Solvent dependence of helix stability in aromatic oligoamide foldamers†‡
Ting
Qi
ab,
Victor
Maurizot
ab,
Hiroki
Noguchi
c,
Thiraporn
Charoenraks
c,
Brice
Kauffmann
def,
Makoto
Takafuji
cg,
Hirotaka
Ihara
cg and
Ivan
Huc
*ab
aUniv. Bordeaux, CBMN, UMR 5248, Institut Européen de Chimie et Biologie, 2 rue Escarpit, F-33600 Pessac, France. E-mail: i.huc@iecb.u-bordeaux.fr; Fax: +33 54000 2215; Tel: +33 54000 2219
bCNRS, CBMN, UMR 5248, Institut Européen de Chimie et Biologie, 2 rue Escarpit, F-33600 Pessac, France
cDepartment of Applied Chemistry and Biochemistry, Kumamoto University, 2-39-1 Kurokami, Kumamoto 860-8555, Japan
dUniv. Bordeaux, IECB, UMS 3033, F-33600 Pessac, France
eCNRS, IECB, UMS 3033, F-33600 Pessac, France
fINSERM, IECB, US 0001, F-33600 Pessac, France
gKumamoto Institute for Photo-Electro Organics (PHOENICS), Japan
First published on 11th April 2012
Abstract
A new helical aromatic oligoamide foldamer, bearing triethyleneglycol side chains for solubility in a broad range of media, was prepared. The stability of the helical conformation was assessed in various solvents and shown to vary greatly and unexpectedly. Stability was remarkably enhanced in methanol–water mixtures.
Synthetic foldamers1 are artificial oligomers, produced by step-wise synthesis, that adopt folded conformations resembling the structures of biopolymers. They may be constructed from chemically varied backbones, but they have shown less variety in terms of their folded structures, with helices being a highly prevalent motif. Helical folding is governed by a combination of internal and external parameters such as monomer shape and rigidity, intramolecular attractive and repulsive interactions that may be local or between units remote in a sequence, rotational restrictions, solvophobic effects, and aggregation or the presence of guest molecules. All these parameters, and thus folding, are sensitive to the nature of the solvent and foldamers are often found to fold in a limited range of media. Examples include the typical use of trifluoroethanol to stabilize short α-helices2 and the strong solvent dependence of oligophenylethynylenes3 and other so called “solvophobic foldamers”.4 Solvents are also critical in the case of guest-induced folding.5 As an exception, helical aromatic amide foldamers6 have been recognized as particularly robust folded architectures and qualitative evidence showed that they may fold in solvents as diverse as chloroform,7 toluene,8 methanol,9 DMSO,5a,10 and water.11 This apparent lack of solvent dependence is atypical and we thus set to undertake a systematic quantitative investigation of helix stability in these systems. As described below, we discovered that helix stability of aromatic oligoamides in fact varies greatly with solvent, but not in an expected way. Chloroform, the solvent in which these foldamers are generally studied and found to have very stable conformations, is one of the least favourable solvents. In contrast, protic solvents lead to a dramatic stabilization of helical conformations.
The folding of aromatic oligoamide foldamers rests primarily on backbone rigidity due to local electrostatic repulsions and hydrogen bonds between adjacent units and, for multiturn helices, on intramolecular aromatic stacking which may include electrostatic and solvophobic components. We selected an octameric oligoamide of 2-quinolinecarboxylic acid 1, one of the most robust helical aromatic amide foldamers (Scheme 1).5a,6 In this series, an octamer forms a helix spanning over three turns with right-handed (P) or left-handed helicity (M).
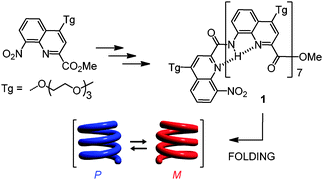 | ||
| Scheme 1 | ||
For the purpose of this study, a new monomer was equipped with a tri-ethyleneglycol side chain at position 4 in order to provide solubility in a wide range of solvents.‡ As detailed in the ESI‡, the synthesis proceeded in a classical manner through dimeric, tetrameric and hexameric intermediates before giving octamer 1. A crystal structure§ of the dimeric intermediate was obtained and is shown in Fig. 1 to illustrate the relative size and position of each monomer and its side-chain in a more or less extended form. Most ethylene glycol units are found in a gauche conformation.
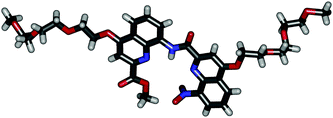 | ||
| Fig. 1 Crystal structure of a dimeric intermediate in the synthesis of 1. | ||
In order to assess the helix stability of 1, we used a previously described method based on the separation of its P and M conformational enantiomers by HPLC on a chiral stationary phase (CHIRALPACK IA).12 This separation is allowed by the exceptional stability of these helices which do not significantly racemize during chromatographic separation carried out at low temperature. The solvent conditions previously used for similar oligomers bearing isobutoxy side chains (n-hexane/chloroform 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25, v/v) did not separate the P and M conformers of 1. After some optimization we found that using chloroform/2-propanol (25:75, v/v) at −5 °C as the mobile phase results in a good separation (Fig. 2a). The positive sign of the circular dichroism (CD) trace at 385 nm indicates that the positive peak which elutes first corresponds to the P helix.13 This is opposite to previous results on oligomers having isobutoxy side chains for which the first fraction to elute was that of the M helix. Changing the side chains to triethylene-glycol and the solvent composition of the mobile phase thus resulted in a reversal of retention times. The P-1 fraction can be collected and evaporated at −5 °C, during which the P helix does not racemize significantly, yielding a solid which can be stored.
:25, v/v) did not separate the P and M conformers of 1. After some optimization we found that using chloroform/2-propanol (25:75, v/v) at −5 °C as the mobile phase results in a good separation (Fig. 2a). The positive sign of the circular dichroism (CD) trace at 385 nm indicates that the positive peak which elutes first corresponds to the P helix.13 This is opposite to previous results on oligomers having isobutoxy side chains for which the first fraction to elute was that of the M helix. Changing the side chains to triethylene-glycol and the solvent composition of the mobile phase thus resulted in a reversal of retention times. The P-1 fraction can be collected and evaporated at −5 °C, during which the P helix does not racemize significantly, yielding a solid which can be stored.
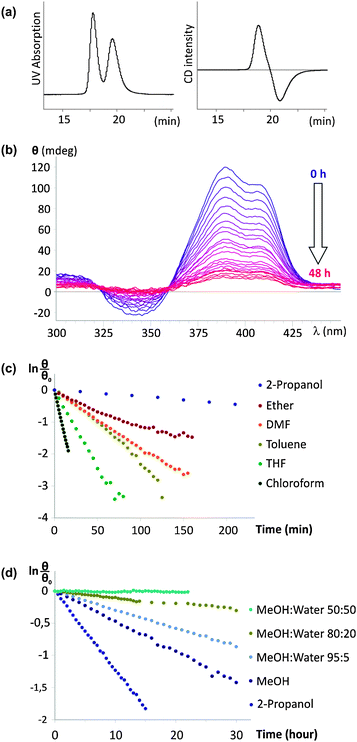 | ||
| Fig. 2 Racemization of P-1 as a function of solvent. (a) Chromatographic separation of P-1 and M-1 on a chiral stationary phase. The UV detector trace (350 nm) is shown on the left and the CD detector trace (385 nm) is shown on the right; (b) example of the variation of the CD spectrum over time in MeOH; (c, d) linear plots of CD intensity at 385 nm fitted to a first order decay. The horizontal scale is in minutes in (c) and in hours in (d). | ||
Samples of P-1 were then redissolved in various solvents, and its racemization was monitored over time using CD spectroscopy. A typical example is shown in Fig. 2b. A slow decay of CD intensity was observed in MeOH; meanwhile the UV-vis absorption spectrum remains constant. As explained in our earlier report,12 the equilibrium between P-1 and M-1 must involve at least partially unfolded intermediates. The activation barrier of helix handedness inversion thus reflects the energy difference between a folded and a partially unfolded state. The racemization kinetics are a direct and quantitative indication of helix stability. As shown in Fig. 2c and d, the decay of CD intensity can be fitted to single order kinetics and the half-life of helix handedness inversion can be extracted. Table 1 summarizes data obtained for the racemization of 1 at 30 °C.
| Solvent | K a/min−1 | t 1/2 a/min |
|---|---|---|
| a Determined by monitoring the decay of CD intensity of P-1. b vol/vol. c Too slow to be determined. | ||
| CHCl3 | 1.2 × 10−1 | 6 |
| THF | 4.4 × 10−2 | 16 |
| Toluene | 2.5 × 10−2 | 28 |
| DMF | 1.7 × 10−2 | 40 |
| Et2O | 9.0 × 10−3 | 77 |
| 2-Propanol | 2.0 × 10−3 | 350 |
| MeOH | 7.7 × 10−4 | 900 |
| 95:5 MeOH/H2Ob |
4.8 × 10−4 | 1445 |
| 80:20 MeOH/H2Ob |
1.7 × 10−4 | 4080 |
| 50:50 MeOH/H2Ob |
NDc | NDc |
The results show that the kinetics of handedness inversion of 1 span about four orders of magnitude which reflects a strong solvent dependence. In this respect, aromatic amides seem not to differ from most foldamers. However, the trend is unexpected: 1 is found to be the least stable in chloroform, the solvent in which the helical structures of aromatic amides including 8-amino-2-quinolinecarboxylic acid oligomers have most often been characterized. It remains that, even in chloroform, the half-life of helix handedness inversion is still 6 min, which is several orders of magnitude more stable than e.g. peptidic helices such as AiB oligomers which interconvert on the millisecond timescale.14 Less polar solvents (toluene, Et2O) enhance helix stability, which can be understood as a reinforcement of the effect of polar interactions such as hydrogen-bonding and electrostatic repulsions. Yet a polar solvent such as DMF leads to a similar enhancement.
The most striking effect is that of protic media. The half-life of helix handedness inversion jumps by about two orders of magnitude in alcohols and by another one in 80:20 MeOH/H2O. In 50:50 MeOH/H2O, P-1 shows marginal racemization after one day at 30 °C. Reliable measurements could not be carried out in the presence of larger amounts of water because of precipitation, as indicated by a decrease of UV absorbance over time. Protic solvents are known to compete with intramolecular hydrogen-bonding and generally destabilize structures held by hydrogen bonds. Protic solvents also induce solvophobic effects, which are presumably the reason for the enhanced conformational stability of 1 in these media. Indeed, the three helical turns of 1 give rise to extensive intramolecular π−π stacking between quinoline rings which may result in hydrophobic effects.
Aggregation, for example double helix formation,15 could also be a factor that influences helix handedness inversion rates. However, this can be ruled out in all but one of the solvents used here. Indeed, 1H NMR spectra show very sharp lines with little solvent or concentration dependence of chemical shift values, characteristic of a unimolecular species. The only exception is 50:50 CD3OD/D2O vol/vol for which some line broadening and upfield shifts of the signals suggest some micellization, possibly due to the hydrophobic character of the terminal quinolines that are exposed to the solvent (Fig. 3).
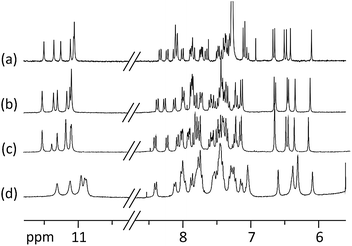 | ||
| Fig. 3 Part of the 300 MHz 1H NMR spectra of 1 at 0.5 mM showing amide (10.5–11.5 ppm) and aromatic (6–8.5 ppm) resonances in: (a) CDCl3; (b) CD3OD; (c) 80:20 CD3OD/D2O vol/vol; (d) 50:50 CD3OD/D2O vol/vol. Note the lack of H/D exchange of most amide protons in deuterated protic solvents. | ||
In summary, the results above delineate an extreme conformational stability of helical aromatic oligoamides in protic solvents. They also call for additional investigations to determine whether the effect of the solvent on helix stability varies with the surface involved in intramolecular π−π stacking (i.e. oligomer length), and with interactions between stacked quinoline units (e.g. presence of electron withdrawing substituents). These studies are in progress and will be reported in due course.
This work was supported by an FP7 Marie Curie Post-Doctoral Fellowship (PIIF-2009-254156) to T.Q. and by a CNRS-JSPS collaborative grant.
Notes and references
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC.
- M. K. Luidens, J. Figge, K. Breese and S. Vajda, Biopolymers, 1996, 39, 367 CrossRef CAS; S. R. Lehrman, J. L. Tuls and M. Lund, Biochemistry, 1990, 29, 5590 CrossRef.
- D. J. Hill and J. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5053 CrossRef CAS.
- J.-L. Hou, M.-X. Jia, X.-K. Jiang, Z.-T. Li and G.-J. Chen, J. Org. Chem., 2004, 69, 6228 CrossRef CAS; A. J. Zych and B. L. Iverson, J. Am. Chem. Soc., 2000, 122, 8898 CrossRef.
- (a) Y.-X. Xu, X. Zhao, X.-K. Jiang and Z.-T. Li, J. Org. Chem., 2009, 74, 7267 CrossRef CAS; (b) J.-m. Suk and K.-S. Jeong, J. Am. Chem. Soc., 2008, 130, 11868 CrossRef CAS.
- I. Huc, Eur. J. Org. Chem., 2004, 17 CrossRef CAS.
- (a) H. Jiang, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2003, 125, 3448 CrossRef CAS; (b) B. Gong, H. Zeng, J. Zhu, L. Yua, Y. Han, S. Cheng, M. Furukawa, R. D. Parra, A. Y. Kovalevsky, J. L. Mills, E. Skrzypczak-Jankun, S. Martinovic, R. D. Smith, C. Zheng, T. Szyperski and X. Cheng Zeng, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11583 CrossRef CAS; (c) C. Dolain, A. Grélard, M. Laguerre, H. Jiang, V. Maurizot and I. Huc, Chem.–Eur. J., 2005, 11, 6135 CrossRef CAS; (d) V. Berl, I. Huc, R. Khoury, M. J. Krische and J.-M. Lehn, Nature, 2000, 407, 720 CrossRef CAS; (e) H.-P. Yi, X.-B. Shao, J.-L. Hou, C. Li, X.-K. Jiang and Z.-T. Li, New J. Chem., 2005, 29, 1213 RSC.
- C. Dolain, J.-M. Léger, N. Delsuc, H. Gornitzka and I. Huc, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16146 CrossRef CAS; N. Delsuc, J.-M. Léger, S. Massip and I. Huc, Angew. Chem., Int. Ed., 2007, 46, 214 CrossRef.
- Z.-Q. Hu, H.-Y. Hu and C.-F. Chen, J. Org. Chem., 2006, 71, 1131 CrossRef CAS; E. Berni, B. Kauffmann, C. Bao, J. Lefeuvre, D. M. Bassani and I. Huc, Chem.–Eur. J., 2007, 13, 8463 CrossRef.
- L. Yuan, H. Zeng, K. Yamato, A. R. Sanford, W. Feng, H. S. Atreya, D. K. Sukumaran, T. Szyperski and B. Gong, J. Am. Chem. Soc., 2004, 126, 16528 CrossRef CAS.
- R. W. Sinkeldam, M. H. C. J. van Houtem, K. Pieterse, J. A. J. M. Vekemans and E. W. Meijer, Chem.–Eur. J., 2006, 12, 6129 CrossRef CAS; I. Huc, V. Maurizot, H. Gornitzka and J.-M. Léger, Chem. Commun., 2002, 578 RSC; E. Gillies, C. Dolain, J.-M. Léger and I. Huc, J. Org. Chem., 2006, 71, 7931 CrossRef.
- N. Delsuc, T. Kawanami, J. Lefeuvre, A. Shundo, H. Ihara, M. Takafuji and I. Huc, ChemPhysChem, 2008, 9, 1882 CrossRef CAS.
- C. Dolain, H. Jiang, J.-M. Léger, P. Guionneau and I. Huc, J. Am. Chem. Soc., 2005, 127, 12943 CrossRef CAS.
- R.-P. Hummel, C. Toniolo and G. Jung, Angew. Chem., Int. Ed. Engl., 1987, 26, 1150 CrossRef.
- B. Baptiste, J. Zhu, D. Haldar, B. Kauffmann, J.-M. Léger and I. Huc, Chem.–Asian J., 2010, 5, 1364 CAS.
Footnotes |
| † This article is part of the ChemComm ‘Chirality’ web themed issue. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures and characterization of new compounds for the preparation of 1; procedures for chromatographic and crystallographic experiments. CCDC 869654. See DOI: 10.1039/c2cc31533e |
| § Crystal data: (C40H40N6O10), M = 764.78, T = 213(2) K, orthorhombic, space group Pbc21, a = 7.3608 (15), b = 24.153 (5), c = 40.044 (8) Å, V = 7119 (2) Å3, Dc = 1.228 g cm−3, Z = 8, F(000) = 3216, 19954 reflections measured, 5345 independent reflections (Rint = 0.05), refinement on F2 against all reflections. The weighted R-factor wR and goodness of fit GOF are based on F2, GOF = 1.07, R[F2 > 2σ(F2)] = 0.059, wR(F2) = 0.163. Hydrogen site locations were inferred from neighbouring sites, H-atom parameters constrained, 5345 reflections used for refinement, 933 parameters and 2 restraints. |
| This journal is © The Royal Society of Chemistry 2012 |