc., 2000, 122, 7386 CrossRef CAS; (d) B. List, J. Am. Chem. Soc., 2000, 122, 9336 CrossRef CAS; (e) W. S. Jen, J. J. M. Wiener and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 9874 CrossRef CAS. For pioneering reports see, for example: (f) G. Bredig and P. S. Fiske, Biochem. Z., 1913, 46, 7 CAS; (g) W. Langenbeck, Angew. Chem., 1928, 41, 740 CrossRef CAS; (h) H. Pracejus, Justus Liebigs Ann. Chem., 1960, 634, 9 CrossRef CAS; (i) U. Eder, G. Sauer, and R. Wiechert, Ger. Offen. 2,014,757, Schering A.-G., DE, 1971, 20 pp; (j) U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496 CrossRef CAS; (k) Z. G. Hajos, D. R. Parrish, DE 2102623, 1971 Search PubMed; (l) U. Eder, G. R. Sauer and R. Wiechert, DE 2014757, 1971 Search PubMed; (m) Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615 CrossRef CAS; (n) H. Wynberg and R. Helder, Tetrahedron Lett., 1975, 46, 4057 CrossRef. For recent discussions of historic background see, for example: (o) B. List, Angew. Chem., Int. Ed., 2010, 49, 1730 CrossRef CAS; (p) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS; (q) C. F. Barbas III, Angew. Chem., Int. Ed., 2008, 47, 42 CrossRef.
- For a recent discussion of the essential attributes associated with green chemistry, see: E. Negishi, J. Synth. Org. Chem., Jpn., 2011, 69, 1201 CrossRef CAS.
- (a) D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816 CrossRef CAS; (b) R. Breslow, Acc. Chem. Res., 1991, 24, 159 CrossRef CAS; (c) R. Breslow, Acc. Chem. Res., 2004, 37, 471 CrossRef CAS.
- For an instructive discussion about terminology of reactions performed in aqueous media, i.e. “in water”, “on water”, “in the presence of water”, or “in the presence of a large excess amount of water”, see: (a) S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS; (b) Y. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 8103 CrossRef CAS; (c) A. P. Brogan, T. J. Dickerson and K. D. Janda, Angew. Chem., Int. Ed., 2006, 45, 8100 CrossRef CAS.
- (a) U. M. Lindström, Chem. Rev., 2002, 102, 2751 CrossRef; (b) S. Kobayashi and K. Manabe, Acc. Chem. Res., 2002, 35, 209 CrossRef CAS.
- D. G. Blackmond, A. Armstrong, V. Coombe and A. Wells, Angew. Chem., Int. Ed., 2007, 46, 3798 CrossRef CAS.
- Although the first aldol reaction carried out in aqueous media was reported by Wiechert and co-workers, the role of water was not discussed (ref. 1i–j). By contrast, a thorough discussion can be found in K. Sakthivel, W. Notz, T. Bui and C. F. Barbas III, J. Am. Chem. Soc., 2001, 123, 5260 CrossRef CAS.
- For recent reviews on aqueous organocatalytic reactions, see: (a) J. Mlynarski and J. Paradowska, Chem. Soc. Rev., 2008, 37, 1502 RSC; (b) M. Gruttadauria, F. Giacalone and R. Noto, Adv. Synth. Catal., 2009, 351, 33 CrossRef CAS; (c) M. Raj and V. K. Singh, Chem. Commun., 2009, 6687 RSC; (d) N. Mase and C. F. Barbas III, Org. Biomol. Chem., 2010, 8, 4043 RSC; (e) S. Bhowmick and K. C. Bhowmick, Tetrahedron: Asymmetry, 2011, 22, 1945 CrossRef CAS . For recent reviews on aqueous asymmetric aldol reaction, see: ; (f) L. M. Geary and P. G. Hultin, Tetrahedron: Asymmetry, 2009, 20, 131 CrossRef CAS; (g) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC; (h) X.-H. Chen, J. Yu and L.-Z. Gong, Chem. Commun., 2010, 6437 RSC; (i) M. Bhanushali and C.-G. Zhao, Synthesis, 2011, 1815 CAS.
- N. Mase, Y. Nakai, N. Ohara, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2006, 128, 734 CrossRef CAS.
- (a) Y. Hayashi, T. Sumiya, J. Takahashi, H. Gotoh, T. Urushima and M. Shoji, Angew. Chem., Int. Ed., 2006, 45, 958 CrossRef CAS; (b) Y. Hayashi, S. Aratake, T. Okano, J. Takahashi, T. Sumiya and M. Shoji, Angew. Chem., Int. Ed., 2006, 45, 5527 CrossRef CAS.
- (a) Z. Jiang, H. Yang, X. Han, J. Luo, M. W. Wong and Y. Lu, Org. Biomol. Chem., 2010, 8, 1368 RSC; (b) W. Huang, H. Tian, H. Xu, L. Zheng, Q. Liu and S. Zhang, Catal. Lett., 2011, 141, 872 CrossRef CAS; (c) Z. Tang, Z.-H. Yang, L.-F. Cun, L.-Z. Gong, A.-Q. Mi and Y.-Z. Jiang, Org. Lett., 2004, 6, 2285 CrossRef CAS; (d) T. Miura, M. Ina, K. Imai, K. Nakashima, A. Masuda, N. Tada, N. Imai and A. Itoh, Synlett, 2011, 410 CrossRef CAS; (e) M. Raj, V. Maya, S. K. Ginotra and V. K. Singh, Org. Lett., 2006, 8, 4097 CrossRef CAS; (f) A. Russo, G. Botta and A. Lattanzi, Tetrahedron, 2007, 63, 11886 CrossRef CAS; (g) V. Bisai and V. K. Singh, Synlett, 2011, 481 CAS; (h) Y.-J. An, Y.-X. Zhang, Y. Wu, Z.-M. Liu, C. Pi and J.-C. Tao, Tetrahedron: Asymmetry, 2010, 21, 688 CrossRef CAS; (i) N. Mase, K. Watanabe, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2006, 128, 4966 CrossRef CAS; (j) S. Zhu, S. Yu and D. Ma, Angew. Chem., Int. Ed., 2008, 47, 545 CrossRef CAS; (k) Y.-J. Cao, Y.-Y. Lai, X. Wang, Y.-J. Li and W.-J. Xiao, Tetrahedron Lett., 2007, 48, 21 CrossRef CAS; (l) Y.-C. Teo, J.-J. Lau and M.-C. Wu, Tetrahedron: Asymmetry, 2008, 19, 186 CrossRef CAS; (m) Y. Hayashi, S. Samanta, H. Gotoh and H. Ishikawa, Angew. Chem., Int. Ed., 2008, 47, 6634 CrossRef CAS; (n) A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 2458 CrossRef CAS; (o) H. Tian, J.-I. Gao, H. Xu, L.-Y. Zheng, W.-B. Huang, Q.-W. Liu and S.-Q. Zhang, Tetrahedron: Asymmetry, 2011, 22, 1074 CrossRef CAS; (p) G. Guillena, M. C. Hita and C. Nájera, Tetrahedron: Asymmetry, 2006, 17, 1493 CrossRef CAS; (q) G. Guillena, C. Nájera and S. F. Viózquez, Synlett, 2008, 3031 CrossRef CAS; (r) W.-P. Huang, J.-R. Chen, X.-Y. Li, Y.-J. Cao and W.-J. Xiao, Can. J. Chem., 2007, 85, 208 CrossRef CAS; (s) X. Wu, Z. Jiang, H.-M. Shen and Y. Lu, Adv. Synth. Catal., 2007, 349, 812 CrossRef CAS; (t) S. S. V. Ramasastry, K. Albertshofer, N. Utsumi and C. F. Barbas III, Org. Lett., 2008, 10, 1621 CrossRef CAS; (u) M.-K. Zhu, X.-Y. Xu and L.-Z. Gong, Adv. Synth. Catal., 2008, 350, 1390 CrossRef CAS; (v) F.-Z. Peng, Z.-H. Shao, X.-W. Pu and H.-B. Zhang, Adv. Synth. Catal., 2008, 350, 2199 CrossRef CAS; (w) M. R. Vishnumaya and V. K. Singh, J. Org. Chem., 2009, 74, 4289 CrossRef CAS; (x) Y.-J. An, C.-C. Wang, Z.-P. Liu and J.-C. Tao, Helv. Chim. Acta, 2012, 95, 43 CrossRef CAS.
- Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492 CrossRef CAS.
- M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415 RSC.
- P. Walden, Bull. Acad. Imp. Sci. St.–Petersbourg, 1914, 1800 Search PubMed.
- (a) P. Kotrusz, I. Kmentová, B. Gotov, Š. Toma and E. Solčániová, Chem. Commun., 2002, 2510 RSC; (b) T.-P. Loh, L.-C. Feng, H.-Y. Yang and J.-Y. Yang, Tetrahedron Lett., 2002, 43, 8741 CrossRef CAS.
- For recent reviews on organocatalysis with ionic liquids, see: (a) Š. Toma, R. Šebesta and M. Mečiarová, Curr. Org. Chem., 2011, 15, 2257 CrossRef; (b) Š. Toma, M. Mečiarová and R. Šebesta, Eur. J. Org. Chem., 2009, 321 CrossRef; (c) P. Domínguez de María, Angew. Chem., Int. Ed., 2008, 47, 6960 CrossRef; (d) M. H. G. Prechtl, J. D. Scholten, B. A. D. Neto and J. Dupont, Curr. Org. Chem., 2009, 13, 1259 CrossRef CAS , and references therein.
- (a) M. Mečiarová, Š. Toma, A. Berkessel and B. Koch, Lett. Org. Chem., 2006, 3, 437 CrossRef; (b) H.-M. Guo, L.-F. Cun, L.-Z. Gong, A.-Q. Mi and Y.-Z. Jiang, Chem. Commun., 2005, 1450 RSC; (c) D. E. Siyutkin, A. S. Kucherenko and S. G. Zlotin, Tetrahedron, 2010, 66, 513 CrossRef CAS; (d) F.-F. Yong, C.-Y. Poh, G.-L. Chua and Y.-C. Teo, Chem. Lett., 2010, 490 CrossRef CAS; (e) D. E. Siyutkin, A. S. Kucherenko, M. I. Struchkova and S. G. Zlotin, Tetrahedron Lett., 2008, 49, 1212 CrossRef CAS; (f) S. Luo, X. Mi, L. Zhang, S. Liu, H. Xu and J.-P. Cheng, Tetrahedron, 2007, 63, 1923 CrossRef CAS.
- (a) P. Kotrusz, Š. Toma, H.-G. Schmalz and A. Adler, Eur. J. Org. Chem., 2004, 1577 CrossRef CAS; (b) D. Sarkar, R. Bhattarai, A. D. Headley and B. Ni, Synthesis, 2011, 1993 CAS; (c) D.-Z. Xu, Y. Liu, S. Shi and Y. Wang, Tetrahedron: Asymmetry, 2010, 21, 2530 CrossRef CAS; (d) B. Ni, Q. Zhang, K. Dhungana and A. D. Headley, Org. Lett., 2009, 11, 1037 CrossRef CAS; (e) D.-Q. Xu, B.-T. Wang, S.-P. Luo, H.-D. Yue, L.-P. Wang and Z.-Y. Xu, Tetrahedron: Asymmetry, 2007, 18, 1788 CrossRef CAS.
- (a) P. Kotrusz, S. Alemayehu, Š. Toma, H.-G. Schmalz and A. Adler, Eur. J. Org. Chem., 2005, 4904 CrossRef CAS; (b) X. Ding, H.-L. Jiang, C.-J. Zhu and Y.-X. Cheng, Tetrahedron Lett., 2010, 51, 6105 CrossRef CAS.
- (a) K. Huang, Z.-Z. Huang and X.-L. Li, J. Org. Chem., 2006, 71, 8320 CrossRef CAS; (b) H.-M. Guo, H.-Y. Niu, M.-X. Xue, Q.-X. Guo, L.-F. Cun, A.-Q. Mi, Y.-Z. Jiang and J.-J. Wang, Green Chem., 2006, 8, 682 RSC.
- (a) N. S. Chowdari, D. B. Ramachary and C. F. Barbas III, Synlett, 2003, 1906 CAS; (b) F.-F. Yong and Y.-C. Teo, Synth. Commun., 2011, 41, 1293 CrossRef CAS.
- (a) J. K. Park, P. Sreekanth and B. M. Kim, Adv. Synth. Catal., 2004, 346, 49 CrossRef CAS; (b) A. De Nino, O. Bortolini, L. Maiuolo, A. Garofalo, B. Russo and G. Sindona, Tetrahedron Lett., 2011, 52, 1415 CrossRef CAS.
- (a) B. Pégot, G. Vo-Thanh, D. Gori and A. Loupy, Tetrahedron Lett., 2004, 45, 6425 CrossRef; (b) S. Garre, E. Parker, B. Ni and A. D. Headley, Org. Biomol. Chem., 2008, 6, 3041 RSC; (c) D. B. Ramachary, N. S. Chowdari and C. F. Barbas III, Angew. Chem., Int. Ed., 2003, 42, 4233 CrossRef CAS; (d) D. D. Steiner, N. Mase and C. F. Barbas III, Angew. Chem., Int. Ed., 2005, 44, 3706 CrossRef CAS; (e) O. V. Maltsev, A. S. Kucherenko, A. L. Chimishkyan and S. G. Zlotin, Tetrahedron: Asymmetry, 2010, 21, 2659 CrossRef CAS.
- W. Miao and T. H. Chan, Adv. Synth. Catal., 2006, 348, 1711 CrossRef CAS.
- (a) M. Lombardo, F. Pasi, S. Easwar and C. Trombini, Adv. Synth. Catal., 2007, 349, 2061 CrossRef CAS; (b) L. Zhou and L. Wang, Chem. Lett., 2007, 36, 628 CrossRef CAS.
- (a) P. G. Jessop, Green Chem., 2011, 13, 1391 RSC; (b) B. Clare, A. Sirwardana and D. R. MacFarlane, Top. Curr. Chem., 2010, 290, 1 CrossRef; (c) S. Keskin, D. Kayrak-Talay, U. Akman and Ö. Hortaçsu, J. Supercrit. Fluids, 2007, 43, 150 CrossRef CAS; (d) J. Ranke, S. Stolte, R. Strmann, J. Arning and B. Jastorff, Chem. Rev., 2007, 107, 2183 CrossRef CAS; (e) J. R. Harjani, J. Farrell, M. T. Garcia, R. D. Singer and P. J. Scammells, Green Chem., 2009, 11, 821 RSC.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554 CrossRef.
- (a) M. North, F. Pizzato and P. Villuendas, ChemSusChem, 2009, 2, 862 CrossRef CAS; (b) W. Clegg, R. W. Harrington, M. North, F. Pizzato and P. Villuendas, Tetrahedron: Asymmetry, 2010, 21, 1262 CrossRef CAS.
- (a) M. North and P. Villuendas, Org. Lett., 2010, 12, 2378 CrossRef CAS; (b) M. Morcillo, M. North and P. Villuendas, Synthesis, 2011, 1918 CAS.
- C. Beattie, M. North and P. Villuendas, Molecules, 2011, 16, 3420 CrossRef CAS.
- (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS; (b) J. Sun, J. Wang, W. Cheng, J. Zhang, X. Li, S. Zhang and Y. She, Green Chem., 2012, 14, 654 RSC; (c) M. Tudorache, L. Protesescu, S. Coman and V. L. Parvulescu, Green Chem., 2012, 14, 478 RSC; (d) C. J. Whiteoak, E. Martin, M. Martínez-Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469 CrossRef CAS; (e) M. Lombardo and C. Trombini, in Eco-Friendly Synthesis of Fine Chemicals, ed. R. Ballini, The Royal Society of Chemistry, Cambridge, UK, 2009, ch. 1, pp. 1–79 Search PubMed.
- See for example: (a) Z. Tang, Z.-H. Yang, X.-H. Chen, L.-F. Cun, A. -Q. Mi, Y.-Z. Jiang and L.-Z. Gong, J. Am. Chem. Soc., 2005, 127, 9285 CrossRef CAS; (b) M. Jiang, S.-F. Zhu, Y. Yang, L.-Z. Gong, X.-G. Zhou and Q.-L. Zhou, Tetrahedron: Asymmetry, 2006, 17, 384 CrossRef CAS; (c) S. Samanta, J. Liu, R. Dodda and C.-G. Zhao, Org. Lett., 2005, 7, 5321 CrossRef CAS; (d) T. Kano, O. Tokuda and K. Maruoka, Tetrahedron Lett., 2006, 47, 7423 CrossRef CAS; (e) J.-R. Chen, X.-L. An, X.-Y. Zhu, X.-F. Wang and W.-J. Xiao, J. Org. Chem., 2008, 73, 6006 CrossRef CAS; (f) J. Agarwal and R. K. Peddinti, Tetrahedron: Asymmetry, 2010, 21, 1906 CrossRef CAS.
- See for example: (a) T. Ishino and T. Oriyama, Chem. Lett., 2007, 550 CrossRef CAS; (b) A. Scettri, A. Massa, L. Palombi, R. Villano and M. R. Acocella, Tetrahedron: Asymmetry, 2008, 19, 2149 CrossRef CAS; (c) G. Guillena, M. C. Hita, C. Nájera and S. F. Viózquez, J. Org. Chem., 2008, 73, 5933 CrossRef CAS; (d) C. Worch and C. Bolm, Synlett, 2009, 2425 CAS; (e) X. Zeng and G. Zhong, Synthesis, 2009, 1545 CrossRef CAS; (f) D. Almaşi, D. A. Alonso, A.-N. Balaguer and C. Nájera, Adv. Synth. Catal., 2009, 351, 1123 CrossRef; (g) D. R. Magar, C. Chang, Y.-F. Ting and K. Chen, Eur. J. Org. Chem., 2010, 2062 CrossRef CAS; (h) J.-M. Liu, X. Wang, Z.-M. Ge, Q. Sun, T.-M. Cheng and R.-T. Li, Tetrahedron, 2011, 67, 636 CrossRef CAS; (i) B. Lygo, C. Davison, T. Evans, J. A. R. Gilks, J. Leonard and C.-E. Roy, Tetrahedron, 2011, 67, 10164 CrossRef CAS; (j) A. Martínez-Castañeda, B. Poladura, H. Rodríguez-Solla, C. Concellón and V. Del Amo, Org. Lett., 2011, 13, 3032 CrossRef.
- For previously reported aldol reaction under “real” solvent-free conditions, in the absence of stirring, see for example: D. Almaşi, D. A. Alonso and C. Nájera, Adv. Synth. Catal., 2008, 350, 2467 CrossRef.
- For a recent review, see: B. Bradshaw and J. Bonjoch, Synlett, 2012, 337 CrossRef CAS.
- D. Rajagopal, K. Rajagopalan and S. Swaminathan, Tetrahedron: Asymmetry, 1996, 7, 2189 CrossRef CAS.
- (a) B. Bradshaw, G. Etxebarria-Jardí, J. Bonjoch, S. F. Viózquez, G. Guillena and C. Nájera, Adv. Synth. Catal., 2009, 351, 2482 CrossRef CAS; (b) B. Bradshaw, G. Etxebarria-Jardí, J. Bonjoch, S. F. Viózquez, G. Guillena and C. Nájera, Org. Synth., 2011, 88, 330 CAS; (c) P. Zhou, L. Zhang, S. Luo and J.-P. Cheng, J. Org. Chem., 2012, 77, 2526 CrossRef CAS.
- For recent reviews, see: (a) A. Bruckmann, A. Krebs and C. Bolm, Green Chem., 2008, 10, 1131 RSC; (b) G. Kaupp, CrystEngComm, 2009, 11, 388 RSC; (c) A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317 RSC; (d) M. O’Brien, R. Denton and S. V. Ley, Synthesis, 2011, 1157 Search PubMed; (e) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413 RSC; (f) R. B. Nasir-Baig and R. S. Varma, Chem. Soc. Rev., 2012, 41, 1559 RSC.
- For recent applications of HSBM, see: (a) R. Thorwirth, A. Stolle, B. Ondruschka, A. Wild and U. S. Schubert, Chem. Commun., 2011, 4370 RSC; (b) R. Thorwirth, A. Stolle and B. Ondruschka, Green Chem., 2010, 12, 985 RSC; (c) W. C. Shearouse, C. M. Korte and J. Mack, Green Chem., 2011, 13, 598 RSC; (d) J. G. Hernández and E. Juaristi, J. Org. Chem., 2010, 75, 7107 CrossRef; (e) W. Su, J. Yu, Z. Li and Z. Jiang, J. Org. Chem., 2011, 76, 9144 CrossRef CAS; (f) R. Schmidt, R. Thorwirth, T. Szuppa, A. Stolle, B. Ondruschka and H. Hopf, Chem.–Eur. J., 2011, 17, 8129 CrossRef CAS; (g) P. Nun, C. Martin, J. Martinez and F. Lamaty, Tetrahedron, 2011, 67, 8187 CrossRef CAS; (h) R. Thorwirth and A. Stolle, Synlett, 2011, 2200 CAS; (i) F. Ravalico, I. Messina, M. V. Berberian, S. L. James, M. E. Migaud and J. S. Vyle, Org. Biomol. Chem., 2011, 9, 6496 RSC.
- (a) B. Rodríguez, T. Rantanen and C. Bolm, Angew. Chem., Int. Ed., 2006, 45, 6924 CrossRef; (b) B. Rodríguez, A. Bruckmann and C. Bolm, Chem.–Eur. J., 2007, 13, 4710 CrossRef.
- G. Guillena, M. C. Hita, C. Nájera and S. F. Viózquez, Tetrahedron: Asymmetry, 2007, 18, 2300 CrossRef CAS.
- J. G. Hernández and E. Juaristi, J. Org. Chem., 2011, 76, 1464 CrossRef.
- J. G. Hernández and E. Juaristi, Tetrahedron, 2011, 67, 6953 CrossRef.
- J. G. Hernández, V. García-López and E. Juaristi, Tetrahedron, 2012, 68, 92 CrossRef.
- Y.-F. Wang, R.-X. Chen, K. Wang, B.-B. Zhang, Z.-B. Li and D.-Q. Xu, Green Chem., 2012, 14, 893 RSC.
- P. Nun, V. Pérez, M. Calmès, J. Martinez and F. Lamaty, Chem.–Eur. J., 2012, 18, 3773 CrossRef CAS.
- J. D. Moseley and C. O. Kappe, Green. Chem., 2011, 13, 794 RSC.
- (a) Y. Hayashi, K. Okado, I. Ashimine and M. Shoji, Tetrahedron Lett., 2002, 43, 8683 CrossRef CAS; (b) Y. Hayashi, W. Tsuboi, M. Shoji and N. Suzuki, J. Am. Chem. Soc., 2003, 125, 11208 CrossRef CAS; (c) Y. Hayashi, W. Tsuboi, M. Shoji and N. Suzuki, Tetrahedron Lett., 2004, 45, 4353 CrossRef CAS.
- A. Procopio, A. De Nino, M. Nardi, M. Oliverio, R. Paonessa and R. Pasceri, Synlett, 2010, 1849 CrossRef CAS.
- For a recent review see: F. Giacalone, M. Gruttadauria, P. Agrigento and R. Noto, Chem. Soc. Rev., 2012, 41, 2406 RSC . See also references therein.
- G. Rulli, N. Duangdee, K. Baer, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2011, 50, 7944 CrossRef CAS.
- (a) P. Krattiger, R. Kovasy, J. D. Revell, S. Ivan and H. Wennemers, Org. Lett., 2005, 7, 1101 CrossRef CAS; (b) V. Maya, M. Raj and V. K. Singh, Org. Lett., 2007, 9, 2593 CrossRef CAS; (c) T. Kano, Y. Yamaguchi, O. Tokuda and K. Maruoka, J. Am. Chem. Soc., 2005, 127, 16408 CrossRef CAS; (d) B. Rodríguez and C. Bolm, J. Org. Chem., 2006, 71, 2888 CrossRef; (e) M. Wiesner, J. D. Revell, S. Tonazzi and H. Wennemers, J. Am. Chem. Soc., 2008, 130, 5610 CrossRef CAS; (f) M. Wiesner, J. D. Revell and H. Wennemers, Angew. Chem., Int. Ed., 2008, 47, 1871 CrossRef CAS; (g) M. Wiesner, G. Upert, G. Angelici and H. Wennemers, J. Am. Chem. Soc., 2010, 132, 6 CAS.
- For selected examples, see: (a) G. Guillena, M. C. Hita and C. Nájera, Tetrahedron: Asymmetry, 2006, 17, 729 CrossRef CAS; (b) G. Guillena, M. C. Hita and C. Nájera, Tetrahedron: Asymmetry, 2006, 17, 1027 CrossRef CAS; (c) R. Chinchilla, C. Nájera, F. J. Ortega and S. Tarí, Tetrahedron: Asymmetry, 2009, 20, 2279 CrossRef CAS; (d) S. Tarí, R. Chinchilla and C. Nájera, Tetrahedron: Asymmetry, 2009, 20, 2651 CrossRef; (e) D. Almaşi, D. A. Alonso, E. Gómez-Bengoa and C. Nájera, J. Org. Chem., 2009, 74, 6163 CrossRef; (f) Y. Yang, Y.-H. He, Z. Guan and W.-D. Huang, Adv. Synth. Catal., 2010, 352, 2579 CrossRef CAS; (g) C. Wu, X. Fu and S. Li, Eur. J. Org. Chem., 2011, 1291 CrossRef CAS; (h) B. Tan, G. Hernández-Torres and C. F. Barbas III, J. Am. Chem. Soc., 2011, 133, 12354 CrossRef CAS; (i) S. Li, X. Fu and C. Wu, Res. Chem. Intermed., 2012, 38, 195 CrossRef CAS.
- For recent reviews, see: (a) F. Cozzi, Adv. Synth. Catal., 2006, 348, 1367 CrossRef CAS; (b) M. Gruttadauria, F. Giacalone and R. Noto, Chem. Soc. Rev., 2008, 37, 1666 RSC; (c) T. E. Kristensen and T. Hansen, Eur. J. Org. Chem., 2010, 3179 CrossRef CAS . See also references therein.
- J. D. Revell, D. Gantenbein, P. Krattiger and H. Wennemers, Biopolymers, 2006, 84, 105 CrossRef CAS.
- Q. Gao, S.-M. Lu, Y. Liu and C. Li, Tetrahedron Lett., 2011, 52, 3779 CrossRef CAS.
- J. Yan and L. Wang, Synthesis, 2008, 2065 CAS.
- A. Bañón-Caballero, G. Guillena and C. Nájera, Green Chem., 2010, 12, 1599 RSC.
- Ł. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492 CrossRef.
- (a) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS; (b) C. Vaxelaire, P. Winter and M. Christmann, Angew. Chem., Int. Ed., 2011, 50, 3605 CrossRef CAS.
- For recent examples, see: (a) K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248 CrossRef CAS; (b) Ł. Albrecht, L. K. Ransborg, A. Albrecht, L. Lykke and K. A. Jørgensen, Chem.–Eur. J., 2011, 17, 13240 CrossRef; (c) M. Rueping, C. M. R. Volla, M. Bolte and G. Raabe, Adv. Synth. Catal., 2011, 353, 2853 CrossRef CAS; (d) V. Coeffard, A. Desmarchelier, B. Morel, X. Moreau and C. Greck, Org. Lett., 2011, 13, 5778 CrossRef CAS.
- (a) Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051 CrossRef CAS; (b) B. Simmons, A. M. Walji and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2009, 48, 4349 CrossRef CAS; (c) S. B. Jones, B. Simmons and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 13606 CrossRef CAS.
- (a) M. W. Paixão, N. Holub, C. Vila, M. Nielsen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2009, 48, 7338 CrossRef; (b) S. Sternativo, A. Calandriello, F. Costantino, L. Testaferri, M. Tiecco and F. Marini, Angew. Chem., Int. Ed., 2011, 50, 9382 CrossRef CAS.
- (a) H. Uehara, R. Imashiro, G. Hernández-Torres and C. F. Barbas III, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20672 CrossRef CAS; (b) R. Imashiro, H. Uehara and C. F. Barbas III, Org. Lett., 2010, 12, 5250 CrossRef CAS.
- (a) S. Rajkumar, K. Shankland, G. D. Brown and A. J. A. Cobb, Chem. Sci., 2012, 3, 584 RSC; (b) S. Bai, X. Liang, B. Song, P. S. Bhadury, D. Hu and S. Yang, Tetrahedron: Asymmetry, 2011, 22, 518 CrossRef CAS.
- For a recent review, see: H. Pellissier, Adv. Synth. Catal., 2012, 354, 237 CrossRef CAS . See also references therein.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC.
- R. Noyori, Tetrahedron, 2010, 66, 1028 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |