Electrical conductive coordination polymers
Gonzalo
Givaja
a,
Pilar
Amo-Ochoa
a,
Carlos J.
Gómez-García
b and
Félix
Zamora
*a
aDepartamento de Química Inorgánica, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: felix.zamora@uam.es; Fax: +34 91 4974833
bICMol. Parque Científico de la Universidad de Valencia, 46980 Paterna, Valencia, Spain. E-mail: carlos.gomez@uv.es
First published on 29th June 2011
Abstract
Coordination polymers are currently one of the hottest topics in Inorganic and Supramolecular Chemistry. This critical review summarizes the current state-of-the-art on electrical conductive coordination polymers (CPs), also named metal–organic frameworks (MOFs). The data were collected following two sort criteria of the CPs structure: dimensionality and bridging ligands (151 references).
 Gonzalo Givaja | Gonzalo Givaja studied chemistry at Universidad San Pablo-CEU in Madrid (1995–2000). Afterwards he finished his MSci in polymeric materials at CSIC institute. In 2006 he finished his PhD thesis on polypyrrolic macrocycles and their late transition metal complexes under the guidance of Dr J. B. Love and Prof. Dr M. Schröder at the University of Nottingham. In 2007 he was awarded a Juan de la Cierva postdoctoral grant from the Spanish Ministerio de Educacion at the Universidad Autónoma de Madrid. |
 Pilar Amo-Ochoa | Pilar Amo-Ochoa received a BSc in Chemistry and a PhD in Inorganic Chemistry from Autónoma University of Madrid (Spain). As a postdoctoral researcher she worked on Metal-Nucleobases coordination chemistry with Prof. B. Lippert at Dortmund University (Germany). Actually she has a permanent position in the Inorganic Department of Autónoma University of Madrid. Currently, her research focuses on coordination polymers as possible nanoelectronic materials. She is the author of ∼30 papers based on coordination chemistry. |
 Carlos J. Gómez-García | Carlos J. Gómez-García was born in 1964 in Valladolid (Spain). In 1991 he obtained the PhD in Inorganic Chemistry at the University of Valencia under the supervision of Prof. E. Coronado. He moved to the University of Rennes in 1993 and to the CRPP in Bordeaux (France) to work with Prof. P. Delhaès in 1993–1995. He is currently a full Professor in Inorganic Chemistry and works in the Institute of Molecular Science of the University of Valencia. His main research area is focused on the design, synthesis and characterization of multifunctional molecular materials presenting magnetic and electrical properties. |
 Félix Zamora | Félix Zamora was born in 1967 in Cuenca (Spain). In 1994 he obtained the PhD in Inorganic Chemistry at Universidad Autónoma de Madrid. He moved to University of Dortmund (Germany) to work with Professor B. Lippert. He is currently “Profesor Titular” at the Inorganic Chemistry Department at the Universidad Autónoma de Madrid. From 2004 he focused on new nanomaterials with electrical properties based on inorganic systems such as coordination polymers. |
1. Introduction
Coordination polymers (CPs) are a family of compounds that can be considered as the natural extension of coordination compounds towards polymerization. Accordingly, CPs are formed by the assembly of two different building blocks: the metal entities (metal ion or metal complex fragment) and the bridging ligands. Their architectures and dimensionalities are defined by the coordinative geometry and capabilities of these two building-blocks. The definitions found in the literature for coordination polymers (CPs) slightly differ. The broader definition considers that a CP is formed connecting the metal building blocks by either organic or inorganic bridging ligands. However, some authors limit it to metal entities linked just by inorganic ligands and consider as organic–inorganic hybrid material those polymers formed by metals connected through organic ligands. The justification for this differentiation is somehow based on the properties found between both types of polymers. Albeit, the general physical and chemical properties of CPs essentially depend on many factors besides the nature of the bridging ligand. In this scenario, other supramolecular polymers using metal complexes as building-blocks can be found, for instance those connected by H-bonds (H-bonded frameworks). However these are rather different and more difficult to predict and design and will not be considered in the scope of this review. Along this review we will use the term CP from the broader definition above mentioned.In the last few years, a large amount of work has been carried out in the synthesis and structural characterisation of CPs. Structural investigation under the term of crystal engineering, dealing with structure design and control of the architecture, has become a hot topic in this field. This structural control has produced a considerable improvement of the properties associated with these materials. Most of these works have focused on the study of properties such as catalysis, chirality, luminescence, magnetism, spin-transition (spin-crossover), non-linear optics (NLO), porosity or zeolitic-like behaviour. In fact, the search for porosity and catalytic properties has resulted in a specific extension of CPs, named metal–organic frame-works (MOFs) or porous coordination polymers (PCPs). These PCPs or MOFs can be considered as CPs showing large cavities in the structure.
It is well-known that electrical conductivity of classical covalent polymers has attracted the interest of many researchers in material science. However, the search of this property in CPs is still very scarce. Based on recent results, and in part motivated by the high potential of CPs towards nanotechnological applications,1,2 this tendency is being reversed. In addition, the gradual incorporation of theoretical calculations in CPs seems to be a powerful tool for understanding the experimental measurements and for the rational design of new electrically conductive CPs.
Applications in catalysis3 and gas storage4 or gas/molecules separation5 are some of the most current active aspects of this discipline. However, in the near horizon new amazing applications are being developed connecting this field with nanotechnology.6,7 There is no doubt of the technological impact expected for coordination polymers within the next coming years, in fact some of them are already used in industrial applications.8
However, despite the potential implications of high electrical conductivity in CPs in applications such as porous electrodes for batteries, fuel cells, capacitors, sensors, molecular wires, etc., the literature on this field is quite disperse and needs to be rationalized in order to understand the data available and to put into a general perspective their potential. The aim of this review is to summarize the current state-of-the-art of the electrical conductive coordination polymers and provide a personal point of view towards perspectives of these materials and their potential applications.
2. Tools: electrical conductivity measurements. Conductivity mechanism and low temperature ground states
2.1. Measuring electrical conductivity: methods, problems and solutions
Electrical conductivity of a material is a measure of its ability to conduct electric current under certain conditions (as temperature, pressure, applied current,…). From the simple Ohm's law on conductivity (V = R × I; where V is the voltage, I the current, and the proportionality constant, R is the resistance), it is straightforward that a measurement of R may simply be done by applying a current (I) and measuring a potential (V). Since R depends on geometrical parameters (and, therefore, it is sample-dependent) the magnitude usually given is the resistivity (ρ), which is defined as ρ = R × (A/l), where A is the cross-sectional area of the conductor (A = a × d) and l is the distance between the voltage-drop measuring points (Scheme 1). For historical reasons the resistivity is usually measured in Ω cm instead of Ω m, its International System unit. Besides the resistivity, the other magnitude that is usually provided is its inverse, called the conductivity, σ (=1/ρ), which is measured in Ω−1 cm−1 = S cm−1 (Ω−1 = S = Siemens).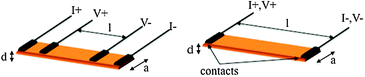 | ||
| Scheme 1 The four (left) and two (right) contacts methods for measuring electrical conductivity. Dimensions l, a and d are the voltage probes distance, the sample width and thickness, respectively. | ||
Note that not all conductors obey Ohm's law. Thus, semiconductors and most of the low-dimensional conductors often deviate from Ohm's law, except in a low current range. Furthermore, even metals only follow Ohm's law in a limited current or voltage range and, therefore, when resistivity or conductivity values are quoted, the current or voltage range in which these values were obtained has to be specified. Besides the value of the electrical resistivity at room temperature, it is much more useful to provide its thermal variation since its analysis allows the determination of its electronic nature as will be shown below.
As stated above, the measurement of the resistance (and, hence, the resistivity) of a sample requires the application of a current (I) and the measurement of a voltage drop across the sample (V). Unfortunately, these measurements are not so simple in most cases since there are different difficulties to be solved: the values of the resistivity may vary many orders of magnitude from sample to sample (in fact, the electrical resistivity is the physical magnitude showing the largest range of values, with differences of ca. 24 orders of magnitude from silver to quartz). Even more, for a given sample, the thermal variation may induce changes of several orders of magnitude in the resistivity. A second problem associated with the conductivity measurements deals with the effect of the electrical contacts with the sample. Thus, if the sample resistivity is very low, the instrument will mainly measure the sample-wire contact resistance. On the contrary, if the sample's resistivity is high, the contact resistance will be negligible (as observed in most CPs) but if it is very high the experiment may only measure the internal impedance of the voltmeter and leakage currents. An additional problem affecting the conductivity measurements is the lack of homogeneity and the anisotropy of many samples. This anisotropy may be inherent (low dimensional structures) or due to the shape of the sample (single crystals, pressed pellets, thin films,…).
All these difficulties have led to the design of different measuring methods depending on the nature and geometry of the sample and on the magnitude of its conductivity. Thus, when the resistivity is very low, to avoid the effects of contacts resistance, the best way is to use the four contacts method (Scheme 1).
This method involves the connection of four parallel in-line contacts to the sample. The two external contacts are used to apply a current (I+ and I−) and the two internal ones are used to measure the voltage drop across the sample (V+ and V−). The connection between the thin metallic wires (usually tempered Cu, Ag, Au or Pt wires with diameters of 25–100 μm) and the sample are done with the help of some metallic emulsion of fine powders in an organic solvent (silver, gold, platinum or graphite pastes) or simply by pressing the wires against the sample (if the mechanical resistance of the sample allows this last method). The resistivity is calculated from the cross-sectional area (A = a × d) and the distance between the internal contacts (l) as ρ = R × (a × d/l). The four contacts method is more accurate since it only measures the resistance of the sample (Rx) but neither the resistance of the wires nor that of the contacts as long as the internal impedance of the voltmeter is higher than the sample resistance. In contrast, in the two probe method the resistance of the sample and those of the wires and contacts are added in the measured resistance. Since these wire and contact resistances are usually of a few Ohms, only for highly conducting samples these resistances have to be considered. The only (sometimes important) advantage of the two-probe method is the smaller limiting sample size that can be measured with two contacts (typically above ca. 50 μm vs. ca. 250 μm for the four contacts method). When the samples are very thin or narrow or are not regular, the measurement of the sample dimensions may imply an important error, leading to errors in the calculated resistivity. In these cases, the four-points (different from the four contacts) and the van der Pauw methods are more convenient. In the first case, the four in-line contacts are very small equidistant points, separated by a distance (l) (Scheme 2). Again the two external contacts are used to apply a current (I) and the two internal ones to measure the voltage drop (V). The resistivity can be easily calculated as: ρ = 2π × l × (V/I).
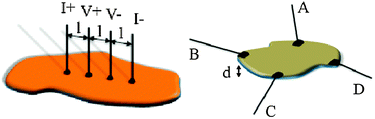 | ||
| Scheme 2 The four points (left) and the van der Pauw (right) methods used for measuring electrical conductivity in very thin samples. | ||
The other method used for thin samples is the van der Pauw configuration where the four contacts are glued in the periphery of the thin plate-like sample (whose thickness is d) approximately arranged in the vertex of a square (Scheme 2). The resistivity in this case is calculated using the formula:
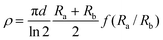
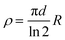
As already mentioned, an additional problem deals with the lack of homogeneity in many samples (either arising from the shape anisotropy or from the structure of the compound). The shape anisotropy can be well resolved with the van der Pauw and the four points methods.11 Albeit, the inherent anisotropy of the sample requires the use of the Montgomery method or, if possible, the measurement of the resistivity along different directions or on different faces of the crystals in order to have a complete idea of the electrical anisotropy. The Montgomery method allows the determination of the resistivity along two different directions in anisotropic samples with a rectangular prismatic shape (Scheme 3).
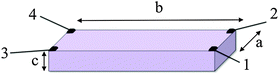 | ||
| Scheme 3 The Montgomery method used for measuring the electrical conductivity in anisotropic rectangular prismatic samples. | ||
This method requires four contacts in the four corners of one of the faces of the prisms (with dimensions a and b). The resistivities in the two directions (ρa and ρb) are calculated with the equations:
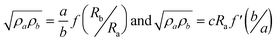
Another source of errors in the determination of the exact value of the conductivity is its high sensitivity to the quality of the crystals. This problem may lead to significant differences in the conductivity measured in different crystals of the same sample. Furthermore, in many molecular superconductors the quality of the crystal may determine the critical temperature and even the presence or absence of the superconducting transition. Of course, the use of several crystals (at least three of four) allows a more precise determination of the value, and the necessary reliability in the observed value.
Finally, it is worth to mention that there are other less common problems such as the conduction through humidity layers in the sample and the possible reaction of the organic solvent or the paste with the sample that may affect the conductivity measurements. These and other problems are, fortunately, very scarce and, together with their possible solutions, have already been discussed in detail.12
2.2. Mechanisms of the electrical conductivity in CPs and low dimensional conductors
Usually resistivity measurements are performed as a function of temperature (from room temperature down to a few K) since the thermal variation of the conductivity is more informative and more reliable than the room temperature value. Thus, although in a first approach the different electrical behaviours can be distinguished looking at the room temperature value of the conductivity (which is usually in the range 101–105 S cm−1 for metals, 10−10–101 S cm−1 for semiconductors and below ca. 10−10 S cm−1 for insulators), the limits between the different regimes are not clear.12 The only reliable way to obtain a clear picture of the electrical behaviour of a compound is to perform the thermal variation of the electrical conductivity inside the Ohmic regime. These measurements show an increase of the conductivity (or decrease of the resistivity) with decreasing temperature in metals and the opposite behaviour in semiconductors (and insulators). Note that although semiconductors show all a thermally activated conductivity (the conductivity increases with increasing temperature), the temperature dependence is not the same in all cases since there are different conduction mechanisms.In general, thermally activated semiconductors present a small gap between the conduction and valence bands (in fact, the classical insulator definition is applied when the band gap is above 3 eV). When this gap is very small, in a first approach we can assume that the number of carriers and, hence, the conductivity, should show an exponential dependence with temperature (σ ∝ Ta). This is the case for some conducting polymers with low doping levels, where values as high as 14 can be found for the exponent a.13 In classical semiconductors the thermal dependence of the conductivity follows the Arrhenius law: σ(T) = σ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/kT), where Ea represents the activation energy (corresponding to half of the band gap, Ea = Eg/2) and σ0 is a pre-exponential factor. Classical semiconductors are quite common among crystalline semiconductors but when dealing with amorphous materials, conducting polymers or coordination polymers, the conduction mechanism may change from a band model to a localized one. In this localized model charges move through a so-called hopping mechanism, which is the term used to describe the “jump” of charges through a phonon-assisted quantum tunnelling mechanism.12,14 The so-called hopping model describes the conductivity with a modified exponential law: σ = σ0exp[−(T0/T)a], where the exponent a is related to the electronic dimensionality of the sample, a = 1/(1 + d), being d the dimensionality. Thus, for one, two and three dimensional systems the a value is 1/2, 1/3 and 1/4, respectively. Note that for three dimensional systems a = 1/4 and we obtain Mott's law.15 In this expression, σ0 and T0 are related to the electronic density of the states at the Fermi level, N(F), to the so-called localization length and to the phonons frequency. This variable range hopping (VRH) model has been used to explain conductivity data of many conducting polymers as well as coordination polymers (the term “variable” arises from the fact that the hopping range varies with temperature, since the thermal energy of the electrons and the phonon frequency change with temperature). Unfortunately in some cases the data can be fit to different a values (precluding a precise determination of the electronic dimensionality) or the obtained a value is located between two limiting cases (i.e., when the hopping mechanism appears mainly, but not exclusively, in one or two dimensions).
exp(−Ea/kT), where Ea represents the activation energy (corresponding to half of the band gap, Ea = Eg/2) and σ0 is a pre-exponential factor. Classical semiconductors are quite common among crystalline semiconductors but when dealing with amorphous materials, conducting polymers or coordination polymers, the conduction mechanism may change from a band model to a localized one. In this localized model charges move through a so-called hopping mechanism, which is the term used to describe the “jump” of charges through a phonon-assisted quantum tunnelling mechanism.12,14 The so-called hopping model describes the conductivity with a modified exponential law: σ = σ0exp[−(T0/T)a], where the exponent a is related to the electronic dimensionality of the sample, a = 1/(1 + d), being d the dimensionality. Thus, for one, two and three dimensional systems the a value is 1/2, 1/3 and 1/4, respectively. Note that for three dimensional systems a = 1/4 and we obtain Mott's law.15 In this expression, σ0 and T0 are related to the electronic density of the states at the Fermi level, N(F), to the so-called localization length and to the phonons frequency. This variable range hopping (VRH) model has been used to explain conductivity data of many conducting polymers as well as coordination polymers (the term “variable” arises from the fact that the hopping range varies with temperature, since the thermal energy of the electrons and the phonon frequency change with temperature). Unfortunately in some cases the data can be fit to different a values (precluding a precise determination of the electronic dimensionality) or the obtained a value is located between two limiting cases (i.e., when the hopping mechanism appears mainly, but not exclusively, in one or two dimensions).
Besides the hopping mechanism, in the case of doped conducting polymers and π-conjugated systems there are two additional different mechanisms to explain conductivity: polarons/bipolarons and solitons. Polarons are charged carriers that bear a spin whereas bipolarons are pairs of compensated spins. When the doping level is low, the main carriers are polarons in conducting polymers with non-degenerate ground states. If the doping increases, then the number of polarons reaches a maximum value and begins to pair in bipolarons. This is easily followed by magnetic susceptibility measurements that show an increase with increasing doping to reach a maximum followed by a decrease indicative of spin pairing in bipolarons.16 The second possible mechanism to explain electrical conductivity in conducting polymers is solitons. Solitons are localized conformational defects that can easily move coupled with the lattice vibrations since the energy of the solitons is the same everywhere (in contrast to electrons whose energy varies from site to site).12,17,18 In one-dimensional conjugated systems at low temperatures this mechanism gives rise to an insulating state since solitons are trapped, given the low dimensionality of the lattice. In the case of solitons generated by doping (as in the case of slightly doped conducting polymers) the charge born by the solitons interact with the doping counterions, reducing their mobility. When the numbers of solitons increase (as in highly doped conducting polymers) the solitons form a partially filled band leading to a band-type conductivity. Finally, when charged and neutral solitons coexist, the trapped charged solitons may hop if the neutral solitons pass near the charged ones. This mechanism is called the intersoliton hopping and constitutes a particular case of hopping conductivity.
Other factors that may determine and explain the conductivity in one-dimensional CPs are the localization effects. These effects are very important when dealing with very small wires (molecular wires in some cases) and are attributed to the presence of unavoidable defects in the lattice.19–21 In these cases, the level of impurities determines the localization of the electrons and if the impurities level is high enough, the electrons will only present a hopping mechanism between localized states. This localization explains the trend towards an insulating state in strictly one-dimensional conductors when the temperature tends to zero, as observed in one dimensional CPs, even if they present, in some cases, weak inter-chain interactions.
2.3. Electronic transitions in CPs and low dimensional conductors
Although the main conducting mechanisms in doped polymers and CPs are now well established, there are many systems (specially molecular and polymeric) that undergo electronic transitions which may be accompanied by structural ones when the temperature and/or the pressure are changed. These electronic transitions are usually driven by the relatively strong electron–lattice or spin–lattice interactions present in these systems due to the low dimensionality of the lattice. Among these transitions the most frequent are the Peierls and spin-Peierls transitions observed in one dimensional systems as KCP (K2Pt(CN)4Br0.303.2H2O) and TTF–TCNQ (TTF = tetrathiafulvalene, TCNQ = tetracyanoquinodimethane).22–25 The Peierls transition can be described as a charge density wave (CDW, see below) that opens a gap at the Fermi level giving rise to an insulating state. This CDW is originated by a periodic molecular displacement that breaks the regularity of the chain, resulting in a dimerization of the chain. The driving force for this transition is the gain in electronic energy due to the resulting lowering of the Fermi level.18,26If the electronic Coulomb repulsion is strong, then the localized spins may suffer a magnetic transition to an antiferromagnetic (AF) ground spin state27,28 or a spin-Peierls (SP) transition giving rise to a diamagnetic dimerized system.29 The AF transition occurs when the system presents non-negligible inter-chain interactions that increase the electronic dimensionality to two or three. The spin-Peierls transition is observed when the localized spins (that interact along the chain) suffer a spin pairing (i.e., each couple of equally spaced spins in the chain suffers an antiferromagnetic ordering to generate paired spins due to a dimerization of the chain). Here the driving force is the magnetic energy resulting from the antiferromagnetic coupling of the spins in the dimers.
In one dimensional metals the Fermi surface (that in a partially filled band represents the limiting area between the occupied and empty wave vector regions) appears as an open surface with a rectangular shape. As the dimensionality of the metal increases this rectangle bends more and more until it closes to form a cylinder for two-dimensional metals and a sphere for three-dimensional metals. When one side of the Fermi surface can be translated in such a way that it overlaps with the other side, then the Fermi surface is said to be nested. The vector describing this translation is called the nesting vector. In this situation the metallic state is not the stable one30 because the one dimensional metal is expected to suffer a metal insulator transition in the form of a charge density wave (CDW) or a spin density wave (SDW). These two insulating states are obtained due to the mixing of the empty and occupied orbitals near the Fermi surface. This mixing of orbitals is important since the energy difference between them is small and gives rise to modified empty and occupied orbitals. When the temperature is lowered, there are two possibilities: (1) if the originally occupied orbitals are now doubly occupied then the metal will present a CDW. This state is driven by the lattice vibrations (phonons). (2) If the on-site electronic repulsion is strong, then the originally occupied and unoccupied orbitals will bear one electron each (with anti-parallel spins) in order to minimize the on-site electron–electron repulsion. This situation gives rise to a spin density wave (SDW). Both states (CDW and SDW) are insulating because they open a gap at the Fermi level.
Other possible electronic transitions observed in conducting molecular materials and CPs are the Mott insulators (MI), the charge disproportionation (CD) or charge ordering (CO), the Wigner solids (a special case of CO) and the spin-gapped (SG) state (Scheme 4).31
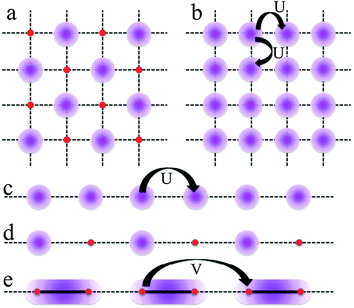 | ||
| Scheme 4 Electronic states observed for filled and half-filled band in one and two dimensional CPs. Electron density is represented as a violet shadow. | ||
CD or CO appears when the charge distribution along the conducting chain or layer is not uniformly distributed (due to the polarising effect of counterions or to structural dissimilarities, Scheme 4a and d).32–36 The Mott insulators are non-degenerate half-filled band systems where the on-site Coulomb repulsion (U) is stronger than the kinetic energy of the electrons (corresponding to the bandwidth, W). This situation corresponds to those systems where the number of electrons is equal to the number of sites (no mixed valence exists) and each electron is located in one site to avoid the on-site repulsions, U (Scheme 4b and 4c for two and one dimensional systems, respectively). In the Winger solids besides the on-site Coulomb repulsion (U), the first neighbour Coulomb repulsion (V) also plays an important role when the kinetic energy of the electrons is low (W ≪ U). If these systems present a quarter filled band then the electrons will be localized on alternating sites to minimize both, U and V (Scheme 4a and d). This special case of CO (the charge orders in alternating sites) is called the Winger solid. If these systems present a dimerization then the electron will occupy the bonding molecular orbital formed inside the dimer. This is the so-called dimer-Mott state, where a localized and uniform charge distribution is present along the chain (Scheme 4e). The spin-gapped state is a special case of a Mott localized insulator where the spins not only are localized on adjacent sites but are antiferromagnetically coupled to generate a non-magnetic ground state at low temperatures.
A special case of conductivity in CP is the neutral linear-chain mixed-valence Rh(I/II) semiquinonato/catecholato complex [Rh(3,6-dbdiox-4,5-Cl2)(CO)2]n, where an intramolecular electron transfer between the semiquinonate and catecholate forms of the ligand accounts for the electron delocalization in the CP.37
Finally, it is worth to mention that although very scarce, there are CPs that present proton conductivity. This conductivity has been attributed to the presence of water clusters,38 –OH groups in the coordinated ligands39 and even to one dimensional nanoarrays of water molecules,40 although there are no definitive proofs of these mechanisms yet.
3. One-dimensional coordination polymers
3.1. Halides bridging metal entities
One extensively studied class of quasi-one-dimensional polymers encompass species with the abridged formulation MX and MMX where M is a metal cation, commonly, Ni, Pd or Pt and X is a halogen, like Cl, Br or I, acting as a bridge between sequentially spaced metallic centres. These two akin families are of great interest, not only for their potential electrical transport behaviour, but also for their wide scope of physical properties. Prime examples are luminescence spectra with large Stokes shifts,41 large third-order nonlinear optical properties, gigantic third-order optical nonlinear susceptibility42 or progressive resonance Raman scattering. Along with those, these materials also exhibit the characteristic features of quasi-one-dimensional polymers such as spin density wave (SDW), charge density wave (CDW), solitons, polarons, and bipolarons.According to their valence ordering, two different series of MX complexes are described as depicted in Fig. 1.
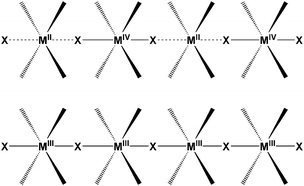 | ||
| Fig. 1 Schematic representation of mixed-valence MX, charge density wave (CDW) for Pd and Pt complexes (up); and spin density wave (SDW) for Ni (down). | ||
3.1.1.1. Pd and Pt MX chains. The Pd and Pt MX chain compounds present a CDW ground state and a lineal structure formed by alternating square planar MIIL2 with octahedral MIVL2X2 subunits, rendering infinite MIV–X⋯MII chains. The halogen is displaced from the midpoint between metal centres as a consequence of large electron–phonon coupling. These interactions may be tuned by a judicious selection of the in-plane ligands, bridging halides, counterions and metal centres. The interchain interactions can also be increased by introducing H-bond nets that give rise to expanded dimensionalities.
Generally, their synthetic procedure encompasses an equimolecular mixture of both single-valence components, i.e. MII and MIV complexes or controlled oxidation of solutions containing the MII component.
In a series of papers during the 70's, Interrante and collaborators studied, among other physical properties, the electrical conductivity and its pressure dependence of selected mixed-valence MX chains,43–45 some of them, like Magnus' green salts derivatives, which may be regarded as MX systems, are out of the scope of this review, but they also conducted the first electrical characterisation of [Pt(en)2][Pt(en)2X2]X4 (en = ethylenediamine; X = Cl, Br, I) derivatives.43 The synthetic methods for these compounds had been previously reported.46–48 At ambient temperature and pressure, the conductivity of these sort of complexes along the chain direction is rather low, <10−8 S cm−1, both in single-crystal and polycrystalline samples. Significantly, their conductivity rapidly increases under pressure, with a fully reversible and reproducible behaviour. A polycrystalline sample of [Pt(en)2][Pt(en)2I2]I4 had an electrical conductivity of almost 1 S cm−1 at 160 kbar whilst that found for [Pt(en)2][Pt(en)2Br2]Br4 analogue was 10−3 S cm−1, the least conductor species of the series was [Pt(en)2][Pt(en)2Cl2]Cl4 displaying a 10−7 S cm−1 value. The trend I > Br > Cl could be explained in terms of better metal–halogen orbital overlaps. The authors assumed that pressure played a role in reducing the geometrical inequivalency of the metal environments and increasing the intrachain metal–halogen orbital overlaps. They also studied the electrical conductivity of Wolffram's red and Reilhlen's green salts ([Pt(C2H5NH2)4][Pt(C2H5NH2)4Cl2]Cl4 and [Pt(C2H5NH2)4][Pt(C2H5NH2)4Br2]Br4, respectively), but their conductivities were below the limit of detection even above 140 kbar.
On a latter report, Interrante and Browall continued studying [Pt(en)2][Pt(en)2X2]X4 systems.43 Four-probe conductivity measurements were conducted on both single crystals and pressed pellets. Electrical contacts were made with 2 mm ∅ platinum wires for the pellet samples and 0.5 mm ∅ gold coated tungsten wires for single crystals and the wires were attached with conducting silver paint. Results of dc conductivity measurements at room temperature were reported as follows: <10−11 and 10−10 S cm−1 for single crystals of [Pt(en)2][Pt(en)2Cl2]Cl4 and [Pt(en)2][Pt(en)2Br2]Br4, respectively, suitable crystals of the [Pt(en)2][Pt(en)2I2]I4 species could not be grown. Values found for pressed pellet samples were ∼10−15, 5 × 10−13 and 1 × 10−9 S cm−1 for [Pt(en)2][Pt(en)2Cl2]Cl4, [Pt(en)2][Pt(en)2Br2]Br4 and [Pt(en)2][Pt(en)2I2]I4, respectively.
In 1981, the research group of Kida reported the electrical conductivity of several [ML2][ML2X2]A4 MX chains;49 the measurements were carried on single crystals using a two-probe method with silver wires contacted with carbon paste along the needle direction. The complexes [Pt(en)2][Pt(en)2I2](BF4)4 and [Pt(en)2][Pt(en)2X2](ClO4)4 (X = Cl, Br, I) exhibited electrical conductivity values of 1.6 × 10−9, 2.0 × 10−15, 3.0 × 10−11 and 1.8 × 10−8 S cm−1, respectively (Table 1 and Fig. 2). They also found that larger conductivity values could be observed on single crystal samples.50
| Compound | Conductivity/S cm−1 | Ref. | |
|---|---|---|---|
| Pressed pellet | Single crystal | ||
| tn = triethylenediamine, en = ethylenediamine, chxn = 1R,2R-diaminocyclohexane. a RT and 1 bar.b RT and 160 kbar.c Four probe method. | |||
| [Pt(en)2][Pt(en)2Cl2]Cl4 | 10−15a,c |
<10−11a,c |
43 |
| 10−7b |
|||
| [Pt(en)2][Pt(en)2Br2]Br4 | 5 × 10−13a,c |
<10−10a,c |
43 |
| 10−3b |
|||
| [Pt(en)2][Pt(en)2I2]I4 | 10−9a,c |
<10−8a |
43 |
| 1b | |||
| [Pt(en)2][Pt(en)2I2](BF4) | 1.6 × 10−9a |
49 | |
| [Pt(en)2][Pt(en)2Cl2](ClO4)4 | 2.0 × 10−15a |
49 | |
| [Pt(en)2][Pt(en)2Br2](ClO4)4 | 3.0 × 10−11a |
49 | |
| [Pt(en)2][Pt(en)2I2](ClO4)4 | 1.8 × 10−8a |
49 | |
| [Pd(en)2][Pd(en)2Cl2](ClO4)4 | 10−12a |
2 × 10−12a |
51,53 |
| [Pd(en)2][Pd(en)2Br2](ClO4)4 | 1.2 × 10−8a |
53 | |
| [Pd(en)2][Pd(en)2I2](ClO4)4 | 2 × 10−12a |
5 × 10−7a |
54 |
| 1.1 × 10−8a |
51,52 | ||
| [Pt(tn)2][Pt(tn)2Br2](ClO4)4 | 2.0 × 10−14a |
49 | |
| [Pt(tn)2][Pt(tn)2I2](ClO4)4 | 7 × 10−7a |
49 | |
| [Pt(chxn)2I]I2 | 10−4a |
53 | |
![Room temperature solid-state structures of: (a) [Pt(en)2][Pt(en)2Cl2](ClO4)4 (displaying disordered bridging chloride positions), (b) [Pt(en)2][Pt(en)2Br2](ClO4)4 and (c) [Pt(en)2][Pt(en)2I2](ClO4)4. Counter-ions and hydrogen atoms are omitted for clarity.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f2.gif) | ||
| Fig. 2 Room temperature solid-state structures of: (a) [Pt(en)2][Pt(en)2Cl2](ClO4)4 (displaying disordered bridging chloride positions), (b) [Pt(en)2][Pt(en)2Br2](ClO4)4 and (c) [Pt(en)2][Pt(en)2I2](ClO4)4. Counter-ions and hydrogen atoms are omitted for clarity. | ||
As a logical extension of these pioneering works, the Pd analogues were also prepared and studied in the 80's. Thus, in 1982, Papavassiliou et al. recorded the electrical conductivity of pressed pellets of [Pd(en)2][Pd(en)2X2](ClO4)4 (X = Cl and I) and found values of 10−12 and 2 × 10−12 S cm−1 for the chloride and iodide derivatives, respectively. Afterwards, Kurita et al., in 1987, studied the photoconductivity of the iodine derivative and reported an improved electrical conductivity value of 1.1 × 10−8 S cm−1.51 Some of the Pd analogues were also electrically characterised by Kida. Thus, [Pd(en)2][Pd(en)2X2](ClO4)4, (X = Cl and Br) showed conductivities of 2.0 × 10−12 and 1.2 × 10−8 S cm−1, respectively, higher than those of the corresponding Pt analogues (Table 1). For instance, Yamashita reported for a single crystal of [Pd(en)2][Pd(en)2I2](ClO4)4 a conductivity of 5 × 10−7 S cm−1,52ca. 300 times higher than the corresponding Pt derivative (1.6 × 10−9 S cm−1).
Kida et al. also reported that [Pt(tn)2][Pt(tn)2X2](ClO4)4 (tn = triethylenediamine, X = Br and I)49 show similar conductivity values (2.0 × 10−14 and 7 × 10−7 S cm−1, respectively) to those reported for the en analogues (3.0 × 10−11 and 1.8 × 10−8 S cm−1, respectively, Table 1).49
The slow diffusion of I2 vapour into a methanolic solution of Pt(chnx)2I2 afforded single-crystals of [Pt(chnx)2I]I2 (chnx = 1R,2R-diaminocyclohexane) as reported by Yamashita and Takaishi in 2006.53Fig. 3 depicts a view of the solid-state structure evidencing the planar environment of the [Pt(chnx)2] moieties which are bridged by half-occupied midpoint-displaced bridging iodine, forming linear chains. The authors studied an anomalous valence state behaviour caused by the fast fluctuation between PtII⋯I_PtIV and PtIV_I⋯PtII in the double well potential, by using EPR, X-ray diffuse scattering and STM images and recorded the electrical conductivity of the complex using the four-probe method with carbon paste. The observed value was of the order of 10−4 S cm−1 at room temperature. They also claimed the smallest charge-transfer energy observed for this complex compared with all the other halogen-bridged MX Pt compounds.
![Crystal structures of [Pd(en)2][Pd(en)2Br2](ClO4)4 (a), [Pt(tn)2][Pt(tn)2Cl2] (b) and [Pt(chnx)2I]I2 (c). All hydrogen atoms along with counterions have been removed for clarity.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f3.gif) | ||
| Fig. 3 Crystal structures of [Pd(en)2][Pd(en)2Br2](ClO4)4 (a), [Pt(tn)2][Pt(tn)2Cl2] (b) and [Pt(chnx)2I]I2 (c). All hydrogen atoms along with counterions have been removed for clarity. | ||
3.1.1.2. Ni MX chains. Conversely to Pt and Pd species, due to the strong electron correlation ascribed to Ni metal centres, their charge distribution along a MX compound is best described as infinite NiIII–X–NiIII chains. The strong ligand field forces the NiIII ion to be in a low spin state (NiIIId7; S = 1/2) with an unpaired electron occupying the dz2 orbital. In this situation, the halide sits on the midpoint of neighbouring Ni pairs conforming a Mott–Hubbard semiconductor with a SDW ground state. Standard synthetic preparations of Ni-based MX chains include previous electrolysis of the divalent discrete compounds in the presence of a pertinent electrolyte. In this way, many [NiL2][NiL2X2]A4 chains have been isolated but only a few selected ones have been the subject of electrical conductivity studies.
In 1981, Kida et al. also characterised single crystals of the compound [Ni(en)2][Ni(en)2Cl2](ClO4)4.54 The contacts were done with silver wires and carbon paste in a two-probe experiment, the observed electrical conductivity was 1.3 × 10−8 S cm−1.
A new series of 1D Ni MX chains were reported in 1999 by the group of Yamashita,55 both chemical and electrochemical methods were used for the isolation of [Ni(chxn)2][Ni(chxn)2X2]Y4 species (X = Cl, Br and (Cl/Br), Y = Cl−, Br−, (Cl/Br), NO3−, BF4− and ClO4−), as expected, X-ray crystallography confirmed a planar N4 donor set for the two in-plane diamine ligands with the Ni(chxn)2 fragments bridged and stacked along the b axis in all cases. The slow diffusion of Br2 and Cl2 into 2-methoxypropanol solutions of [Ni(chxn)2]Cl2 and [Ni(chxn)2]Br2 led to the isolation of Ni(chxn)2Cl2.459Br0.541 and Ni(chxn)2Cl1.28Br1.72, respectively, X-ray diffraction proved to be a powerful tool in order to determine the Cl and Br ratio in both mixed-halide compounds. Although their structures were described previously,56 no electrical characterisation experiments were undertaken at that moment. NO3−, BF4− and ClO4− derivatives were obtained by both chemical and electrochemical methods.
The electrical conductivities of some selected compounds were measured on single-crystals using gold wires and carbon paste with the four-probe method.57 The room temperature conductivities decreased in the order of Ni(chxn)2Br3 > Ni(chxn)2Cl1.28Br1.72 > Ni(chxn)2Br(NO3)2 > Ni(chxn)2Cl3 and presented the following values: 6.92 × 10−4, 1.86 × 10−5, 2.45 × 10−6 and 2.45 × 10−7 S cm−1, respectively (Table 2). Crystals of Ni(chxn)2Cl2.459Br0.541 were too small to be measured. Fig. 4 shows the electrical conductivity dependence with temperature for both Ni(chxn)2Cl(NO3)2 and Ni(chxn)2Br(NO3)2.
| Compounds | Conductivity/S cm−1 | Ref. | |
|---|---|---|---|
| Two probe | Four probe | ||
| chxn = 1R,2R-diaminocyclohexane, S,S-bn = 2S,3S-diaminobutane, en = ethylenediamine. | |||
| [Ni(en)2][Ni(en)2Cl2](ClO4)4 | 1.3 × 10−8 | 54 | |
| Ni(chxn)2Br3 | 6.92 × 10−4 | 55 | |
| Ni(chxn)2Cl1.28Br1.72 | 1.86 × 10−5 | 55 | |
| Ni(chxn)2Br(NO3)2 | 2.45 × 10−6 | 55 | |
| Ni(chxn)2Cl3 | 2.45 × 10−7 | 57 | |
| [Ni(S,S-bn)2Br]Br2 | 10−1 | 58 | |
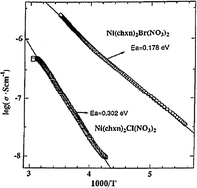 | ||
| Fig. 4 Single-crystal electrical conductivities of Ni(chxn)2Cl(NO3)2 and Ni(chxn)2Br(NO3)2. (Data extracted from ref. 57. Reproduced with permission of the American Chemical Society.) | ||
Takaishi et al. published in 2008 that the electrolysis of [Ni(S,S-bn)2]Br2 (S,S-bn = 2S,3S-diaminobutane) in anhydrous 1-propanol under a constant 10 μA current yielded novel [Ni(S,S-bn)2Br]Br2 MX species using tetramethylammonium bromide as the electrolyte.59 Suitable crystals for X-ray diffraction studies were grown over a two-week period. The evaluation of its solid-state structure showed a stronger Ni–Br bond than that of the chxn derivative. Thus, in [Ni(S,S-bn)2Br]Br2 the Ni⋯Ni contact along the 1D chain is 5.129(1) Å at 120 K, whereas in [Ni(chxn)2Br]Br2 this distance is 5.157(1) Å at the same temperature. The electrical conductivity of the [Ni(S,S-bn)2Br]Br2 complex, measured with the four contact method, showed a quite high room temperature conductivity value of 10−1 S cm−1, decreasing to almost 2.0 × 10−4 S cm−1 at ca. 133 K (Table 2). Since the X-ray diffraction showed the presence of some NiII impurities (due to the synthetic procedure), the conductivity may be attributed to these Ni(II) impurities.58
![Electrical conductivity profiles of [Pd(en)2][Pt(en)2Cl2](ClO4)4 (1), [Ni(en)2][Pt(en)2Cl2](ClO4)4 (2), [Pd(en)2][Pt(en)2Br2](ClO4)4 (3), [Ni(en)2][Pt(en)2Br2](ClO4)4 (4) and [Pd(en)2][Pt(en)2I2](ClO4)4. Measurements performed on single crystals along the needle axis by the two probe method using silver paste. (Data extracted from ref. 53, Reproduced with permission of The Chemical Society of Japan.)](/image/article/2012/CS/c1cs15092h/c1cs15092h-f5.gif) | ||
| Fig. 5 Electrical conductivity profiles of [Pd(en)2][Pt(en)2Cl2](ClO4)4 (1), [Ni(en)2][Pt(en)2Cl2](ClO4)4 (2), [Pd(en)2][Pt(en)2Br2](ClO4)4 (3), [Ni(en)2][Pt(en)2Br2](ClO4)4 (4) and [Pd(en)2][Pt(en)2I2](ClO4)4. Measurements performed on single crystals along the needle axis by the two probe method using silver paste. (Data extracted from ref. 53, Reproduced with permission of The Chemical Society of Japan.) | ||
Recent attention has been focused on a novel non-stoichiometrical hetero-metal [Ni1−xPdx(chxn)2X]X2 (X = Cl, Br) backbone. Yamashita et al. reported in 1999, that combined methanolic solutions containing different ratios of Ni(chxn)2Br2 and Pd(chxn)2Br2 precursors were electrochemically oxidised with tetramethylammonium halides as electrolytes, after several weeks, good quality single crystals of [Ni1−xPdx(chxn)2Br]Br2 were formed on H-shaped cells.57 The Ni–Pd ratio was determined by inductively coupled plasma emission spectrometry, X-ray powder patterns of the [Ni1−xPdx(chxn)2Br]Br2 complex revealed an isomorphous phase with the same I222 space group of the parent starting materials Ni(chxn)2Br2 and Pd(chxn)2Br2, hence a halogen-bridged linear structure was assumed for the mixed-metal Ni–Pd species. Interestingly, the PdII–X–PdIV mixed-valence state gradually changed to the unusual PdIII–X–PdIII state on increasing the NiIII content. This observation indicates that the electron–phonon interaction in PdII–X–PdIV systems is destabilized by strong electron correlation of the NiIII ion that promotes the change from a CDW state to a SDW state with a crossover between both at x ≈ 0.7. IR, Raman, XPS and Auger spectrometry corroborated these results. Three years later, in 2002, the same authors informed on the electrical resistance of [Ni1−xPdx(chxn)2Br]Br2 and its dependence with increasing pressure, however, no explicit conductivity results were communicated.64
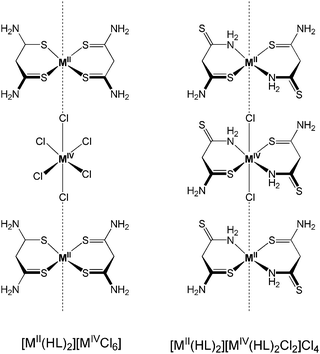
The lack of X-ray diffraction data precluded a concise determination of the solid state structure of all these complexes; however, elemental analyses were consistent with the proposed molecular formulae, while detailed coordination environments were unambiguously resolved by 195Pt NMR spectroscopy for the Pt analogues and inferred for the Pd ones.
Powder conductivity measurements were determined on pressed pellets evidencing the semiconductor nature of the samples (Table 4).
| Compound | M | Conductivity/S cm−1 | Ref. |
|---|---|---|---|
| HL = dithiomalonamide. | |||
| [MII(HL)2][MIVCl6] | Pt | 3.25 × 10−8 | 67 |
| Pd | 7.63 × 10−8 | ||
| [MII(HL)2][MIVBr6] | Pt | 3.25 × 10−8 | 66 |
| Pd | 7.63 × 10−8 | ||
| [MII(HL)2][MIVBr6] | Pt | 8.67 × 10−8 | 65 |
| Pd | 2.99 × 10−7 | ||
| [MII(HL)2][MIVI6] | Pt | 2.23 × 10−7 | 65 |
| Pd | 6.51 × 10−7 | ||
| [MII(HL)2][MIV(HL)2Cl2]Cl4 | Pt | 1.43 × 10−6 | 66 |
| Pd | 3.52 × 10−6 | ||
| [MII(HL)2][MIV(HL)2Br2]Br4 | Pt | 0.86 × 10−5 | 67 |
| Pd | 1.72 × 10−5 | ||
| [MII(HL)2][MIV(HL)2I2]I4 | Pt | 7.96 × 10−5 | 65 |
| Pd | 9.77 × 10−5 | ||
The electrical properties of the complexes depend on the metal centre, on the coordination donor set and on the linking halogen (X). As observed for parent MX chains, the conductivity increases in the sequence Cl < Br < I. Pd derivatives have higher values than the Pt ones, and a significant increase, circa two orders of magnitude, is observed on moving from the [MII(HL)2][MIVX6] series to the [MII(HL)2][MIV(HL)2X2]X4 one. The authors postulated this trend as a consequence of a better overlap of dz2 orbitals which may lead to a M(III)-like behaviour.
The coordination chemistry of the dibenzyl sulfide ligand towards gold was firstly explored by Herrmann by the turn of the 19th century.68 At that time he isolated an orange precipitate with a [Au(DBS)Cl2] (DBS = dibenzyl sulfide) formulation. Following this study, Smith reported on the bromide derivative obtained during the study of the addition compounds of gold halides with benzyl sulfide.69 It was in 1952 when the first structural assumption for these complexes was made by Brain and collaborators.70 Their preliminary X-ray study suggested the presence of a 1D Au(III)–Cl⋯Au(I) mixed-valence linear chain in the solid state structure, analogous to that observed in MX chains. However, the disordered crystal structure of [Au(DBS)Cl2] species was not resolved until 1988 by Takahashi and Tanino,71 who evidenced the infinite Au(III)–Cl⋯Au(I) chain with a positional disorder for the bridging chlorine atom, a common feature observed in MX chains (Fig. 7). Another remarkable characteristic of the structure is the pairing of two 1D chains, the shortest interchain contact being 3.32 Å, shorter than the average intrachain Au–Cl (bridging) distance of 3.41 Å. The structure of the isomorphous [Au(DBS)Br2] derivative was also reported by the same group three years later (Fig. 7).72 The electrical conductivities of both chain compounds were measured by Interrante and Bundy in 1977,45 as for most MX chains, these complexes are described as poor conductors at room temperature with σ < 10−10 S cm−1 but their conductivities increase when applying pressure, reaching a broad maximum at ca. 250 kbar with recorded conductivities of 10−6 and 10−3 S cm−1 for the chlorine and bromine derivatives, respectively. The measurements were performed using the four-probe method with 25 μm ∅ gold wires and silver paint. Measurements under pressure were carried out on a Drickamer apparatus with diamond compact-tipped pistons.45
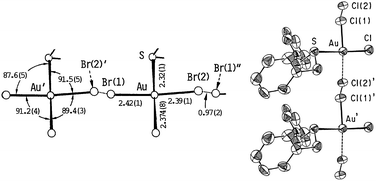 | ||
| Fig. 7 Structures of the compound Au(DBS)X2 (X = Br and Cl) showing the two possible disordered positions for the halogen atoms: X(1) and X(2). (Data collected from ref. 71 and 72. Reproduced with permission of The Chemical Society of Japan.) | ||
An important consequence of passing from MX to MMX chains is the gain of internal degrees of freedom from the charge–spin–lattice point of view. Thus, now there are new valence ordered states different from those inherent to MX compounds. These states can be summarized as average valence (AV), charge polarisation (CP), charge density wave (CDW) and alternate-charge polarisation (ACP) states (Scheme 5).
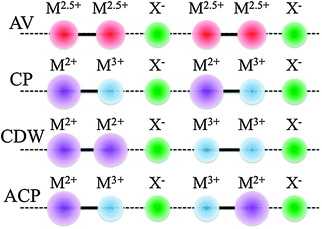 | ||
| Scheme 5 Possible electronic states in MMX chains. | ||
As previously stated, some of the MX compounds are best described as CDW systems, in particular those containing Pd or Pt, whereas those with Ni are characterised as being Mott–Hubbard semiconductors. The pop MMX's are reported to have a CDW ground state (closely related to MX) while SCS have an ACP metal-sublattice dimerisation at low temperature. Nonetheless, fine tuning of ground state transitions can be achieved applying pressure or by replacing halogen bridges, counterions and ligands. In most cases, the nature of the metallic phases of these chains is attributable to AV or CP electronic structure stabilisation.
Two main routes can be followed in order to isolate the desired mixed-valence species, the first method consist in co-crystallising equimolar solutions of the MM and MMX2 precursors. A second procedure implies partial oxidation through halogen addition. In certain cases, as for pop systems, a precipitating agent is also combined in the reaction mixtures. Both dta (CH3CS2−) and pop derivatives give rise to 1D chains; the dta derivatives are neutral with ligand helical arrangements wrapping the bimetallic core, while the diphosphite derivatives are negatively charged linear chains with almost no twist and the need of a counter-ion.
3.1.4.1. Dithioacids with platinum. The first report of the electrical properties of Pt2(CH3CS2)4I, published by C. Bellitto et al. in 1983, started the quest for metallic conduction involving dta MMX. At room temperature, a compressed pellet of Pt2(CH3CS2)4I showed a maximum electrical conductivity of 7 × 10−3 S cm−1.73 Yamashita et al. reported in 1989 that the electrical conductivity performed on single crystals with the two contact method was as high as 2 S cm−1 at atmospheric pressure and 10 S cm−1 at 7 GPa. This increase with applied pressure is probably due to an increase in the energy transfer or a decrease in the band gap and Peirls distortion as pressure rises up.74 They also claimed that thin films of the material could be prepared in vacuum with a conductivity of 0.2 S cm−1 at room temperature.74 Finally, in 1997, Kitagawa and co-workers provided a new value of 13 S cm−1,75 reinforced by another report of the same group in 1999.76 This discrepancy in the reported values reflects the already mentioned fact that the exact value of the conductivity may be affected by the quality and purity of the single crystals (and by the accuracy in the determination of the dimensions of the crystals). The dc electrical conductivity measurements were carried out along the crystallographic b axis (i.e. through the Pt–Pt chain using a standard four-probe technique with gold paint and 20 μm ∅ gold wires, between 50 and 380 K. Metallic behaviour was recorded in the 300–340 K range.
Increasing the number of carbon atoms in the lateral chain of the dithiocarboxylic acid may tune the observed Pt–Pt and Pt–I distances. It can be anticipated that bulkier aliphatic chains will impose a demanding steric repulsive interaction resulting in elongated Pt–Pt contacts. In 2001, Mitsumi et al. reported the synthesis and characterisation of novel Pt2(EtCS2)4I species.77 Single crystal X-ray analyses confirmed the 1D nature of the structure. The Pt–Pt (2.684(1) Å) and Pt–I (average 2.980 Å) distances measured at 293 K were slightly longer than those observed for the parent Pt2(CH3CS2)4I compound (Pt–Pt 2.677(2) Å, Pt–I (average) 2.978 Å). Electrical transport measurements concluded that the polymer was a metallic conductor showing an electrical conductivity between 5 and 30 S cm−1 at room temperature (again the values differ for different crystals). The results were tested in several single crystals using the four-probe technique with gold paint contacts and 25 μm ∅ gold wires. The temperature was varied from 80 to 400 K and a metallic-semiconductor transition was observed at 205 K.77
Several MMX chains were reported later in the following year by the same authors.78,79 In these compounds the dimetallic Pt–Pt units were built with n-PrCS2H and n-BuCS2H dithioacid ligands to form the corresponding Pt2(n-PrCS2)4I and Pt2(n-BuCS2)4I chain polymers. These compounds were examined with classical inorganic techniques and further characterised through four-probe electrical transport measurement on single crystals. The n-BuCS2 derivative displayed at room temperature an electrical conductivity in the range 17–83 S cm−1 and a phase transition at 325 K. The same year Nakazawa and co-workers published a heat capacity study of the complex and reported an approximate electrical conductivity of 30 S cm−1 (in the range already observed by Mitsumi and Toriumi) with weak temperature dependence.80 In 2002, Mitsumi et al. reported that Pt2(n-PrCS2)4I showed an electrical conductivity value of ca. 0.23 S cm−1 at room temperature, a value similar to those of other dta family members.79
On a later report concerning the heat capacity of Pt2(n-PenCS2)4I, published in 2005, Saito's group reported a room temperature conductivity value of 0.84 S cm−1 with a metal–insulator transition at 235 K.81
The obvious difference between these MMX derivatives is the alkyl chain length, which governs the interchain distances, responsible for the observed physical properties. These properties are, at the same time, intimately related to the valence-ordered state. The RT metallic phases of both Pt2(MeCS2)4I and Pt2(EtCS2)4I are associated with AV states, the crystal structure of Pt2(n-PrCS2)4I at RT showed three well differentiated Pt–I distances as well as alternatively disordered dta ligands. When temperature increases, a phase transition occurs at 359 K, giving rise to a pure AV state for the chain. Similar features are observed for Pt2(n-BuCS2)4I.
DC electrical conductivity measurements carried out on crystals of [Pt2(n-pentylCS2)4I] showed room temperature conductivities in the range 0.3–1.4 S cm−1.82 Two different experiments were used for the conductivity measurements.
In the first experiment, the crystals were cooled from 300 to 100 K and then warmed to 300 K. The cooling run shows a metallic behaviour with a rounded minimum at around 255–270 K, followed by an abrupt increase at ca. 200 K (Fig. 8b). The thermally activated regime follows the 1D variable range hopping model (σ = σ0*exp[−(T0/T)1/2]), 1D-VRH, used to explain conductivity in many conducting polymers. This fact suggests that the conductivity in [Pt2(n-pentylCS2)4I] is mainly one-dimensional, as expected from its linear structure. However, this model does not reproduce the conductivity in the intermediate temperature range 225 < T < 255 K, which suggests the presence of an intermediate regime between the metallic and semiconducting regimes. In the intermediate regime, the coexistence of metallic and semiconducting regimes may be attributed to a progressive distortion of the Pt–I distance along the chain.
![(a) A single polymer chain of [Pt2(n-pentylCS2)4I]. (b) Thermal variation of the electrical resistivity of [Pt2(n-pentylCS2)4I]. The dashed line shows the behaviour of a non-heated sample. The inset shows the derivative of the resistivity vs. temperature around the RT-HT transition. (From ref. 82. Reproduced with permission of Wiley-VCH.)](/image/article/2012/CS/c1cs15092h/c1cs15092h-f8.gif) | ||
| Fig. 8 (a) A single polymer chain of [Pt2(n-pentylCS2)4I]. (b) Thermal variation of the electrical resistivity of [Pt2(n-pentylCS2)4I]. The dashed line shows the behaviour of a non-heated sample. The inset shows the derivative of the resistivity vs. temperature around the RT-HT transition. (From ref. 82. Reproduced with permission of Wiley-VCH.) | ||
In the second experiment, the crystals were heated from 300 to 400 K and then cooled to 100 K, followed by a final heating scan from 100 to 400 K (Fig. 8b). An irreversible RT-HT transition was confirmed. The heated crystals also show an abrupt metal–insulator transition at ca. 200 K and after this M–I transition, the crystals again suffered important micro-cracks and showed a resistivity 103–104 times higher than that observed before the transition. The structural changes that cause the electrical behaviour observed for [Pt2(n-pentylCS2)4I] were confirmed by X-ray diffraction analyses (Fig. 8). Studies on crystals of [Pt2(n-pentylCS2)4I] carried out at three temperatures (100 (LT), 298 (RT) and 350 (HT) K) showed the existence of three different phases for [Pt2(n-pentylCS2)4I].
The last MMX chain incorporating dta ligands came out in 2009.83 The group of Saito conducted a study concerning the heat capacity, absorption spectrum, spin susceptibility and electrical conductivity of Pt2(n-HexCS2)4I. The four-probe method revealed a value of 2 × 10−3 S cm−1 at room temperature, a value slightly lower than those typically observed for Pt MMX derivatives (Table 5).
The data collected in Table 5 summarized the excellent electrical conductive properties found in these polymers.
3.1.4.2. Dithioacids with nickel. In addition to Pt2(CH3CS2)4I, Bellitto et al. also reported the synthetic procedure, isolation and characterisation of Ni2(CH3CS2)4I back in 1985.84 Its solid-state structure consisted of linear Ni2(CH3CS2)4–I–Ni2(CH3CS2)4–I– chains stacked along the b crystallographic axis. They also measured an electrical conductivity of 5 × 10−6 S cm−1 for what at that time was the first example of a conductive nickel mixed-valence 1D-chain. The electrical measurements were made on pressed pellets of the compound using the two-probe technique. Later electrical conductivity measurements performed by Yamashita's group on single crystals with the four-probe method gave an improved conductivity of 2.5 × 10−2 S cm−1.85
While continuous reports concerning Pt2(RCS2)4I have appeared in the literature during the last few decades, the number of papers related to the nickel derivatives, Ni2(RCS2)4I, are much more limited. In fact, only very recently, in 2009, the NiMMX family has been enlarged by Mitsumi et al. who successfully isolated and electrically characterised the R = Et, n-Pr and n-Bu derivatives in the series Ni2(RCS2)4I.86 The reported room temperature electrical conductivity values are: 1.6 × 10−3, 7.6 × 10−4 and 6.0 × 10−4 S cm−1 for R = Et, n-Pr and n-Bu, respectively (Table 6). The measurements were performed with the four-probe method on various single crystals using gold paint and 20 μm ∅ gold wires. The compounds were synthesised in low yield (3–7%) by exposing solutions of the corresponding MM dimer to iodine vapours. These solutions yielded black single crystals whose structures show neutral 1D chains built up with repeating Ni–Ni–I units (Fig. 9).
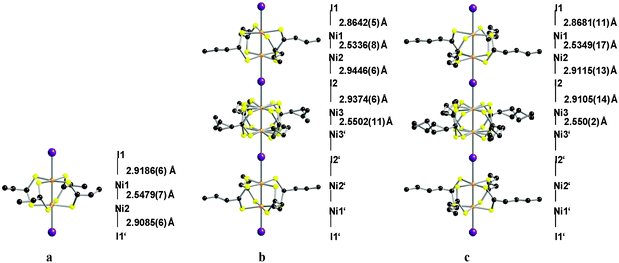 | ||
| Fig. 9 Room temperature solid-state structure and significant distances of Ni2(EtCS2)4I (a), Ni2(n-PrCS2)4I (b) and Ni2(n-BuCS2)4I (c). All hydrogen atoms not displayed for clarity. Disordered ligand chain position shown. | ||
Both Ni2(MeCS2)4I and Ni2(EtCS2)4I derivatives present a CP state at room temperature. In the n-Pr derivative, Ni2(n-PrCS2)4I, there are three significantly different Ni–I distances, the valence order can be regarded as –I⋯Ni3+–Ni2+–I⋯Ni2.5+–Ni2.5+–I⋯Ni2+–Ni3+–I⋯, rendering an approximate AV ground state at RT, attributed to the entropy reservoir of the ligand alkyl chains. A similar behaviour was observed in Ni2(n-BuCS2)4I chains. The four Ni2(dta)4I compounds show semiconducting behaviour as well as an intense sharp absorption band at 5400 cm−1, which is assigned to a Mott-Hubbard gap due to a relatively large on-site Coulomb repulsion energy (U) of the nickel atoms.
Tables 5 and 6 collect the conductivity data of the MMX chains M2(dta)4I (M = Ni and Pt) and show important differences of ca. 3 orders of magnitude in the electrical conductivity values between analogous compounds with different metals. DFT calculations87 suggest that these differences could be associated to the greater localization of the 3d states of Ni compared to the 5d states of Pt and to the electron–lattice dominant interactions in Pt2(dta)4I compared to Ni2(dta)4I, where the solid state properties are governed by the strong electron correlation of the Ni atoms.86
3.1.4.3. Platinum with pop. The A4[Pt2(pop)4X]·nH2O (A = alkali metal, ammonium, alkyl ammonium and alkyl diammonium; pop = P2O5H22−; n = 0, 2 and 3; and X = Cl, Br, I) family contains more than 50 structurally characterised linear chains, all of them described as being insulators and presenting AV, CP or CDW valence states, in contrast with the dta family which, in some cases, can stabilise an ACP phase related to their metallic electrical transport properties.
In 1983, the group of Gray prepared the first example of partially oxidised K4[Pt2(pop)4X] linear chains88 and reported the synthesis of K4[Pt2(pop)4Cl], K4[Pt2(pop)4Br]·3H2O and K4[Pt2(pop)4I], the X-ray single-crystal structure of the bromine derivative, K4[Pt2(pop)4Br]·3H2O and the electrical properties of the bromine and iodine derivatives, K4[Pt2(pop)4Br]·3H2O and K4[Pt2(pop)4I] (Fig. 10). Conductivity measurements carried out on several single crystals of K4[Pt2(pop)4Br]·3H2O with the standard two-probe method revealed at room temperature conductivity values in the range 5 × 10−4–1 × 10−3 S cm−1. The small crystal size of the iodine derivative prevented a detailed examination of the solid-state structure and subsequent measurement of its electrical behaviour; however a compressed pellet showed a value of 2.8 × 10−4 S cm−1 at 298 K. In 1986, Clark and co-workers reported the crystal structure of the chlorine derivative, K4[Pt2(pop)4Cl], and showed that it is constituted by Pt2+–Pt3+–Cl polarised repeating units.
![A fragment of the molecular structure of K4[Pt2(pop)4I] species, counterions and cocrystallised water molecules are not displayed for the sake of clarity.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f10.gif) | ||
| Fig. 10 A fragment of the molecular structure of K4[Pt2(pop)4I] species, counterions and cocrystallised water molecules are not displayed for the sake of clarity. | ||
The data collected in Table 7 show the significant (3–5 orders of magnitude) decrease in conductivity between K4[Pt2(pop)4X] and their Pt2(dta)4I analogues (Table 5).
3.1.4.4. Other MMX systems. Whilst not formally considered as MMX in the literature, there exist a large number of compounds having this formulation. Kawamura et al. reported the first examples of halide-bridged paddle-wheel chains enclosing a Rh25+ core with wrapping acetamide (CH3CONH2 = acam) ligands in 2000.89
Crystals of [Rh2(acam)4Cl]n·7nH2O and [Rh2(acam)4Br]n were isolated by reacting the Rh25+ precursor with sodium halides. Their structures comprise zig-zag chains of repeating (–Rh–Rh–X–)n units assisted via hydrogen-bond formation between amidato-NH donors and amidato-O acceptors of adjacent units for the bromide chain and between amidato-O and interstitial water molecules for the chloride derivative as depicted in Fig. 11. Pressed pellets of both compounds showed a conductivity value below 2 × 10−7 S cm−1 at room temperature. One year after, on a subsequent report, the iodide derivative (Fig. 12) was isolated and crystallographically and electrically characterised.90 This derivative was found to be almost isostructural with the dehydrated chlorine chain, crystallising in the same C2/c space group with similar cell parameters. The observed conductivity of a pressed pellet was 10−7 S cm−1 (Table 8).
![Crystal structure of [Rh2(acam)4Cl]n·7nH2O (up) and [Rh2(acam)4Br] (down) with partial numbering scheme. Hydrogen atoms not shown and only selected water solvent molecules are displayed. Short range contacts depicted as red dotted lines.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f11.gif) | ||
| Fig. 11 Crystal structure of [Rh2(acam)4Cl]n·7nH2O (up) and [Rh2(acam)4Br] (down) with partial numbering scheme. Hydrogen atoms not shown and only selected water solvent molecules are displayed. Short range contacts depicted as red dotted lines. | ||
![Solid-state structure of [Rh2(acam)4I]n with atom label scheme. Short interdimer amide contacts shown as red dotted lines.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f12.gif) | ||
| Fig. 12 Solid-state structure of [Rh2(acam)4I]n with atom label scheme. Short interdimer amide contacts shown as red dotted lines. | ||
| Compounds | Conductivity/S cm−1 | Ref. |
|---|---|---|
| acam = CH3CONH2. a RT and 1 bar.b Four probe, single crystal. | ||
| [Rh2(acam)4Cl]n·7nH2O | 2 × 10−7a |
89 |
| [Rh2(acam)4Br]n | 2 × 10−7a |
90 |
| [Rh2(acam)4I]n | 2 × 10−7a |
90 |
| [Rh2(acam)4(H2O)2][Rh2(acam)4{(μ-Cl)2PtCl2}]·2H2O | 10−7a |
91 |
| [Rh2(acam)4(H2O)2][Rh2(acam)4{(μ-Cl)2PdCl2}]·2H2O | 10−7a |
91 |
| [–Pt–Rh–Pt4–Rh–Pt–Cl–]n | 3.52 × 10−6a,b |
92 |
| {[PPh4]2Te2Ru4(CO)10Cu4Br2Cl2)·THF}∞ | 5 × 10−2a |
93 |
Although the halide bridging two paddle-wheel complex has proven to be a successful strategy, it is not the only one. Thus, another approach for the formation of linear chains may include additional metal halide cores as linkers. In this regard, Ebihara et al. have recently published the compounds formed by reaction of K2MCl4 (M = Pd, Pt) with the cationic complex Rh2(acam)4+.91 Thus, the reaction between [Rh2(acam)4(H2O)2]ClO4 and K2PtCl4 yielded the chain compound formulated as [Rh2(acam)4(H2O)2][Rh2(acam)4{(μ-Cl)2PtCl2}]·2H2O, whereas when K2PdCl4 was used, two different compounds, formulated as [Rh2(acam)4(H2O)2][Rh2(acam)4{(μ-Cl)2PdCl2}]·2H2O and [Rh2(acam)4(H2O)2][Rh2(acam)4{(μ-Cl)2PdCl(H2O)}]·H2O, were obtained (Fig. 13). All of them present infinite (–Rh–Rh–Cl–M–Cl–)n repeating units, forming zig-zag chains in the two first cases and helical in the third one (Fig. 13).
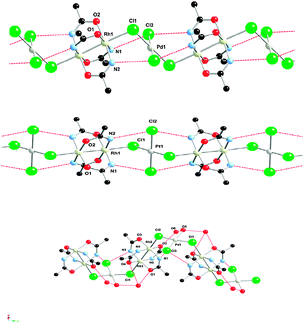 | ||
| Fig. 13 Crystal structure of different infinite (–Rh–Rh–Cl–M–Cl–)n chains M = Pd (top), M = Pt (center) and M = Pd (bottom). | ||
The observed room temperature conductivity values measured on pressed pellets of the two derivatives [Rh2(acam)4(H2O)2][Rh2(acam)4{(μ-Cl)2MCl2}]·2H2O (M = Pt and Pd) were around 10−7 S cm−1. These values are similar to those observed for related Rh25+ chains.
Besides these 1D structures, paddle-wheel complexes constructed with Rh25+ cores and acetamide ligands may also display several coordination modes when bridged by halides, rendering 2D and 3D dimensional structures. These will be discussed in the following sections.
Closely related to MMX polymers, there exists a short number of extended linear metal atom chains bridged by halogen linkers [MnX]∞, however, limited information is reported in terms of their electrical transport properties. A good example of this series was reported in 2005 by Matsumoto's group, who succeeded in the isolation of a mixed-metal mixed-valence quasi 1D system shown in Fig. 14. Its solid-state structure revealed a [Pt6Rh2]19+ repeating octameric core bridged by two-coordinated chloride ions rendering a [–Pt–Rh–Pt4–Rh–Pt–Cl–]n infinite chain.92,94 The octamer can be viewed as being formed by four dimeric units: two inner Pt–Pt dimers (Pt1–Pt2 and Pt3–Pt4) and two outer Pt–Rh dimers (Pt5–Rh1 and Pt6–Rh2), all of them doubly bridged by pivalamidate ligands that warrant both close metal–metal contacts and interdimeric hydrogen bond formation between amide oxygen and amine groups along the chain. Since two ammonia groups complete the coordination requirements of the Pt cations, two monodentate chloride ligands complete the Rh1 and Rh2 environments and balance the charge.
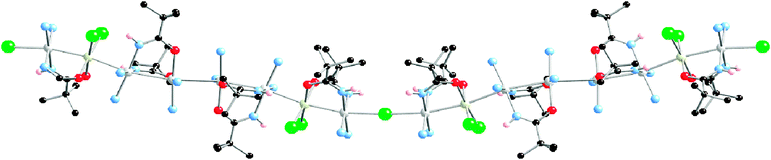 | ||
| Fig. 14 Crystal structure of the octanuclear quasi 1D chain isolated by Matsumoto's group. | ||
The electrical conductivity measured by the four-contacts method shows a semiconducting behaviour with a room temperature conductivity of 3.52 × 10−6 S cm−1 along the perpendicular chain direction, crystal thin plate morphology precluded conductivity measurements along the chain direction.
During their investigation on Te2Rh4 cluster formation, Shieh et al. succeeded in the isolation and characterisation of a number of discrete Te–Ru–Cu carbonyl complexes, including the first Cu(Br)CuCl-bridged Te2Rh4 octahedral cluster chain polymer: {[PPh4]2Te2Ru4(CO)10(Cu4Br2Cl2)·THF}∞. The electrical conductivity measurements of this compound showed a room temperature conductivity of 1–5 × 10−2 S cm−1 and a semiconducting behaviour.93
This polymeric {[PPh4]2Te2Ru4(CO)10Cu4Br2Cl2)·THF}∞ chain was prepared by reaction between K2TeO3 and Ru3(CO)12 followed by treatment with PPh4Br and CuCl. Its crystal structure (Fig. 15) can be described as formed by Te2Ru4 octahedral units, each Ru centre is coordinated to two terminal CO groups, two Cu2Br cores and a bridging CO group connecting opposite pairs of Ru2 units. There is also a short interaction of one Cu centre with an apical Te atom. These repetitive units are further connected by chloride bridges forming an anionic chain, whose charge is balanced with PPh4+ counterions running along the b axis. A solvated THF molecule is enclosed in the asymmetric unit cell.
![Solid-state structure of {[PPh4]2Te2Ru4(CO)10Cu4Br2Cl2)·THF}∞, PPh4 counterions and co-crystallised THF solvent molecules are not displayed for clarity.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f15.gif)
3.2. Cyanopyridines as linkers
Cyanopyridine ligands offer two different donor nitrogen sites located at their ends. These nitrogen donor atoms differ in steric requirements and basic character. Thus the cyano nitrogen atom is a weaker donor than the pyridine one. Indeed this is the reason why organic molecules, and counteranions can connect to the metal centre easier than the cyano nitrogen, resulting in the formation of complexes in which the n-cyanopyridines act as monodentate ligands and the cyano groups are not coordinated to the metal.In a work carried out by Henderson and co-workers,95 several one dimensional coordination polymers of n-cyanopyridines (n = 3, 4) and pyridine-3,4-dicarbonitrile with Ag(I) and Cu(I) have been explored. In some of the polymers reported n-cyanopyridine and pyridine-3,4-dicarbonitrile ligands coordinated in a monodentate fashion while NO3− and SCN− bridge the metal centres (Fig. 16).
![One-dimensional polymeric structures of cyanopyridine polymers with [Cu(pyridine-3,4-dicarbonitrile)2(SCN)]n (top) and [Ag(pyridine-3,4-dicarbonitrile)2(NO3)]n (bottom)). Close non-bonding interaction for [Ag(pyridine-3,4-dicarbonitrile)2(NO3)]n displayed as red dotted lines.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f16.gif) | ||
| Fig. 16 One-dimensional polymeric structures of cyanopyridine polymers with [Cu(pyridine-3,4-dicarbonitrile)2(SCN)]n (top) and [Ag(pyridine-3,4-dicarbonitrile)2(NO3)]n (bottom)). Close non-bonding interaction for [Ag(pyridine-3,4-dicarbonitrile)2(NO3)]n displayed as red dotted lines. | ||
The electrical characterisation carried out with the two-probe method on pressed pellets (Table 9) shows that the coordination polymers [Cu(4-cyanopyridine)2(SCN)]n and [Ag(3-cyanopyridine)2(NO3)]n are insulators in the whole range of temperature (290–350 K).
| Compounds | Conductivity/S cm−1 | Ref. |
|---|---|---|
| TANC = 5,6,11,12-tetraazanaphthacene. a Along a axis.b Along b axis.c Along c axis at 303–393 K.d Pressed pellets and two probe.e Single crystal and four probe. | ||
| [Ag(3-cyanopyridine)2(NO3)]n | 10−11d |
95 |
| [Ag(pyridine-3,4-dicarbonitrile)2(NO3)]n | 7.3 × 10−6d |
95 |
| [Cu(4-cyanopyridine)2(SCN)]n | 10−13d |
95 |
| [Cu(pyridine-3,4-dicarbonitrile)2(SCN)]n | 4.3 × 10−5d |
95 |
| [{Cu(TANC)}F0.5]n | 50a,e | 96 |
| 0.91b,e | ||
| 6.90 × 10−3c,e |
[Ag(pyridine-3,4-dicarbonitrile)2(NO3)]n and [Cu(pyridine-3,4-dicarbonitrile)2(SCN)]n show a semiconductor behaviour although with a different thermal reversibility. Thus, on one hand, the Ag(I) polymer shows a thermal reversible behaviour and its conductivity increases by a factor of 2 (from 7.37 × 10−6 to 15.1 × 10−6 S cm−1) as the temperature increases from 298 K to 358 K. On the other hand, the Cu(I) polymer shows an irreversible thermal dependence and the conductivity increases only by 25% (from 4.29 × 10−5 to 5.39 × 10−5 S cm−1) when the temperature is varied in the same range (298–348 K). This difference can be explained according to their structures. Thus, in [Ag(pyridine-3,4-dicarbonitrile)2(NO3)]n the one dimensional chain shows short and stable Ag–O bonds that form a stable and effective conjugated system in the measured temperature range. The authors explain the difference claiming that in [Cu(pyridine-3,4-dicarbonitrile)2(SCN)]n, the Cu(I) in the crystal is reduced at high temperature by the SCN− resulting in Cu0 doped semiconducting behaviour. It should be also mentioned that none of the two polymers shows metal–metal bonding in their structures.
The 1D-coordination polymer [{Cu(TANC)}F0.5]n was obtained by reaction of 5,6,11,12-tetraazanaphthacene (TANC) and [Cu(MeCN)4]BF4 in MeOH/MeCN. The presence of half an equivalent of non-stoichiometric F− ions was determined by X-ray photoelectron spectroscopy and quantitative chemical analysis of F− ions. These F− ions are generated by the dissociation of BF4− anions in the reaction mixture (Fig. 17).96
![One dimensional structure of [{Cu(TANC)}F0.5]n with alternating linkages between Cu(i) ions and TANC. Only relevant H atoms depicted for clarity.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f17.gif) | ||
| Fig. 17 One dimensional structure of [{Cu(TANC)}F0.5]n with alternating linkages between Cu(I) ions and TANC. Only relevant H atoms depicted for clarity. | ||
Conductivity measurements for this complex, performed with the four-probe method, showed a high room temperature conductivity of 50 S cm−1 along the a axis. This value decreases rapidly to 10−6 S cm−1 at 10 K showing a semiconducting behaviour. Conductivity measurements performed in different crystal directions showed a high anisotropy with room temperature values of 50, 0.91 and 6.90 × 10−3 S cm−1 along the a, b and c directions, respectively. These results are explained based on the disordered F− ions, which affect the stacking of the TANC columns since the particular crystal structure of this compound shows alternating layers of F− ions that disturb the conductivity through the c axis. This means that the crystal behaves as a quasi-1D molecular semiconductor with a relatively good conductivity in the ab plane and a maximum conductivity along the a axis, corresponding to the 1D stacking of the TANC ligands.96Table 9 collects conductivity data reported on these coordination polymers.
3.3. Systems containing nucleobases
The design and construction of coordination polymers containing nucleobases as either terminal or bridging ligands is of interest due to their abilities to act as functional materials given their particular electrical, magnetic, catalytic and non-linear optical properties. Three-dimensional coordination polymers may also show applications in molecular storage (gases, anionic exchange, etc.) and/or molecular separation. The formation of polymers based on biologically relevant ligands, such as nucleobases, may provide additional applications in biological fields. For instance, some of these structures could be used as models of specific forms of DNA (e.g. M-DNA, G4,….).97 It is surprising that there are relatively little number of polymeric structures based only on bridging nucleobases, despite the number of available coordination positions. Recently, one of these examples has attracted particular attention since its structure resembles that initially suggested for M-DNA (a novel type of DNA).98 The initial studies were carried out on the [Cd(6-MP)2·2H2O]n (6-MP = 6-mercaptopurinato) polymer showing an insulating behaviour which could be explained as a consequence of the large HOMO–LUMO band gap.99 Subsequent DFT calculations performed with the aim of designing suitable electrical conductive [M(6-MP)2]n (M = transition metal, 6-MP = 6-mercaptopurinato) one-dimensional coordination polymers pointed out that several metal ions such as Ni(II) could provide suitable materials.100 Thus, direct hydrothermal reactions between 6-mercaptopurine (6-MPH) and the analogous 6-thioguanine (6-ThioGH) with NiSO4·6H2O gave [Ni(6-MP)2·2H2O]n and [Ni(6-ThioG)2·2H2O]n (Fig. 18).![Fragments of the polymeric chains of compounds [Ni(6-MP)2·2H2O]n(left) and [Ni(6-ThioG)2·2H2O]n (right).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f18.gif) | ||
| Fig. 18 Fragments of the polymeric chains of compounds [Ni(6-MP)2·2H2O]n(left) and [Ni(6-ThioG)2·2H2O]n (right). | ||
The structures of these coordination polymers are rather similar to that with Cd(II) and consist of one-dimensional chains in which the deprotonated nucleobases act as the bridging ligands connecting the metal ions with short M–M distances (3.677 and 3.646 Å). Two probe DC electrical conductivity measurements at 300 K show ohmic behaviour, with conductivity values of 1.10 × 10−5 and 1.34 × 10−4 S cm−1, for [Ni(6-MP)2·2H2O]n and [Ni(6-ThioG)2·2H2O]n, respectively (Table 10).101 The small differences found in these measurements were rationalized with the help of additional DFT calculations. The main difference is that the lattice constant obtained by GGA-DFT calculations is 2.2% smaller than that obtained by X-ray analysis. This leads to an increase of ∼14% in the HOMO–LUMO gap (0.77 eV for the X-ray structure) with respect to the value obtained for the theoretical geometry. The calculated electronic structure of [Ni(6-ThioG)2·2H2O]n presents similar electronic properties to those observed for [Ni(6-MP)2·2H2O]n, with a calculated HOMO–LUMO gap of 0.69 eV. By comparing the band structures of [Ni(6-MP)2·2H2O]n and [Ni(6-ThioG)2·2H2O]n, it is observed that the amino group, present in the [Ni(6-ThioG)2·2H2O]n, mainly introduces modifications in the deep level states lying at about −1.25 eV from the Fermi level, and below. The lower gap observed for [Ni(6-ThioG)2·2H2O]n, when compared with [Ni(6-MP)2·2H2O]n, is due to the fact that the former presents a smaller lattice constant (3.65 Å) than the latter (3.68 Å). By using the value we obtained for the difference in band gaps between the two coordination polymers, we have estimated that polymer [Ni(6-ThioG)2·2H2O]n should have about 20 times more thermally activated intrinsic carriers, at room temperature, than [Ni(6-MP)2·2H2O]n which is consistent with the experimental conductivity measurements.
Therefore, it seems that theoretical calculations are a suitable tool to understand experimental measurements but, even more important, to design coordination polymers with the desired property.
3.4. Sulfur ligands as linkers
The field of molecular organic conductors has been widely studied in recent years, focusing mainly on the synthesis of charge transfer complexes based on tetrathiafulvalene (TTF).102 Compounds of this type with intermolecular S⋯S contacts have shown metallic conductivity and, in some few cases, even superconductivity.103 These results have encouraged researchers to select ligands with similar structural features (Fig. 19) and extend the electrical conductivity studies towards coordination compounds and coordination polymers. | ||
| Fig. 19 Structures of some representative tetrathiafulvalene ligands: 2,5-bis(4′,5′-bis(methylthio)-1′,3′-dithiol-2′-ylidene)-1,3,4,6-tetrathiapentalene (TTM-TTP), maleonitriledithiolate (mnt2−), isomaleonitriledithiolate (i-mnt2−), 1-ethoxycarbonyl-1-cyanoethylene-2,2-dithiolate (ecda2−), diethyldithiocarbamate (Et2dtc−), benzothiazole-2-thiolate (bzta−), 4,5-dimercapto-1,3-dithiole-2-thione (dmit2−), bis(ethylenedithio)tetrathiafulvalene (ET) and, 4,5-ethylenedithio-1,3-dithiole-2-thione (C5H4S5). | ||
TTF derivatives are π-electroactive compounds with chelating sites such as the organylthio groups, which could potentially further enhance the bonding and electronic interaction between the metal centre and the organic ligands. These organosulfur groups can act as linkers between metallic centres to form charge transfer complexes that could act as Synth. Met. or semiconductors. The physical properties of these compounds not only depend on the specific molecular properties of the individual components, metal ions and sulfur ligands, but are also strongly influenced by the arrangement of interactions within the crystal lattice.104
The coordination of TTF derivatives to metal ions provides a wide variety of conductive complexes and coordination polymers. These CPs exhibit a broad panel of electrical properties due to the existence of a coordination polymeric network that can be achieved through the coordination of metal ions and/or through the S⋯S contacts. In the following lines we will describe some examples of electrical conductive coordination polymers with this kind of ligands.
A series of heterobimetallic compounds with maleonitriledithiolate (mnt2−),105 isomaleonitriledithiolate (i-mnt2−),105 4,5-dimercapto-1,3-dithiole-2-thione (dmit2−)106 and 1-ethoxycarbonyl-1-cyanoethylene-2,2-dithiolate (ecda2−)107,108 have been prepared by reaction of M2M′L2 (L = mnt2−, i-mnt2−, ecda2−, dmit2; M = Na and K, and M′ = Cu(II), Ni(II), Au(I)) with a solution of different metallic salts (Ag(I), Pd(II), Cd(II), Pb(II), Hg(II), Cu(I) and Au(I)) in the appropriate molar ratio. Their structural characterisation have been carried out by spectroscopic techniques suggesting the formation of one-dimensional coordination polymers in which the metal entities are connected by the sulfur atoms of the organic ligands to generate heterobimetallic layered polymeric structures (Fig. 20). These structures extend the electron delocalization beyond the MS2 group and hence exhibit interesting conductivity in its complexes (Table 11).
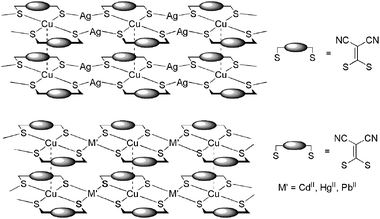 | ||
| Fig. 20 Suggested structures for the two types of heterobimetallic coordination polymers with i-mnt2− ligands. | ||
| Compound | Conductivity/S cm−1 | Ref. |
|---|---|---|
| mnt 2− = maleonitriledithiolate, i-mnt2− = isomaleonitriledithiolate, dmit2− = 4,5-dimercapto-1,3-dithiole-2-thione, ecda2− = 1-ethoxycarbonyl-1-cyanoethylene-2,2-dithiolate, ET = bis(ethylenedithio)tetrathiafulvalene, C4S62− = dithiolato, TTM-TTP = 2,5-bis-4′,5′-bis(methylthio)-1′,3′-dithiol-2′-ylidene-1,3,4,6-tetrathiapentacene, C5H4S5 = 4,5-ethylenedithio-1,3-dithiole-2-thione, (C5H5N)S2 = 2,2′-dipyridyldisulfide, 4-SpyH = zwitterionic pyridium-4-thiolate. a 303–393 K.b 300 K and 1 bar.c Single crystal.d Four probe. | ||
| [Ag[Cu(ecda)2]2]n | 10−11a |
107 |
| [Pd[Cu(ecda)2]2]n | 10−7a |
107 |
| {(NEt4)[CuNi(dmit)2]}n | 10−5b |
106 |
| {(NEt4)[AgNi(dmit)2]}n | 2 10−6b |
106 |
| {(NEt4)[AuNi(dmit)2]}n | 10−2b |
106 |
| [Cu2Ni(dmit)2]n | 6 × 10−4b |
106 |
| [Ag2Ni(dmit)2]n | 5 × 10−4b |
106 |
| [Au2Ni(dmit)2]n | 1.6b | 106 |
| [CuAu(dmit)2]n | 10−6b |
106 |
| [AgAu(dmit)2]n | 3 × 10−7b |
106 |
| [Au2(dmit)2]n | 2 × 10−4b |
106 |
| [Ag2Cu(i-mnt)2]n | 10−6a |
105 |
| [CdCu(i-mnt)2]n | 10−9a |
105 |
| [HgCu(i-mnt)2]n | 10−6a |
105 |
| [PbCu(i-mnt)2]n | 10−7a |
105 |
| [Ag2Ni(mnt)2]n | 10−5a |
105 |
| [PdNi(mnt)2]n | 10−5a |
105 |
| [HgNi(mnt)2]n | 10−5a |
105 |
| (ET)CuI2Br4 | 1.6 × 10−2b,c |
109 |
| (ET)2CuI6Br10 | 2.5b,c |
109 |
| (ET)2[Cu4Br6ET] | 2.5 × 10−2b,c |
109 |
| (ET)3CuI6Br10(H2O)2 | 5.1 × 10−2b,c |
109 |
| [Cu2C4S6] | 0.1b,d |
110 |
| [NiC4S6] | 0.9b,d |
110 |
| [Ag(TTM-TTP)(CF3SO3)] | 7 × 10−6b |
104 |
| [Ag(C5H4S5)2NO3]In | 3 × 10−5b |
104 |
| [Cu9(C5H5NS)8(SH)8]nn+ | 1.6 × 10−3b,c,d |
111 |
| [Cu5(μ-4-SpyH)7(μ-I)I4]n | 6.84 × 10−8b,c |
112 |
| {[Cu2(μ-I)(μ-4-SpyH)3]I}n | 2.74 × 10−9b,c |
112 |
The electrical characterisation showed that the conductivity of these materials increases upon heating and decreases upon cooling showing a typical semiconductor behaviour (Table 11).
In the case of [M′2Ni(dmit)2]n the polymeric structures present additional metal ions that interconnect the [M′Ni(dmit)2] (M′ = Cu, Ag and Au) linear chains through S–M–S bonds (Fig. 21).106 The high value of 1.6 S cm−1 observed for the complex [Au2Ni(dmit)2]n (Table 11) is remarkable. The authors attributed this value to the oxidation state observed in the [Ni(dmit)2] units.106
![Suggested polymeric structures consisting in [MNi(dmit)2] units (M = CuI, AgI and AuI) interconnected by metallic centres.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f21.gif) | ||
| Fig. 21 Suggested polymeric structures consisting in [MNi(dmit)2] units (M = CuI, AgI and AuI) interconnected by metallic centres. | ||
The iodine doping of the [MCu(i-mit)2] derivatives (M = AgI, CdII, HgII and PbII) seems to be an effective strategy for the synthesis of electrically conductive compounds. A remarkable increase in the conductivity, i.e. 10−6–10−5 S cm−1 for the iodine-doped coordination polymers as compared to 10−9–10−6 S cm−1 for the respective undoped compounds, is observed.105 The authors state that this could be due to stronger intermolecular S⋯S stack formation as well as partial oxidation–reduction (mixed valence state) of the metal ions.
In a similar approach, N. Singh and Prasad have synthesized several heterobimetallic coordination polymers (1D and 2D) by reaction of two equivalents of the corresponding mixed metal salts with one equivalent of benzothiazole-2-thiolate (bzta−) or diethylthiocarbamate (Et2dtc−) and one equivalent of chalcogenocyanates (XCN−) (X = S or Se).113 In this case, the chelating-bridging characteristics of the Et2dtc− ligand and the hetero-donor atoms ligands (bzta− and XCN−) yield mixed ligand heterobimetallic 1D polymers as is shown in Fig. 22.
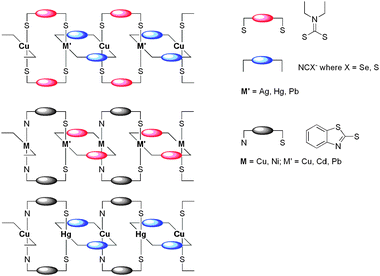 | ||
| Fig. 22 Suggested polymeric structures for the heterobimetallic complexes obtained with benzothiazole-2-thiolate (bzta−), diethylthiocarbamate (Et2dtc−) and chalcogenocyanates (XCN−) (X = S and Se) ligands. | ||
The proposed structures are based on various physico-chemical studies and resemble those previously suggested for similar ligands but, as in the previous cases, there is unfortunately no X-ray structure of these polymers. Basically, the polymeric structures of these heterobimetallic mixed ligands have the Cu(II) and Ni(II) in square planar geometry, Ag(I) in linear geometry and Hg(II), Cu(I) and Pb(II) are preferentially tetrahedrally coordinated.113
The electrical conductivity of these coordination polymers, measured between 300–393 K in pressed pellets using the two probe technique, shows low values of ca. 10−8–10−9 S cm−1. Doping the polymers with iodine produces an increase in their electrical conductivity by 1 or 2 orders of magnitude. The smaller conductivity values found in the undoped polymers are explained by the authors as a result of weaker S⋯S, S⋯Se and M⋯S intermolecular interactions in the solid state.113
Following the idea of preparing new materials combining different physical properties such as conductivity and magnetism, R. Kanehama et al. have studied the reactions of Cu(II) halides with bis(ethylenedithio)tetrathiafulvalene (ET)109 (Fig. 19). They obtained new charge transfer coordination polymers of formula (ET)CuI2Br4,(ET)2CuI6Br10, (ET)2[CuI4Br6ET] and (ET)3CuI6Br10(H2O)2, where the ET ligands coordinated to Cu(I) by the sulfur atoms of the ethylenedithio groups showing different coordination modes. Unfortunately, the reduction of the Cu(II) ions to Cu(I) precludes the coexistence of magnetic and electrical properties in these compounds. The authors classified the compounds into three different groups according to the coordination modes of ET molecules. Group I: the ET coordinates as a monodentate ligand (the authors obtained discrete complexes with this mode of coordination). Group II: the ET coordinates as trans-bidentate ligand. Group III: the ET coordinates as cis-bidentate ligand. In the case of (ET)CuI2Br4, a one-dimensional structure [ET–BrCu–(μ2(Br)2–Cu(Br)–]n is obtained, where the ET ligands are coordinated to two copper ions with the two sulfur atoms in trans mode (Fig. 23). In (ET)2CuI6Br10 complex, all the ET molecules coordinate to two copper ions with the two sulfur atoms in a cis-mode, giving a one dimensional chain running along the c-axis (Fig. 24). Both structures present short interchain contacts established by S⋯S interactions.
![View of the one-dimensional chain [ET–(Br)Cu–(μ2-Br)2–Cu(Br)]∞ of (ET)CuI2Br4.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f23.gif) | ||
| Fig. 23 View of the one-dimensional chain [ET–(Br)Cu–(μ2-Br)2–Cu(Br)]∞ of (ET)CuI2Br4. | ||
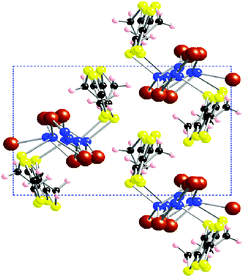 | ||
| Fig. 24 Packing diagram of (ET)2CuI6Br10 projected along the c-axis. | ||
The crystal structure of the (ET)2[Cu4Br6ET] polymer consists of two different parts. On one side the conducting ET layers and, on the other, the ET-copper one-dimensional chains. In the ET layers, the ET molecules stack parallel to the c-axis and are not coordinated to the copper ions. In the ET-copper chains, the ET molecules coordinate to two copper ions with the two sulfurs in a trans mode. Two quasi 1D copper complexes [–Br–Cu–(μ2-Br)2–Cu(Br)–]n bridged by ET molecules form a ladder like structure (Fig. 25). This compound is a hybrid material in which conducting layers and a one-dimensional lattice coexist in the crystal packing.
![View of the ladder like structure of (ET)2[Cu4Br6ET].](/image/article/2012/CS/c1cs15092h/c1cs15092h-f25.gif) | ||
| Fig. 25 View of the ladder like structure of (ET)2[Cu4Br6ET]. | ||
In the complex (ET)3CuI6Br10(H2O)2, the ET molecules coordinate to two Cu ions with the two S atoms in cis mode (Fig. 26).
 | ||
| Fig. 26 View of the zigzag one-dimensional chain of (ET)3CuI6Br10(H2O)2, projected along the a-axis. | ||
The conductivity data obtained for these ET coordination polymers, with the two-probe method on single crystals along the c axis, using 15 μm gold wires and carbon paste, show that these complexes are highly conducting materials (Table 11).
A very interesting work on 1D coordination polymers with dithio ligands has been reported by Szczepura et al.110 They prepared several 1D-coordination polymers by reaction of dithiolato C4S62− ligand with [Ni(H2O)6]Cl2 or [Cu(MeCN)4]BF4. The analysis of the insoluble black solids prompted the authors to propose the empirical formula [Cu2C4S6] and [NiC4S6] with a 1D-polymeric structure. These polymers are diamagnetic and show semiconducting properties that have been attributed to the presence of a delocalized π-electron system. Both [Cu2C4S6] and [NiC4S6] behave as semiconductors with conductivity values of 0.1 and 0.9 S cm−1 (measured at 294 K, using the four contact method). Despite that the measurements were done in pressed pellets, these are some of the better values so far reported. However, to clarify this unusual conductivity their structures should be elucidated.
Following with the series of TTF derivatives, one dimensional homometallic silver(I) coordination polymers obtained with 2,5-bis-4′,5′-bis(methylthio)-1′,3′-dithiol-2′-ylidene-1,3,4,6-tetrathiapentacene (TTM-TTP) and 4,5-ethylenedithio-1,3-dithiole-2-thione (C5H4S5) (Fig. 19) ligands have also been studied.104,114
In the first case, the one dimensional coordination polymer [Ag(TTM-TTP)(CF3SO3)] (Fig. 27) is obtained by slow diffusion of n-hexane into a solution containing a mixture of AgCF3SO3 and TTM-TTP in acetonitrile/benzene. In this polymer, besides the Ag(I)–S coordination bonds, there are extensive intermolecular S⋯S contacts, providing additional interactions across the individual chains.114
![Side view of [Ag(TTM-TTP)(CF3SO3)] with face-to-face S⋯S contacts.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f27.gif) | ||
| Fig. 27 Side view of [Ag(TTM-TTP)(CF3SO3)] with face-to-face S⋯S contacts. | ||
In the second case, the strong electron-donor C5H4S5 ligand forms a one-dimensional silver(I) coordination polymer with two parallel coordinating molecular units (Fig. 28). The ligands coordinate to silver(I) through both sulfur atoms of the thiocarbonyl and through the sulfur atoms of the thioether. An additional extension of the contacts produces a 2D network by strong S⋯S interactions. The iodine doped complex gives a room temperature conductivity of 3 × 10−5 S cm−1 measured in pressed pellets by a conventional two-probe method and a semiconductor behaviour.104 As in other already mentioned examples, the conductivity values of these two polymers also increase when doped with iodine.
![Perspective view of [Ag(C5H4S5)2+]n.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f28.gif) | ||
| Fig. 28 Perspective view of [Ag(C5H4S5)2+]n. | ||
Recently, the use of new synthetic routes as solvothermal (see Section 3.2 nucleobases as nitrogen ligands) and microwave methods has led to the synthesis of interesting coordination polymers with different architectures and dimensionalities. One of these examples was obtained by microwave solvothermal reaction of 2,2′-dipyridyldisulfide and Cu(BF4)2. The reaction proceeds with an unusual cleavage of C–S and S–S bonds of the 2,2′-dipyridyldisulfide ligand leading to a Cu(I) 1D-coordination polymer formation. The structure is generated based on the assembling of Cu9 cages (Fig. 29).111
![Details of the structure of [Cu9(C5H5NS)8(SH)8]nn+ and thermal variation of the electrical conductivity with the scheme of the 2,2′-dipyridyldisulfide ligand. (Data collected from ref. 111; reproduced by permission of the American Chemical Society.)](/image/article/2012/CS/c1cs15092h/c1cs15092h-f29.gif) | ||
| Fig. 29 Details of the structure of [Cu9(C5H5NS)8(SH)8]nn+ and thermal variation of the electrical conductivity with the scheme of the 2,2′-dipyridyldisulfide ligand. (Data collected from ref. 111; reproduced by permission of the American Chemical Society.) | ||
The unusual polymeric structure presenting a large number of S bridges prompted the authors to measure its electrical conductivity. Studies carried out at variable temperature (Fig. 29), using the four probe technique, show a classical semiconductor behaviour with a high room temperature conductivity of 1.6 × 10−3 S cm−1.111
Another polymer obtained under solvothermal conditions was synthesised by reaction of 4,4′-dipyridyl disulfide (dpds), oxalic acid and CuI.112 The products obtained depend on the reaction time. Thus, three different Cu/I/S-based coordination polymers were isolated: [Cu6(μ-4-SpyH)4I6]n, {[Cu2(μ-I)(μ-4-SpyH)3]I}n, and [Cu5(μ-4-SpyH)7(μ-I)I4]n.112
The process involves the in situ formation of the zwitterionic pyridinium-4-thiolate (4-SpyH) generated by S–S cleavage of the dpds ligand. The hexameric compound [Cu6(μ-4-SpyH)4I6]n forms a three dimensional diamond-like net, that will be discussed in Section 5.2. The structure of the dimeric compound {[Cu2(μ-I)(μ-4-SpyH)3]I}n consists of a cationic {[Cu2(μ-I)(μ-4-SpyH)3]n}n+ polymeric chain with disordered iodides embedded between the chains. In contrast, the pentameric compound [Cu5(μ-4-SpyH)7(μ-I)I4]n contains [Cu5(μ-I)I4(μ-4-SpyH)7] units that are interconnected with its neighbouring ones through two μ-4-SpyH bridges to form a neutral chain (Fig. 30). The formation of these three different compounds provided an interesting example that different coordination polymers could be produced from the same components under solvothermal conditions at different time periods, indicating that the often-neglected kinetic factors may be very important in this kind of synthesis.
![Views of {[Cu2(μ-I)(μ-4-SpyH)3]n}n+ (up) and [Cu5(μ-4-SpyH)7(μ-I)I4]n (down).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f30.gif) | ||
| Fig. 30 Views of {[Cu2(μ-I)(μ-4-SpyH)3]n}n+ (up) and [Cu5(μ-4-SpyH)7(μ-I)I4]n (down). | ||
The thermal variation (from 293 to 443 K) of the electrical conductivity measured for several single crystals of the dimeric and pentameric compounds with the two probe technique shows an exponential increase with increasing temperature from 6.84 × 10−8 to 3.96 × 10−6 S cm−1, for the dimeric {[Cu2(μ-I)(μ-4-SpyH)3]I}n compound and from 2.74 × 10−9 to 2.43 × 10−7 S cm−1, for the pentameric one, both displaying a typical semiconducting behaviour. The authors attributed the better conductivity of {[Cu2(μ-I)(μ-4-SpyH)3]I}n to the fact that the density of states or effective mass in this compound is larger than in the other one. In addition, the disordered counter iodides and the weak Cu⋯Cu interactions found in compound {[Cu2(μ-I)(μ-4-SpyH)3]I}n may also contribute to the increase in conductivity.112
3.5. Systems containing metallomacrocycles
The metallomacrocycles such as metallophthalocyanines and metalloporphyrines have long been studied due to their interesting applications like dyes, pigments, catalysts, electrical conductors, etc. (Fig. 31).115 | ||
| Fig. 31 Schematic representation of 1D coordination polymers composed of MPcs (Pc = phthalocyanine) molecules bridged with μ-axial ligands. | ||
Their typical structures provide poor π–π overlap between the adjacent molecules showing low electrical conductivities usually in the range 10−11–10−13 S cm−1.116,117 Collecting these works, excellent reviews and articles about the electrical properties of these molecular materials have been published.118–120
In order to improve the electrical conductivity, new types of low-dimensional compounds have been prepared where the metallomacrocycles achieve a stacked co-facial arrangement with low interplanar distances allowing electron delocalization by π–π overlapping. This can be achieved using several bridging ligands between the metallomacrocycles to generate a linear coordination polymer, where the central metal–axial ligand spine was expected to be a reasonable pathway for conduction.
Among the large variety of semiconducting polymers based on metallophthalocyanines, the most important families are the polyphthalocyaninato-metalloxanes [PcMO]n (M = Si, Ge, Sn), studied in detail by Kenney and Marks,121–123 and those so-called “shish-kebab” developed by Hanack and Dini, based on the self-assembled Pc coordination polymers (Fig. 32).124
![Example of an octacyanometallophthalocyanines polymer [FePcOc(dib)]n (Pc = phthalocyanine, dib = 1,4 diisocyanobenzene).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f32.gif) | ||
| Fig. 32 Example of an octacyanometallophthalocyanines polymer [FePcOc(dib)]n (Pc = phthalocyanine, dib = 1,4 diisocyanobenzene). | ||
A significant amount of these kind of coordination polymers based on metallophthalocyanines and metalloporphyrines with several bridging ligands, [MPc/P(L)]n (Pcs = phthalocyanines, P = porphyrines; M = Fe, Co, Mn, Ru, Os; and L = cyano, pyrazine, tetrazine and many others bidentate ligands), have been investigated as potential electrical conductors.125,126 These compounds showed low conductivity values in the lower part of the semiconducting region. However, doping these polymers with iodine increases conductivity up to values between 10−5–10−2 S cm−1. In contrast to other bridging ligands, for these compounds it seems that the use of tetrazine allows significant increases of the semiconducting properties without the need of external oxidative doping (0.05–0.3 S cm−1).127Table 12 shows a selection of these polymers with the most significant conductivity values.
| Compounds | Conductivity/S cm−1 | Ref. |
|---|---|---|
| Pc = phthalocyanines, pyz = pyrazine, tz = tetrazine, me2tz = 3,6-dimethyl-s-tetrazine, MPcOc = octacyanometallophthalocyanines. a RT and 1 bar.b RT and 108 pa.c Two probe. | ||
| FePc(pyz)]n | 2 × 10−5a |
126 |
| [FePc(tz)]n | 2 × 10−2a |
126 |
| [RuPc(tz)]n | 10−2a |
125 |
| [FePc(me2-tz)]n | 4 × 10−3a |
125 |
| FePcOc | 6.20 × 10−8b,c |
127 |
| [FePcOC(dib)]n | 5.73 × 10−7b,c |
127 |
The improvement of the electrical properties by polymerizing metallomacrocycle units into one-dimensional coordination polymers seems to be a consequence of the partial electron loss in the d(π)-orbital of the metal centre towards the bridging ligand π/π* orbitals of the macrocycle ring.128 However, most of these complexes are insoluble, and their purification towards thin-film fabrication are therefore restricted. Attempts to improve the solubility of these polymers by additional functionalization of the macrocycle rings have been carried out. The case of the octacyanometallophthalocyanines (MPcOc) (Fig. 32) is an interesting example. In this case strong electron acceptor properties have shown that this material could be useful as very rare and stable n-type organic semiconductors or as cathode material of lithium rechargeable batteries.127
Although functionalization leads to an increase in the solubility of the polymers, their electrical properties do not improve. Only a significant increase of the conductivity is observed by doping with lithium127 in both the monomeric (FePcOc) and polymeric [FePcOc(dib)]n (dib = 1,4-diisocyanobenzene) complexes (Table 12).
Therefore, despite the past and current interest in metallophthalocyanines and metalloporphyrines as conductive molecular materials,129 so far the electrical properties found in coordination polymers containing these molecules as building-blocks have shown only moderate electrical conductivity values.
3.6. Aromatic hydrocarbons as linkers
Munakata et al. have been exploring the chemistry of polycyclic aromatic hydrocarbons with different silver(I) metal salts for decades (Table 13). They have described a vast body of literature comprising a good number of structurally characterised frameworks. The first record considering electrical conduction properties came in 1997130 when they isolated two unusual silver(I)-aromatic networks containing pyrene (pyr) and perylene (per) tectons, [Ag2(pyr)(ClO4)2] and [Ag2(per)(ClO4)2] respectively (Fig. 33), both displaying a tetra-η2 coordination mode, linking four metal centres and rendering W-shaped polymeric chains and sheets, in this order.| Compounds | Conductivity/S cm−1 | Ref. |
|---|---|---|
| a btp = 1,2-benztriphenylene. rub = rubrene, bpyr = benzo[a]pyrene, cor = corenene, bphen = benzo[a]phenanthrene, dban = dibenz[a,h]anthracene, ban = benz[a]anthracene, napyr = naphtho(2,3-a)pyrene, btp = 1,2-benztriphenylene, dbchry = dibenzo[b,def]chrysene, bpry = benzo[e]pyrene, dpbd = trans,trans-1,4-diphenyl-1,3-butadiene, bmsb = 1,4-bis(methylstyryl)benzene. | ||
| [Ag4(rub)(ClO4)4(H2O)4], | 1.9 × 10−6 | 131 |
| [Ag4(bpyr)2(ClO4)4(toluene)2] | 1.1 × 10−5 | 131 |
| [Ag4(cor)3(ClO4)4], | 3.1 × 10−3 | 131 |
| [Ag3(bphen)(ClO4)3(H2O)2] | 1.65 × 10−3–6.9 × 10−2 | 132 |
| [Ag2(dban)(ClO4)2] | 1.65 × 10−3–6.9 × 10−2 | 132 |
| [Ag2(ban)(ClO4)2(H2O)] | 1.65 × 10−3–6.9 × 10−2 | 132 |
| [Ag2(napyr)(CF3SO3)2] | 1.65 × 10−3–6.9 × 10−2 | 132 |
| [Ag0.5(btp)0.5(ClO4)0.5] | 1.32a | 133 |
| [Ag2(dbchry)(CF3SO3)2][Ag2(toluene)2(CF3SO3)]n | 5.77 × 10−2 | 134 |
| [Ag4(bpyr)4(p-xylene)(ClO4)4]. | 6.3 × 10−2 | 134 |
| [Ag(dpbd)(ClO4)] | 2.1 × 10−7 | 134 |
| [Ag2(bmsb)(ClO4)2] | 1.0 × 10−6 | 135 |
| [Ag2(bmsb)(H2O)4](BF4)2 | 1.2 × 10−2 | 135 |
![Solid-state molecular structure of [Ag2(pyr)(ClO4)2] (left) and [Ag2(per)(ClO4)2] (right) with partial numbering scheme. No hydrogen atoms are displayed.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f33.gif) | ||
| Fig. 33 Solid-state molecular structure of [Ag2(pyr)(ClO4)2] (left) and [Ag2(per)(ClO4)2] (right) with partial numbering scheme. No hydrogen atoms are displayed. | ||
A compressed powder pellet with the standard two-probe method showed insulator behaviour for both species. However, I2-doping resulted on semiconduction with σ values at room temperature of 1.7 × 10−5 and 4.4 × 10−5 S cm−1 for [Ag2(pyr)(ClO4)2] and [Ag2(per)(ClO4)2], respectively. These values were postulated to be connected with the presence of crystallised organic radicals, as evidenced by ESR spectroscopy.
In a later report, in 1998, the same authors succeeded in the construction of new polymeric forms of organosilver compounds with strained aromatic moieties.131 This time rubrene (rub), benzo[a]pyrene (bpyr) and corenene (cor) were combined with Ag(ClO)4·H2O to form novel polymeric networks formulated as: [Ag4(rub)(ClO4)4(H2O)4], [Ag4(bpyr)2(ClO4)4(toluene)2] and [Ag4(cor)3(ClO4)4]. The most significant features of the crystal structure of [Ag4(rub)(ClO4)4(H2O)4] are a 3D network consisting of twisted rubrene fragments coordinated by eight silver cations with both η1- and η2-bonding modes, loose of planarity of the rubrene fragment and lack of extended π–π interactions (Fig. 34).
![View of the crystal structure of [Ag4(rub)(ClO4)4(H2O)4].](/image/article/2012/CS/c1cs15092h/c1cs15092h-f34.gif) | ||
| Fig. 34 View of the crystal structure of [Ag4(rub)(ClO4)4(H2O)4]. | ||
A single crystal X-ray diffraction study of [Ag4(bpyr)2(ClO4)4(toluene)2] revealed a double helical arrangement sandwiching a toluene solvent molecule, η1- and η2-coordination modes for silver cations and an intricate interweaved network of aromatic π–π interactions (Fig. 35).
![Crystal structure of [Ag4(bpyr)2(ClO4)4(toluene)2].](/image/article/2012/CS/c1cs15092h/c1cs15092h-f35.gif) | ||
| Fig. 35 Crystal structure of [Ag4(bpyr)2(ClO4)4(toluene)2]. | ||
The solid-state structure of [Ag4(cor)3(ClO4)4] can be regarded as a triple-decker polymeric framework with both intra and intermolecular π–π interactions warranting the formation of columnar stacks with very close contacts of 3.23 Å between adjacent corenene planes (Fig. 36).
![The triple-decker motif observed on the molecular structure of [Ag4(cor)3(ClO4)4] evidencing close π–π interactions.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f36.gif) | ||
| Fig. 36 The triple-decker motif observed on the molecular structure of [Ag4(cor)3(ClO4)4] evidencing close π–π interactions. | ||
No doping was needed to observe conductivity and the recorded values measured at room temperature on a conventional two-probe method on pressed pellets were 1.9 × 10−6, 1.0 × 10−5 and 3.1 × 10−3 S cm−1, for [Ag4(rub)(ClO4)4(H2O)4], [Ag4(bpyr)2(ClO4)4(toluene)2] and [Ag4(cor)3(ClO4)4], respectively.
In 1999, Munakata et al. extended this family of conducting organosilver polymeric compounds with benzo[a]phenanthrene (bphen), dibenz[a,h]anthracene (dban), benz[a]anthracene (ban) and naphtho(2,3-a)pyrene (napyr) ligands.132 The reaction of these molecules with silver(I) salts generated herringbone-like architectures reminiscent of those of the free ligands. Thus, the crystalline solid state of [Ag3(bphen)(ClO4)3(H2O)2] consists of aromatic-linked chain strands along the c axis where each ligand adopts di-η2 and tetra-η2 coordination modes bridging two and four metal centres, respectively. The existence of intermolecular π–π interactions renders a supramolecular 2D sheet framework (Fig. 37).
![Atom labelling scheme (left) and herringbone disposition (right) of [Ag3(bphen)(ClO4)3(H2O)2].](/image/article/2012/CS/c1cs15092h/c1cs15092h-f37.gif) | ||
| Fig. 37 Atom labelling scheme (left) and herringbone disposition (right) of [Ag3(bphen)(ClO4)3(H2O)2]. | ||
[Ag2(dban)(ClO4)2] exists in the solid state as a polymeric W-like sandwich of alternating AgClO4 and dban building-blocks repeating along the b axis, each aromatic ring displays a tetra-η2 coordination bridging four metal ions. Extended intermolecular π–π interactions give rise to 2D sheets in the ab plane (Fig. 38).
![Crystal structure of [Ag2(dban)(ClO4)2] (left). View of the 2D multilayer structure along the ab plane (right).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f38.gif) | ||
| Fig. 38 Crystal structure of [Ag2(dban)(ClO4)2] (left). View of the 2D multilayer structure along the ab plane (right). | ||
Similar multilayer 2D sheets are observed in the crystal structure of [Ag2(ban)(ClO4)2(H2O)] where two independent silver ions are bonded to the head and tail of the aromatic rings and to H2O molecules. The perchlorate counterions exhibit mono and tridentate coordination modes and participate in silver coordination through Ag–O–Cl–O–Ag and Ag–O–Ag bonds (Fig. 39).
![Solid state structure of [Ag2(ban)(ClO4)2(H2O)] with selected atom labelling (up) and view of the multilayer framework depicted on the ab plane (down).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f39.gif) | ||
| Fig. 39 Solid state structure of [Ag2(ban)(ClO4)2(H2O)] with selected atom labelling (up) and view of the multilayer framework depicted on the ab plane (down). | ||
Although explicit details of electrical conduction for each polymer were not given, authors claimed that on a two-probe experiment with silver coated contacts the values observed for pressed pellets were in the range 1.65 × 10−3–6.9 × 10−2 S cm−1 at room temperature.
A striking feature in the structure of [Ag2(napyr)(CF3SO3)2] is the presence of an unprecedented CF3SO3− group tricoordinated to a silver(I) ion. The herringbone pattern of the free ligand is mainly retained giving rise to a 2D multilayer framework with aromatic sheets suspended between pairs of infinite ionic chains by Ag–O–S–O–Ag bonds (Fig. 40).
![Crystal structure of [Ag2(napyr)(CF3SO3)2] showing coordination environments for silver centres and atomic labelling scheme (top). Depiction of herringbone pattern retention (bottom).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f40.gif) | ||
| Fig. 40 Crystal structure of [Ag2(napyr)(CF3SO3)2] showing coordination environments for silver centres and atomic labelling scheme (top). Depiction of herringbone pattern retention (bottom). | ||
In a later communication, Munakata et al. reported a new metallocyclophane with a columnar aromatic stacking.133 This time the cyclophane motif was tethered through silver(I) ions rather than the previously reported C, O, S or N atoms, hence the unprecedented metallocyclophane nomenclature. When the T-shaped 1,2-benztriphenylene (btp) ligand reacts with AgClO4 in p-xylene, crystals with a [Ag0.5(btp)0.5(ClO4)0.5] formulation could be isolated and their X-ray structure determined (Fig. 41).
![Argentocyclophane structure of [Ag0.5(btp)0.5(ClO4)0.5] (top) and evidence of intra and intermolecular π–π interactions (down).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f41.gif) | ||
| Fig. 41 Argentocyclophane structure of [Ag0.5(btp)0.5(ClO4)0.5] (top) and evidence of intra and intermolecular π–π interactions (down). | ||
The btp ligand shows a di-η2 coordination and is π–π face-to-face stacked to parent aromatic units by two bridging Ag centres. This repeating unit is further connected via ClO4− counterions affording a chain running along the b axis. Additional intermolecular π–π interactions provide columnar aromatic stacking of the argentocyclophane motifs. Noteworthy, the electrical conductivity of a compressed pellet of [Ag0.5(btp)0.5(ClO4)0.5] with a standard silver coated two-probe method revealed the highest value of all characterised organosilver complexes with σ = 1.32 S cm−1 at room temperature.
In 2001 the same authors reported three new complexes of silver(I) with benzopyrene and two of them were found to be semiconductors.134 Single yellow crystals of [Ag2(dbchry)(CF3SO3)2][Ag2(toluene)2(CF3SO3)]n were obtained combining Ag(CF3SO3) and dibenzo[b,def]chrysene (dbchry). Its crystal structure can be described as a 2D layer presenting cation–π interactions of Ag(I) and dbchry together with a 1D polymer formed by Ag(I) and solvent toluene molecules (Fig. 42).
![Crystal structure of [Ag2(dbchry)(CF3SO3)2][Ag2(toluene)2(CF3SO3)]n co-crystallised species.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f42.gif) | ||
| Fig. 42 Crystal structure of [Ag2(dbchry)(CF3SO3)2][Ag2(toluene)2(CF3SO3)]n co-crystallised species. | ||
On the border limits of the scope of this review stands the stacking polymer formed by the reaction of Ag(ClO4) with benzo[e]pyrene (bpyr): [Ag4(bpyr)4(p-xylene)(ClO4)4]. This polymer presents a tetranuclear core formed by two crystallographically different Ag(I) centres, two symmetry related bpyr molecules sandwiching a p-xylene solvent molecule and two pairs of ClO4− counterions interacting via intermolecular π–π contacts through extra pendant bpyr ligands, leading to a 2D sheet structure together with a 1D columnar stacking (Fig. 43).
![Solid state structure of the tetranuclear species [Ag4(bpyr)4(p-xylene)(ClO4)4] (left) and arrangement of the stacking polymer (right).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f43.gif) | ||
| Fig. 43 Solid state structure of the tetranuclear species [Ag4(bpyr)4(p-xylene)(ClO4)4] (left) and arrangement of the stacking polymer (right). | ||
The room temperature electrical conductivity values, measured with the two probe method, are 6.3 × 10−2 S cm−1 in [Ag4(bpyr)4(p-xylene)(ClO4)4] and 5.8 × 10−2 S cm−1 in [Ag2(dbchry)(CF3SO3)2][Ag2(toluene)2(CF3SO3)]n.
Extending their postulate to multiphenyl dienes, Munakata et al. reported another semiconducting polymer in 2003.134trans,trans-1,4-Diphenyl-1,3-butadiene (dpbd) was reacted with AgClO4 to yield [Ag(dpbd)(ClO4)] as colourless crystals. Its structure shows a trigonal local environment for the silver atom formed by η1-bonds to two phenyl fragments and an oxygen atom from a ClO4− group. The dpbd coordinates two metal atoms building a 1D W-shaped infinite chain that stacks forming a herringbone (Fig. 44). Its electrical conductivity shows a room temperature σ value of 2.1 × 10−7 S cm−1.
![Waved infinite chains of [Ag(dpbd)(ClO4)].](/image/article/2012/CS/c1cs15092h/c1cs15092h-f44.gif) | ||
| Fig. 44 Waved infinite chains of [Ag(dpbd)(ClO4)]. | ||
The last work published by Munaksta's group concerning silver metal–organic frameworks relates to another multiphenyl diene ligand.135 The new complexes [Ag2(bmsb)(ClO4)2] and [Ag2(bmsb)(H2O)4](BF4)2 (bmsb = 1,4-bis(methylstyryl)benzene) displayed σ values of 1.0 × 10−6 and 1.2 × 10−2 S cm−1, respectively (Fig. 45 and 46). Attending to their crystal structure, the presence of columnar stacking in the latter but not in the former might be responsible for the enhanced electrical conductivity observed.
In the solid state structure of [Ag2(bmsb)(H2O)4](BF4)2 (Fig. 45) the ligand face-to-face arrangement is secured via double silver μ-tetra-η2 bridges forming a metallocyclophane moiety with a chain structure. Strong contacts, both intra and interchain, generate a supramolecular 2D network.
2 where both intra and interchain π–π contacts extend in columnar stacking (top). Portion of the molecular structure (bottom).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f45.gif) | ||
| Fig. 45 Supramolecular 2D structure observed on [Ag2(bmsb)(H2O)4](BF4)2 where both intra and interchain π–π contacts extend in columnar stacking (top). Portion of the molecular structure (bottom). | ||
![Pseudo-cyclophane moiety observed in the molecular structure of [Ag2(bmsb)(ClO4)2] (left) and single sheet component along the ab plane (right).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f46.gif) | ||
| Fig. 46 Pseudo-cyclophane moiety observed in the molecular structure of [Ag2(bmsb)(ClO4)2] (left) and single sheet component along the ab plane (right). | ||
On the other hand, a cyclophane motif is also observed for [Ag2(bmsb)(ClO4)2] (Fig. 46). These cyclic structures give rise in the solid state to a supramolecular 2D polymer constructed by 1D chains where equivalent silver(I) centres bond to both phenyl and vinylene fragments of bmsb. The metal coordination sphere is completed with oxygen atoms of different ClO4− groups. Moreover, no π–π intrachain interactions are observed whereas close interchain contacts of 3.42 Å between phenyl and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C are present.
C are present.
4. Two dimensional coordination polymers
4.1. 2D polymers with inorganic linkers
Mixed-valence dinuclear species have proved to be resourceful building-blocks for the preparation of coordination polymers with notable electrical properties. Thus, Kawamura and colleagues have been exploiting the [Rh2(acam)4(H2O)2] (acam = acetamidato) dimer in order to generate networks with different dimensionalities. In 2003 they constructed a 2D honeycomb-like frame employing [Rh2(acam)4] as its neutral and radical cation (Rh24+ and Rh25+) forms linked by μ3-Cl halides.136 The reaction between [Rh2(acam)4(H2O)2]·6H2O or [Rh2(acam)4(H2O)2]ClO4 with excess NaCl gave [{Rh2(acam)4}3(μ3-Cl)2]n·4nH2O. Higher concentration of the radical salt resulted in the formation of the previously reported [Rh2(acam)4Cl]n crystals (Fig. 11). The structure of [{Rh2(acam)4}3(μ3-Cl)2]n·4nH2O consists of honeycomb layers in the bc plane (Fig. 47). The layers are formed by hexagons containing six [Rh2(acam)4] dimers in the sides and six Cl− ions in the corners. Attending to the Rh–O and Rh–N bond distances, there exists both Rh24+ and Rh25+ units in a 1∶2 ratio, giving rise to an average Rh2(14/3)+ oxidation state. Solvent water molecules are sandwiched between layers forming an intricate hydrogen-bond network. The authors postulated that these hydrogen bonds favour a localization in the metal's oxidation state responsible for the low electrical conductivity of the network, with a value of 2 × 10−7 S cm−1 measured on a pressed pellet at room temperature.
![Honeycomb structure of [{Rh2(acam)4}3(μ3-Cl)2]n·4nH2O, some of the hydrogen atoms have been omitted for clarity.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f47.gif) | ||
| Fig. 47 Honeycomb structure of [{Rh2(acam)4}3(μ3-Cl)2]n·4nH2O, some of the hydrogen atoms have been omitted for clarity. | ||
Following their studies with the Rh2(acam)4 building block, Fuma and Ebihara prepared in 2006 a 2D square-sheet comprising this unit linked with ReO4− ions.137 This was the first example of a perrhenate ion bonded to four metal complexes. The structure of [{Rh2(acam)4}2(μ4-ReO4)] consists of equivalent Rh2 units linked by ReO4− ions as stacked square sheets in the bc plane. There are amido-NH and amido-O inter-layer hydrogen bonds (Fig. 48). The intra-dimer Rh–Rh distance is 2.4047(6) Å. The network was assembled mixing equimolar amounts of [Rh2(acam)4(H2O)2]·6H2O and [Rh2(acam)4(H2O)2]ClO4 with excess NH4ReO4, a common procedure for these type of arrays. The electrical conductivity of [{Rh2(acam)4}2(μ4-ReO4)] as a pressed pellet at room temperature was 8.9 × 10−6 S cm−1 (Table 14).
![Top (left) and side (right) view of the square sheet formed by [{Rh2(acam)4}2(μ4-ReO4)]. Only hydrogen atoms involved in hydrogen bonding are displayed.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f48.gif) | ||
| Fig. 48 Top (left) and side (right) view of the square sheet formed by [{Rh2(acam)4}2(μ4-ReO4)]. Only hydrogen atoms involved in hydrogen bonding are displayed. | ||
4.2. 2D polymers with nitrogen-containing ligands
P. Amo-Ochoa et al. have synthesized a 2D multifunctional mixed-valence Cu(I/II) coordination polymer [Cu2Br(IN)2]n (IN = isonicotinate = py–COO−) presenting conducting, magnetic and luminescent properties (Fig. 49).![View of the copper environments in [Cu2Br(IN)2]n (top left). View of a single layer framework (top right). Superposition of layers along the a axis (bottom).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f49.gif) | ||
| Fig. 49 View of the copper environments in [Cu2Br(IN)2]n (top left). View of a single layer framework (top right). Superposition of layers along the a axis (bottom). | ||
Its structure consists of pairs of copper atoms bridged by two isonicotinato ligands and a bromide ion. The fourth coordination site of the copper ion is occupied by an additional terminal N-coordinated isonicotinato ligand. The geometry of this entity forms four-arm units which propagate along the bc plane in a 2D-framework (Fig. 49). Stacking of layers along the a axis results in a 3D organization with π-stacking between isonicotinato aromatic rings (3.36 Å). The electrical characterisation of the crystals, carried out by the two contacts method, showed semiconductor behaviour with a conductivity value of 1.2 × 10−5 S cm−1 at room temperature.
The singular crystal structure and its multifunctional features prompted the authors to exfoliate the crystals by means of ultrasounds. In such a way single-layers were isolated on mica and characterized by atomic force microscopy and XPS. This result suggests that 2D-coordination polymers can be considered as alternative materials to graphene.138
Rao et al. have studied supramolecular assemblies of cyanuric acid (CA) involving both hydrogen bonding and metal ion-coordination capabilities. Thus, during the course of their investigations, Rao et al. isolated a novel silver compound: Ag2(CA) (Fig. 50), whose structure presents two dimensional Ag sheets with the cyanuric acid molecules in the interlayer space, forming linear hydrogen bonding.139
 | ||
| Fig. 50 Structure of Ag2·CA (CA = cyanuric acid) showing the Ag sheets and CA chains. | ||
The presence of two-dimensional Ag sheets is expected to give rise to anisotropic conductivity. Accordingly, the values of the room temperature dc conductivity, parallel and perpendicular to the Ag sheets (bc plane) are ca. 5 × 10−3 and ca. 2 × 10−5 S cm−1, respectively. The conductivity along the sheets is temperature-independent down to 15 K (Table 15).
4.3. 2D polymers containing organosulfur ligands
As it has already been described (Section 3.4) the sulfur rich 4,5-ethylenedithio-1,3-dithiole-2-thione (C5H4S5) presents different available sites that can coordinate to metal ions. Furthermore, thanks to its capacity to act as an electron-donor, it has been considered as a good candidate for the synthesis of donor–acceptor metallic systems. As mentioned before (Section 3.4), Jie Dai et al. used this ligand to prepare 1D coordination polymers and, interestingly, a modification of the reaction conditions between C5H4S5 and AgCF3SO3 lead to the synthesis of the 2D-coordination polymer [Ag(C5H4S5)CF3SO3]n.104 In this polymer, there are two independent silver(I) atoms (types A and B), both showing a five coordinate environment. Type A silver atoms have a S4O donor set and are coordinated by four thiocarbonyl sulfur atoms from four different C5H4S5 molecules and one oxygen atom from a triflate anion (CF3SO3−). Type B silver atoms are surrounded by a S2O3 donor set from two thioether sulfur atoms and three oxygen atoms of triflate anions (Fig. 51).![Schematic representation of the two dimensional network of [Ag(C5H4S5)CF3SO3]n.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f51.gif) | ||
| Fig. 51 Schematic representation of the two dimensional network of [Ag(C5H4S5)CF3SO3]n. | ||
[Ag(C5H4S5)CF3SO3]n presents short S⋯S distances, indicative of an efficient intermolecular π–π orbital interaction, usually related with an increase in the electrical conductivity. Since the mixed-valence oxidation state (partial-oxidation, reduction, or charge-transfer) is a key feature in many conducting compounds, this polymer was partially oxidized by iodine-doping. The studies of the electrical properties of this material showed that although the coordination polymer is an insulator with σ < 10−12 S cm−1 (measured in pressed pellets at 300 K and using the two probe method, Table 16), the iodine-doped product behaves as a semiconductor with a room temperature conductivity of 1.5 × 10−4 S cm−1.104
| Compounds | Conductivity/S cm−1 | Ref. |
|---|---|---|
| C5H4S5 = 4,5-ethylenedithio-1,3-dithiole-2-thione, C4N2H3S = pyrimidine-2-thiol, C5H4NS = pyridine-2-thiolate (PyS)−, HT = 4-hydroxythiophenolate. | ||
| [Ag(C5H4S5)CF3SO3]n | <10−12 | 104 |
| Ni2(C4N2H3S)4 | 5 × 10−3 | 140 |
| [{Ag(C5H4NS)}]n | 2.04 × 10−5 | 141 |
| CuHT | 120 | 142 |
In an attempt to solve the insolubility problems found in metal thiolate compounds, some authors have used pyrimidyl nitrogen-containing thiols as ligands to generate crystalline networks with metal ions such as Ag(I) or Ni(II). Two examples are reported in the literature with these kind of ligands.140 The first one describes a 2D-coordination polymer obtained by a solvothermal reaction between pyrimidine-2-thiol and Ni(OAc)2 in DMF/H2O.143 The lamellar structure of formula Ni2(C4N2H3S)4 (Fig. 52) presents electrical conductivity and ferromagnetic interactions between the Ni(II) centres. The conductivity measurement (in pressed pellets) of 5 × 10−3 S cm−1 at room temperature increases upon heating, indicating typical semiconductor behaviour. The semiconducting properties may be attributed to the presence of an interconnected array of nickel(II) ions with pyrimidine presenting some Ni(II)-pyridine ring interactions.
 | ||
| Fig. 52 View of a layer of compound Ni2(C4N2H3S)4. | ||
The second example with a pyrimidyl nitrogen-containing thiol is an interesting 2D-polymer synthesized by Su et al.141 using pyridine-2-thiolate as building-block. This polymer of formula [{Ag(C5H4NS)}]n (C5H4NS = pyridine-2-thiolate (PyS−)) shows a graphite-like array of silver(I) ions (Fig. 53). Its structure consists of sheets where the silver atoms are connected by PyS− ligands to generate layers with the pyridyl groups of the PyS− ligands inserted into the interlayer region (the interlayer distance is 17.17 Å). Each PyS− ligand acts as a μ3 bridge to link two silver atoms through a S atom, and a third silver atom through a N atom. Each silver atom is coordinated by two S atoms and one N atom. The electrical conductivity measured in a pressed pellet is 2.04 × 10−5 S cm−1 at 298 K, with a semiconducting behaviour that was attributed to the presence of Ag–Ag interactions.141
![A view of the lamella structure of [{Ag(C5H4NS)}]n (left). Graphite-like structure formed by Ag metal centres (right).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f53.gif) | ||
| Fig. 53 A view of the lamella structure of [{Ag(C5H4NS)}]n (left). Graphite-like structure formed by Ag metal centres (right). | ||
On a subsequent study, two years later Su et al. described different coordination modes of the pyridine-2-thiolate (and/or thione) (Spy and/or HPyS, respectively) upon silver(I) complexation.144 Several polymers were described and X-ray diffraction analyses of all compounds were collected.
Although precise data on their electrical conductivity were not provided, the σ values found were typical of insulators, with values in the range 10−12 to 10−15 S cm−1 at 25 °C.
Soaking [{Ag(C5H4NS)}]n in DMF for one month gave rise to the isomer [{Ag6(C5H4NS)6}]n. In the absence of water and using MeCN rather than DMF, [{Ag(C5H5NS)2}(BF4)2]n was obtained. In the same way [{Ag(C5H5NS)2}(ClO4)2]n was synthesised from AgClO4 and [{Ag4(C5H5NS)6}(NO3)4]n from AgNO3. The BF4− and ClO4− salts are best described as isostructural cationic metal–metal-chains whereas the NO3− salt forms a 1D pseudo-ladder chain (Fig. 54).
![Crystal structure of isostructural [{Ag(C5H5NS)2}(BF4)2]n and [{Ag(C5H5NS)2}(ClO4)2]n chains (left). Ladder chain observed in the molecular structure of [{Ag4(C5H5NS)6}(NO3)4]n (right). Non-coordinating counterions and H atoms are not shown for clarity.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f54.gif) | ||
| Fig. 54 Crystal structure of isostructural [{Ag(C5H5NS)2}(BF4)2]n and [{Ag(C5H5NS)2}(ClO4)2]n chains (left). Ladder chain observed in the molecular structure of [{Ag4(C5H5NS)6}(NO3)4]n (right). Non-coordinating counterions and H atoms are not shown for clarity. | ||
Another layered polymer was isolated from the reaction between pyridine-2-thiol (HPyS) and AgBF4 in MeOH∶CH3CN (40∶1), in this way [{Ag5(Spy)4(HPyS)}BF4]n was assembled (Fig. 55).
![2D lamellar structure of [{Ag5(Spy)4(HPyS)}BF4]n.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f55.gif) | ||
| Fig. 55 2D lamellar structure of [{Ag5(Spy)4(HPyS)}BF4]n. | ||
The reaction of K2(i-mnt) with HPyS and AgNO3 furnished the polymeric chain [Ag4(μ4-i-mnt)2(μ-HPyS)2(μ-HPyS)4/2]n (Fig. 56). Finally, when HPyS, NaS2CNEt2 and AgNO3 were combined another polymeric 1D species resulted, [Ag4(μ3-S2CNEt2)2(μ2-SPy)4/2]n (Fig. 56).
![The 1D chains observed in the solid state structures of [Ag4(μ4-i-mnt)2(μ-HPyS)2(μ-HPyS)4/2]n (top) and [Ag4(μ3-S2CNEt2)2(μ2-SPy)4/2]n (bottom).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f56.gif) | ||
| Fig. 56 The 1D chains observed in the solid state structures of [Ag4(μ4-i-mnt)2(μ-HPyS)2(μ-HPyS)4/2]n (top) and [Ag4(μ3-S2CNEt2)2(μ2-SPy)4/2]n (bottom). | ||
Recently, Che et al. have reported an unprecedented 2-D Cu–S coordination polymer formed by self-assembly of Cu(I) ions with 4-hydroxythiophenol. Its X-ray structure solved by using powder X-ray diffraction data agrees with the lamellar architecture of CuHT (HT = 4-hydroxythiophenolate). The self-assembled coordination network displays ionic behaviour with a bulk electrical conductivity value measured on pellets at room temperature of 120 S cm−1.142
5. Three dimensional coordination polymers
5.1. 3D coordination polymers with inorganic linkers
In a short communication in 2004, Kawamura et al. reported the isolation of a 3D diamond-like network built with Rh2(acam)4 paddle-wheel complexes connected via μ4-iodide bridges.145 The slow evaporation of an aqueous solution with [Rh2(acam)4(H2O)2]·6H2O, [Rh2(acam)4(H2O)2]ClO4 and NaI produced the [{Rh2(acam)4}2I]n·6nH2O network (Fig. 57).![Diamond network of [{Rh2(acam)4}2I]n·6nH2O (top left), only Rh and I centres are represented for clarity. Arrangement of [{Rh2(acam)4}2I]n network (bottom left) showing the unusual μ4-I coordination mode. Solvent water molecules are located around the iodine site (top right). Electrical conductivity oscillation observed for a pressed pellet of [{Rh2(acam)4}2I]n during dehydration and rehydration cycles (bottom right). (Data collected from ref. 145; reproduced by permission of the American Chemical Society.)](/image/article/2012/CS/c1cs15092h/c1cs15092h-f57.gif) | ||
| Fig. 57 Diamond network of [{Rh2(acam)4}2I]n·6nH2O (top left), only Rh and I centres are represented for clarity. Arrangement of [{Rh2(acam)4}2I]n network (bottom left) showing the unusual μ4-I coordination mode. Solvent water molecules are located around the iodine site (top right). Electrical conductivity oscillation observed for a pressed pellet of [{Rh2(acam)4}2I]n during dehydration and rehydration cycles (bottom right). (Data collected from ref. 145; reproduced by permission of the American Chemical Society.) | ||
X-Ray diffraction analyses revealed the presence of equivalent Rh2 and μ4-I units arranged on a diamond framework with water solvent molecules occupying the interstitial holes. There are inter-dimer hydrogen bonds formed between all amido-NH and amido-O that extend to the co-crystallised water molecules. Interestingly, the solvent molecules play a key role in the electrical conductivity of the polymer, a pressed pellet of [{Rh2(acam)4}2I]n·6nH2O displayed a room temperature electrical conductivity of 1.4 × 10−3 S cm−1 that decreases to 7.0 × 10−9 S cm−1 when the sample dehydrates (Table 17). Exposing compound to moisture resulted in both the recovery of the [{Rh2(acam)4}2I]n·6nH2O formulation, as inferred by X-ray powder diffraction analyses, and an upturn in the electrical conductivity which scopes over a range of 105 during dehydration–rehydration cycling. The authors claimed that the diamond structure persisted over those cycles.
5.2. 3D coordination polymers with organic linkers
One of the most notable examples of conductivity in coordination polymers are the compounds Cu(R1,R2-DCNQI)2, where R1, R2 = H, Cl, Br, I, Me, OMe and DCNQI = N,N′-dicyanoquinonediimine. Their structures contain seven interpenetrating diamond nets, with infinite stacks of DCNQI radical anions (Fig. 58). The copper atoms have an average oxidation state of +1.33 and the series show metal-like conductivities. The dimethyl derivative, for example, has a conductivity of 103 S cm−1 at room temperature.146![Solid-state structure of [Cu(2,5-dimethyl-DCBQI)2]n projected over the ab plane.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f58.gif) | ||
| Fig. 58 Solid-state structure of [Cu(2,5-dimethyl-DCBQI)2]n projected over the ab plane. | ||
As we already mentioned in Section 3.4, the solvothermal synthetic methods may give rise to new interesting supramolecular structures. For example, reaction of CuI with 4,4′-dipyridyl disulfide (dpds) and oxalic acid at three different time periods allowed the isolation of three Cu/I/S-based coordination polymers, [Cu6(μ-4-SpyH)4I6]n, {[Cu2(μ-I)(μ-4-SpyH)3]I}n, and [Cu5(μ-4-SpyH)7(μ-I)I4]n112 (the two last ones are one dimensional polymers that were already described in Section 3.4). The first complex [Cu6(μ-4-SpyH)4I6]n has an adamantine-type Cu6S4 core. The four equatorial Cu(I) atoms are tetrahedrally coordinated by two bridging I− atoms and two S atoms from two 4-SpyH ligands while the two apical Cu(I) centres are trigonally coordinated by one terminal iodide and two sulfur atoms from one 4-SpyH ligand. Each [Cu6S4] core acts as a tetrahedral 4-connecting node and is linked with four other equivalent [Cu6S4] cores through four pairs of iodide bridges coordinated to the equatorial Cu(I) ions, forming a 3D diamond-like network (Fig. 59). All these features suggest that this 3D network may not be robust enough to survive at solvothermal conditions and may dissociate into other more stable species if the time of this reaction is deliberately prolonged. The conductivity of the crystal increased exponentially from 1.97 × 10−9 S cm−1 to 6.32 × 10−7 S cm−1, upon heating from 293 to 443 K.
![Several views of the structure of [Cu6(μ-4-SpyH)4I6]n.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f59.gif) | ||
| Fig. 59 Several views of the structure of [Cu6(μ-4-SpyH)4I6]n. | ||
Cao et al. reported in 2002 a novel 3D polymer constructed from silver(I) metal centres and benzene-1,3,5-tricarboxylic acid (H3btc).147 Slow diffusion of methanolic AgNO3 over aqueous H3btc gave rise to the formation of the polymeric [{Ag(H2btc)2}{Ag2(Hbtc)}]n compound. Its crystal structure shows the existence of two interpenetrated distinct building blocks: Ag8(Hbtc)12/3 (A unit) and Ag2(H2btc)2 (B unit) (Fig. 60). The intricate structure can be rationalised as distorted ladder-like chains of Ag atoms (Fig. 61) bridged by both H2btc−and Hbtc2− spacers. Remarkably, the whole structure is sustained by direct Ag–Ag interactions.
![Structure of [{Ag(H2btc)2}{Ag2(Hbtc)}]n showing the A units Ag8(Hbtc)12/3 (up) and the B units Ag2(H2btc)2 (down).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f60.gif) | ||
| Fig. 60 Structure of [{Ag(H2btc)2}{Ag2(Hbtc)}]n showing the A units Ag8(Hbtc)12/3 (up) and the B units Ag2(H2btc)2 (down). | ||
![Silver chain network formed by direct Ag–Ag interactions in [{Ag(H2btc)2}{Ag2(Hbtc)}]n.](/image/article/2012/CS/c1cs15092h/c1cs15092h-f61.gif) | ||
| Fig. 61 Silver chain network formed by direct Ag–Ag interactions in [{Ag(H2btc)2}{Ag2(Hbtc)}]n. | ||
The electrical conductivity, measured on pressed pellet showed a σ value of 1.06 × 10−6 S cm−1 at room temperature that increased with increasing temperature in a semiconducting regime.
In 2004, the group of Wu described a new 3D silver organodiphosphonate framework: [Ag4(O3PCH2CH2PO3)],148 with a zeolite-like structure sustaining a sub-lattice with direct Ag–Ag contacts (Fig. 62). The electrical conductivity of a polycrystalline pellet showed maximum values at 170 °C of ca. 7 × 10−7 (Fig. 63).
![The zeolite-like arrangement of [Ag4(O3PCH2CH2PO3)] showing its channel structure (left). Substructural network constructed through Ag–Ag bonding interactions (right).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f62.gif) | ||
| Fig. 62 The zeolite-like arrangement of [Ag4(O3PCH2CH2PO3)] showing its channel structure (left). Substructural network constructed through Ag–Ag bonding interactions (right). | ||
![Temperature dependence of the electrical conductivity found for [Ag4(O3PCH2CH2PO3)] (Reproduced from reference 148 with permission of Elsevier).](/image/article/2012/CS/c1cs15092h/c1cs15092h-f63.gif) | ||
| Fig. 63 Temperature dependence of the electrical conductivity found for [Ag4(O3PCH2CH2PO3)] (Reproduced from reference 148 with permission of Elsevier). | ||
Table 18 collects the conductivity data reported on 3D-CPs with organic linkers.
6. Conclusions and perspectives
In the past few decades, the synthesis and characterisation of low-dimensional crystalline conducting materials has attracted much interest due to their highly tuneable structures and their potential applications in electronics and optoelectronic devices.149Coordination polymers are currently one of the hottest topics in Inorganic and Supramolecular Chemistry (see for instance the excellent review by C. Janiak that is now receiving over 200 citations a year and the special volume devoted in this journal to MOFs).150 There is no doubt of the technological impact expected for coordination polymers within the next coming years, in fact some of them are already being used in industrial applications.
Despite the electrical conductivity not being the prime focus of research, several conductive coordination polymers have been reported in the last few decades. We have shown that this tendency seems to be changing due to the current interest in the development of highly conducting coordination polymers aimed for novel technological applications: porous electrode for batteries, fuel cells, capacitors, sensors, nanomaterials, etc.6,7,151 Thus, from the pioneering works of Interrante et al. in the 70's on the MX chains and of Bellitto et al. in the 80's on the MMX chains, the number of conducting CPs of these and other families has shown an exponential increase with time. This huge rise is the logical consequence of the increase in the number of used metals and ligands (either as linkers or as chelating units). Thus, besides the initial examples with Pt, Pd and Ni, here we have seen that many other metals such as Ag, Rh, Ru, Cu, Au, Fe,… can also be used to prepare CPs. The number of bridging ligands has also increased from the initial simple halides to more complex ones such as CN−, NO3−, SCN−, SeCN−, N3−, pyrazines, nucleobases, diverse S-containing ligands and aromatic hydrocarbons,… The chelating ligands stabilising the structure and completing the coordination environment of the metal atoms have also increased from the initial alkyldiamines (en, tn, bn and chxn) and carboxylates to more complex ones such as dithiomalonamide, thioureas, thioamides, dibenzyl sulfide, diphosphites, dithiocarboxylates, acetamides, TTF-type donors, dithiolates,…
In this review we have tried to summarize the state-of-the-art in this topic collecting the vast number of compounds structurally and electrically characterized that were disperse in the literature (in some cases there were no clear relationship between the reported examples). This compilation has also shown a large dispersion of the experimental conductivity values (from highly conducting CPs with room temperature conductivities above 102 S cm−1 to very poor semiconductors with room temperature conductivities below 10−14 S cm−1). The experimental setups and conditions in the conductivity measurements are also very disperse since they depend in many cases on the availability of large enough single crystals and on the conductivity values and thermal behaviours. Thus, there are conductivity measurements made on single crystals, pressed pellets, thin films or even nanowires with just a few molecular chains. The thermal variations cover a wide range of temperatures from liquid helium to values well above room temperature (even above 440 K) and there are also measurements done with different applied pressures up to 250 kbar. Finally, although this is not expected to have an influence on the reported values since the resistivities are usually quite high, there are measurements done with the two and four contact methods. All these experimental differences have also to be taken into account when comparing values and behaviours of the electrical conductivity in these CPs. More additional experimental studies need to be done. Indeed this is necessary to gain understanding towards the redesign of better conductors. In any case the aspects collected here will need a further revision with additional new incoming experiments.
Moreover, the data currently available suggest that the number of electrical conductive 1D coordination polymers is higher than 2D- and 3D-CPs. This is probably motivated by the interest in the preparation of low-dimensional materials but in fact at this point we cannot conclude about whether this is just experimental evidence or real data. We believe that along the next years the number of CPs will not depend on the dimensionality but rather on other more important aspects related with the structure and composition.
Despite the dispersion of data reported, some insights can be inferred. Thus, we and others agree that the mixed-valence oxidation state of the metal ions seems to affect the electronic aspects including electrical conductivity. Probably one of the most relevant examples in CPs is the case of the MMX chains with platinum and dithiocarboxylate linkers.
The metal-to-metal direct interactions are important but not essential for conduction. In this respect it is obvious that short metal-to-metal distances may give rise to high conductivity when the metals are close enough to form a metal–metal bond. Nevertheless, when the metal–metal distance increases, the conductivity decreases but does not vanish completely.
Other aspects concerning the specific use of metal ions and ligands in the design of CPs are of high relevance in terms of conductivity. In fact the experimental data already available suggest that CPs containing metals such as platinum, nickel, silver and copper combined with sulfur- and halides-containing ligands may be a good strategy.
Few theoretical calculations have been used to explain experimental results and conducting mechanism.1,83,88,100–102 Albeit, we need to go one step forward in order to predict successful strategies leading to the design of new CPs.
Acknowledgements
This work was supported by the MICINN (MAT2010-20843-C02-01, ACI2009-0969, S-0505/MAT/0303), Comunidad de Madrid (S-0505/MAT/0303), Generalitat Valenciana (PROMETEO/2009/095) and EU (FP6-029192).References and notes
- L. Welte, A. Calzolari, R. di Felice, F. Zamora and J. Gómez-Herrero, Nat. Nanotechol., 2010, 5, 110–115 CrossRef CAS.
- R. Mas-Balleste, O. Castillo, P. J. S. Miguel, D. Olea, J. Gomez-Herrero and F. Zamora, Eur. J. Inorg. Chem., 2009, 2885–2896 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS.
- D. Tanaka, K. Nakagawa, M. Higuchi, S. Horike, Y. Kubota, L. C. Kobayashi, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 3914–3918 CrossRef CAS.
- A. Carne, C. Carbonell, I. Imaz and D. Maspoch, Chem. Soc. Rev., 2011, 40, 291–305 RSC.
- R. Mas-Balleste, J. Gomez-Herrero and F. Zamora, Chem. Soc. Rev., 2010, 39, 4220–4233 RSC.
- A. U. Czaja, N. Trukhan and U. Muller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- L. J. van der Pauw, Philips Res. Rep., 1958, 13, 1–9 Search PubMed.
- L. J. van der Pauw, Philips Tech. Rev., 1958, 20, 220–224 Search PubMed.
- H. C. Montgome, J. Appl. Phys., 1971, 42, 2971–2975 CrossRef.
- S. Roth and D. Carrol, One Dimensional Metals, Willey-VCH, Weinheim, 2004 Search PubMed.
- A. J. Epstein, H. Rommelmann, M. Abkowitz and H. W. Gibson, Mol. Cryst. Liq. Cryst., 1981, 77, 81–96 CrossRef CAS.
- C. Bellitto, G. Dessy and V. Fares, Inorg. Chem., 1985, 24, 2815–2820 CrossRef CAS.
- N. F. Mott and E. A. Davis, Electronic Proceses in Non-Crystalline Materials, Clarendon Press, Oxford, 2nd edn, 1979 Search PubMed.
- M. Nechtschein, F. Devreux, F. Genoud, E. Vieil, J. M. Pernaut and E. Genies, Synth. Met., 1986, 15, 59–78 CrossRef CAS.
- J. L. Bredas and G. B. Street, Acc. Chem. Res., 1985, 18, 309–315 CrossRef CAS.
- IUPAC Compendium of Chemical Terminology, 2006.
- R. Krishnan and V. Srivastava, Phys. Rev. B: Condens. Matter, 1999, 59, R12747–R12750 CrossRef CAS.
- R. Krishnan and V. Srivastava, Microelectron. Eng., 2000, 51–52, 317–326 CrossRef CAS.
- D. J. Thouless, Phys. Rev. Lett., 1977, 39, 1167–1169 CrossRef CAS.
- J. Bernasco, P. Bruesch, D. Kuse and H. R. Zeller, J. Phys. Chem. Solids, 1974, 35, 145–157 CrossRef.
- L. B. Coleman, M. J. Cohen, D. J. Sandman, F. G. Yamagish, A. F. Garito and A. J. Heeger, Solid State Commun., 1973, 12, 1125–1132 CrossRef CAS.
- J. Ferraris, V. Walatka, Jh. Perlstei and D. O. Cowan, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef CAS.
- F. Wudl, G. M. Smith and E. J. Hufnagel, Chem. Commun., 1970, 1453–1454 RSC.
- R. E. Peierls, Quantum Theory of Solids, Clarendon Press, Oxford, 1955 Search PubMed.
- S. Inagaki and H. Fukuyama, J. Phys. Soc. Jpn., 1983, 52, 2504–2513 CrossRef CAS.
- S. Inagaki and H. Fukuyama, J. Phys. Soc. Jpn., 1983, 52, 3620–3629 CrossRef CAS.
- J. W. Bray, L. W. Interrante, I. S. Jacobs and J. C. Bonner, Extended Linear Chain Compounds, Plenum Press, 1983 Search PubMed.
- J. M. Williams, J. R. Ferraro, R. J. Thorn, K. D. Carlson, U. Geiser, H. H. Wang, A. M. Kini and M. H. Whangbo, Organic Superconductors (Including Fullerenes). Synthesis, Structure, Properties and Theory, Prentice Hall, Englewood Cliffs, NJ, 1992 Search PubMed.
- S. Kagoshima, R. Kato, H. Fukuyama, H. Seo and H. Kino, Advances in Synth. Met. Twenty years of Porgress in Science and Technology, Elsevier, 1999 Search PubMed.
- E. Coronado, J. R. Galan-Mascaros, C. Gimenez-Saiz, C. J. Gomez-Garcia and S. Triki, J. Am. Chem. Soc., 1998, 120, 4671–4681 CrossRef CAS.
- J. R. Galanmascaros, C. Gimenezsaiz, S. Triki, C. J. Gomezgarcia, E. Coronado and L. Ouahab, Angew. Chem., Int. Ed., 1995, 34, 1460–1462 CrossRef CAS.
- C. J. Gomezgarcia, C. Gimenezsaiz, S. Triki, E. Coronado, P. Lemagueres, L. Ouahab, L. Ducasse, C. Sourisseau and P. Delhaes, Inorg. Chem., 1995, 34, 4139–4151 CrossRef CAS.
- C. J. Gomezgarcia, L. Ouahab, C. Gimenezsaiz, S. Triki, E. Coronado and P. Delhaes, Angew. Chem., Int. Ed., 1994, 33, 223–226 CrossRef.
- P. LeMagueres, L. Ouahab, N. Conan, C. J. GomezGarcia, P. Delhaes, J. Even and M. Bertault, Solid State Commun., 1996, 97, 27–32 CrossRef CAS.
- M. Mitsumi, H. Goto, S. Umebayashi, Y. Ozawa, M. Kobayashi, T. Yokoyama, H. Tanaka, S. Kuroda and K. Toriumi, Angew. Chem., Int. Ed., 2005, 44, 4164–4168 CrossRef CAS.
- Y. Nagao, M. Fujishima, R. Ikeda, S. Kanda and H. Kitagawa, Synth. Met., 2003, 133, 431–432 CrossRef.
- Y. Nagao, T. Kubo, K. Nakasuji, R. Ikeda, T. Kojima and H. Kitagawa, Synth. Met., 2005, 154, 89–92 CrossRef CAS.
- T. Yamada, M. Sadakiyo and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 3144–3145 CrossRef CAS.
- H. Tanino and K. Kobayashi, J. Phys. Soc. Jpn., 1983, 52, 1446–1456 CrossRef CAS.
- H. Kishida, H. Matsuzaki, H. Okamoto, T. Manabe, M. Yamashita, Y. Taguchi and Y. Tokura, Nature, 2000, 405, 929–932 CrossRef CAS.
- L. V. Interrante and K. W. Browall, Inorg. Chem., 1974, 13, 1162–1166 CrossRef CAS.
- L. V. Interrante, K. W. Browall and F. P. Bundy, Inorg. Chem., 1974, 13, 1158–1162 CrossRef CAS.
- L. V. Interrante and F. P. Bundy, J. Inorg. Nucl. Chem., 1977, 39, 1333–1337 CrossRef CAS.
- G. W. Watt and R. E. McCarley, J. Am. Chem. Soc., 1957, 79, 3315–3317 CrossRef CAS.
- G. W. Watt and R. E. McCarley, J. Am. Chem. Soc., 1957, 79, 4585–4589 CrossRef CAS.
- G. L. Johnson, Inorg. Synth., 1966, 8, 242–246 CrossRef CAS.
- Y. Hamaue, R. Aoki, M. Yamashita and S. Kida, Inorg. Chim. Acta, 1981, 54, L13–L14 CrossRef CAS.
- M. Yamashita, N. Matsumoto and S. Kida, Inorg. Chim. Acta, 1978, 31, L381–L382 CrossRef CAS.
- M. Haruki, M. Tanaka and S. Kurita, Synth. Met., 21, 373–378 CrossRef CAS.
- M. Yamashita, I. Murase, T. Ito, Y. Wada, T. Mitani and I. Ikemoto, Bull. Chem. Soc. Jpn., 1985, 58, 2336–2339 CrossRef CAS.
- M. Yamashita and S. Takaishi, Bull. Chem. Soc. Jpn., 2006, 79, 1820–1833 CrossRef CAS.
- M. Yamashita, Y. Nonaka, S. Kida, Y. Hamaue and R. Aoki, Inorg. Chim. Acta, 1981, 52, 43–46 CrossRef CAS.
- M. Yamashita, T. Manabe, K. Inoue, T. Kawashima, H. Okamoto, H. Kitagawa, T. Mitani, K. Toriumi, H. Miyamae and R. Ikeda, Inorg. Chem., 1999, 38, 1894–1899 CrossRef CAS.
- M. Yamashita, E. Tsuruta, K. Inoue, T. Furuta, H. Okamoto, T. Mitani, K. Toriumi, H. Ohki and R. Ikeda, Synth. Met., 1993, 56, 3461–3468 CrossRef CAS.
- M. Yamashita, T. Ishii, H. Matsuzaka, T. Manabe, T. Kawashima, H. Okamoto, H. Kitagawa, T. Mitani, K. Marumoto and S. Kuroda, Inorg. Chem., 1999, 38, 5124–5130 CrossRef CAS.
- S. Takaishi, Y. Tobu, H. Kitagawa, A. Goto, T. Shimizu, T. Okubo, T. Mitani and R. Ikeda, J. Am. Chem. Soc., 2004, 126, 1614–1615 CrossRef CAS.
- S. Takaishi, M. Yamashita, H. Matsuzaki, H. Okamoto, H. Tanaka, S. I. Kuroda, A. Goto, T. Shimizu, T. Takenobu and Y. Iwasa, Chem.–Eur. J., 2008, 14, 472–477 CrossRef CAS.
- G. C. Papavassiliou and D. Layek, Z. Naturforsch., B: J. Chem. Sci., 1982, 37, 1406–1409 Search PubMed.
- G. C. Papavassiliou and D. Layek, Z. Naturforsch., B: J. Chem. Sci., 1982, 37, 1406–1409 Search PubMed.
- N. Matsumoto, M. Yamashita and S. Kida, Bull. Chem. Soc. Jpn., 1978, 51, 2334–2337 CrossRef CAS.
- N. Matsumoto, M. Yamashita and S. Kida, Bull. Chem. Soc. Jpn., 1978, 51, 2334–2337 CrossRef CAS.
- H. Ito, M. Sunata, S.-I. Kuroda, T. Manabe and M. Yamashita, Mol. Cryst. Liq. Cryst., 2002, 379, 285–290 CrossRef CAS.
- P. Arrizabalaga, P. Castan, J. P. Laurent and A. Salesse, Chem. Phys. Lett., 1980, 76, 548–552 CrossRef CAS.
- J. M. Bret, P. Castan and J. P. Laurent, Inorg. Chim. Acta, 1981, 51, 103–107 CrossRef CAS.
- J. M. Bret, P. Castan and J. P. Laurent, J. Chem. Soc., Dalton Trans., 1984, 1975–1980 RSC.
- F. Herrmann, Ber. Dtsch. Chem. Ges., 1905, 38, 2813–2825 CrossRef CAS.
- G. M. P. Smith, J. Am. Chem. Soc., 1922, 44, 1769–1775 CrossRef CAS.
- F. H. Brain, C. S. Gibson, J. A. J. Jarvis, R. F. Phillips, H. M. Powell and A. Tyabji, J. Chem. Soc., 1952, 3686–3694 RSC.
- K. Takahashi and H. Tanino, Chem. Lett., 1988, 641–644 CrossRef CAS.
- K. Takahashi and H. Tanino, Chem. Lett., 1991, 1977–1980 CrossRef CAS.
- C. Bellitto, A. Flamini, L. Gastaldi and L. Scaramuzza, Inorg. Chem., 1983, 22, 444–449 CrossRef CAS.
- I. Shirotani, Y. Inagaki and M. Yamashita, Physics and Chemistry of Organic Superconductors, 1989 Search PubMed.
- H. Kitagawa, N. Onodera, J. S. Ahn, T. Mitani, K. Toriumi and M. Yamashita, Synth. Met., 1997, 86, 1931–1932 CrossRef CAS.
- H. Kitagawa and T. Mitani, Coord. Chem. Rev., 1999, 190–192, 1169–1184 CrossRef CAS.
- M. Mitsumi, T. Murase, H. Kishida, T. Yoshinari, Y. Ozawa, K. Toriumi, T. Sonoyama, H. Kitagawa and T. Mitani, J. Am. Chem. Soc., 2001, 123, 11179–11192 CrossRef CAS.
- M. Mitsumi, K. Kitamura, A. Morinaga, Y. Ozawa, M. Kobayashi, K. Toriumi, Y. Iso, H. Kitagawa and T. Mitani, Angew. Chem., Int. Ed., 2002, 41, 2767–2771 CrossRef CAS.
- M. Mitsumi, S. Umebayashi, Y. Ozawa, K. Toriumi, H. Kitagawa and T. Mitani, Chem. Lett., 2002, 258–259 CrossRef CAS.
- S. Ikeuchi, K. Saito, Y. Nakazawa, A. Sato, M. Mitsumi, K. Toriumi and M. Sorai, Phys. Rev. B: Condens. Matter, 2002, 66, 115110 CrossRef.
- K. Saito, S. Ikeuchi, Y. Nakazawa, A. Sato, M. Mitsumi, T. Yamashita, K. Toriumi and M. Sorai, J. Phys. Chem. B, 2005, 109, 2956–2961 CrossRef CAS.
- A. Guijarro, O. Castillo, L. Welte, A. Calzolari, P. J. S. Miguel, C. J. Gomez-Garcia, D. Olea, R. di Felice, J. Gomez-Herrero and F. Zamora, Adv. Funct. Mater., 2010, 20, 1451–1457 CrossRef CAS.
- S. Ikeuchi, Y. Yamamura, M. Mitsumi, K. Toriumi, H. Saitoh, T. Atake and K. Saito, Chem. Lett., 2009, 1190–1191 CrossRef CAS.
- C. Bellitto, G. Dessy and V. Fares, Inorg. Chem., 1985, 24, 2815–2820 CrossRef CAS.
- M. Yamashita, Y. Wada, K. Toriumi and T. Mitani, Mol. Cryst. Liq. Cryst., 1992, 216, 207–212 CrossRef CAS.
- M. Mitsumi, Y. Yoshida, A. Kohyama, Y. Kitagawa, Y. Ozawa, M. Kobayashi, K. Toriumi, M. Tadokoro, N. Ikeda, M. Okumura and M. Kurmoo, Inorg. Chem., 2009, 48, 6680–6691 CrossRef CAS.
- A. Calzolari, S. S. Alexandre, F. Zamora and R. Di Felice, J. Am. Chem. Soc., 2008, 130, 5552–5562 CrossRef CAS.
- C. M. Che, F. H. Herbstein, W. P. Schaefer, R. E. Marsh and H. B. Gray, J. Am. Chem. Soc., 1983, 105, 4604–4607 CrossRef CAS.
- Z. Yang, T. Fujinami, M. Ebihara, K. Nakajima, H. Kitagawa and T. Kawamura, Chem. Lett., 2000, 1006–1007 CrossRef CAS.
- Z. Yang, M. Ebihara, T. Kawamura, T. Okubo and T. Mitani, Inorg. Chim. Acta, 2001, 321, 97–106 CrossRef CAS.
- Z. Yang, M. Ebihara and T. Kawamura, Inorg. Chim. Acta, 2006, 359, 2465–2471 CrossRef CAS.
- K. Uemura, K. Fukui, H. Nishikawa, S. Arai, K. Matsumoto and H. Oshio, Angew. Chem., Int. Ed., 2005, 44, 5459–5464 CrossRef CAS.
- M. Shieh, M. H. Hsu, W. S. Sheu, L. F. Jang, S. F. Lin, Y. Y. Chu, C. Y. Miu, Y. W. Lai, H. L. Liu and J. L. Her, Chem.–Eur. J., 2007, 13, 6605–6616 CrossRef CAS.
- K. Uemura, K. Fukui, K. Yamasaki and K. Matsumoto, Sci. Technol. Adv. Mater., 2006, 7, 461–467 CrossRef CAS.
- P. Lin, R. A. Henderson, R. W. Harrington, W. Clegg, C.-D. Wu and X.-T. Wu, Inorg. Chem., 2004, 43, 181–188 CrossRef CAS.
- M. Tadokoro, S. Yasuzuka, M. Nakamura, T. Shinoda, T. Tatenuma, M. Mitsumi, Y. Ozawa, K. Toriumi, H. Yoshino, D. Shiomi, K. Sato, T. Takui, T. Mori and K. Murata, Angew. Chem., Int. Ed., 2006, 45, 5144–5147 CrossRef CAS.
- P. J. Sanz Miguel, P. Amo-Ochoa, O. Castillo, A. Houlton and F. Zamora, Supramolecular Chemistry of Metal-Nucleobase Complexes, Wiley-VCH, Chichester, UK, 2009 Search PubMed.
- P. Aich, S. L. Labiuk, L. W. Tari, L. J. T. Delbaere, W. J. Roesler, K. J. Falk, R. P. Steer and J. S. Lee, J. Mol. Biol., 1999, 294, 477–485 CrossRef CAS.
- D. Olea, S. S. Alexandre, P. Amo-Ochoa, A. Guijarro, F. de Jesus, J. M. Soler, P. J. de Pablo, F. Zamora and J. Gomez-Herrero, Adv. Mater., 2005, 17, 1761–1765 CrossRef CAS.
- S. S. Alexandre, J. M. Soler, P. J. S. Miguel, R. W. Nunes, F. Yndurain, J. Gomez-Herrero and F. Zamora, Appl. Phys. Lett., 2007, 90, 193107 CrossRef.
- P. Amo-Ochoa, O. Castillo, S. S. Alexandre, L. Welte, P. J. de Pablo, M. I. Rodriguez-Tapiador, J. Gomez-Herrero and F. Zamora, Inorg. Chem., 2009, 48, 7931–7936 CrossRef CAS.
- M. R. Bryce, Chem. Soc. Rev., 1991, 20, 355–390 RSC.
- J. M. Williams, A. J. Schultz, U. R. S. Geiser, K. D. Carlson, A. M. Kini, H. H. Wang, W.-K. Kwok, M.-H. Whangbo and J. E. Schirber, Science, 1991, 252, 1501–1508 CAS.
- J. Dai, M. Munakata, T. Kuroda-Sowa, Y. Suenaga, L. Ping Wu and M. Yamamoto, Inorg. Chim. Acta, 1997, 255, 163–166 CrossRef CAS.
- N. Singh, S. Gupta and R. K. Sinha, Inorg. Chem. Commun., 2003, 6, 416–422 CrossRef CAS.
- E. Cerrada, M. C. Diaz, C. Diaz, M. Laguna and A. Sabater, Synth. Met., 2001, 119, 91–92 CrossRef CAS.
- N. Singh and S. Gupta, Synth. Met., 1999, 107, 167–174 CrossRef CAS.
- N. Singh and S. Gupta, Synth. Met., 1999, 107, 167–174 CrossRef CAS.
- R. Kanehama, M. Umemiya, F. Iwahori, H. Miyasaka, K. Sugiura, M. Yamashita, Y. Yokochi, H. Ito, S. Kuroda, H. Kishida and H. Okamoto, Inorg. Chem., 2003, 42, 7173–7181 CrossRef CAS.
- L. F. Szczepura, C. P. Galloway, Y. F. Zheng, P. D. Han, A. L. Rheingold, S. R. Wilson and T. B. Rauchfuss, Angew. Chem., Int. Ed., 1995, 34, 1890–1892 CrossRef CAS.
- S. Delgado, P. J. Sanz Miguel, J. L. Priego, R. Jimenez-Aparicio, C. J. Gomez-García and F. Zamora, Inorg. Chem., 2009, 47, 9128–9130 CrossRef.
- Y. Chen, Z.-O. Wang, Z.-G. Ren, H.-X. Li, D.-X. Li, D. Liu, Y. Zhang and J.-P. Lang, Cryst. Growth Des., 2009, 9, 4963–4968 CAS.
- N. Singh and A. Prasad, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2008, 47, 650–656 Search PubMed.
- J. C. Zhong, Y. Misaki, M. Munakata, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga and H. Konaka, Inorg. Chem., 2001, 40, 7096–7098 CrossRef CAS.
- G. de la Torre, C. G. Claessens and T. Torres, Chem. Commun., 2007, 2000–2015 RSC.
- H. Schultz, H. Lehmann, M. Rein and M. Hanack, Struct. Bonding, 1991, 74, 41–46 CAS.
- M. Hanack and L. Subramanian, in Handbook of Organic Conductive Molecules and Polymer, ed. H. S. Nalwa, John Wiley & Sons, 1997, vol. 1 Search PubMed.
- M. F. Craciun, S. Rogge, M. J. L. den Boer, S. Margadonna, K. Prassides, Y. Iwasa and A. F. Morpurgo, Adv. Mater., 2006, 18, 320–324 CrossRef CAS.
- N. B. McKeown, Phthalocyanine Materials: Synthesis, Structure and Function, Cambridge University Press, Cambridge, 1998 Search PubMed.
- G. de la Torre, M. Nicolau and T. Torres, in Supramolecular Photosensitive and Electroactive Materials, ed. H. Nalwa, Academic Press, New York, 2001, pp. 1–111 Search PubMed.
- R. S. Nohr, P. M. Kuznesof, K. J. Wynne, M. E. Kenney and P. G. Siebenman, J. Am. Chem. Soc., 1981, 103, 4371–4377 CrossRef CAS.
- T. J. Marks, Science, 1985, 227, 881–889 CAS.
- P. J. Toscano and T. J. Marks, J. Am. Chem. Soc., 1986, 108, 437–444 CrossRef CAS.
- M. Hanack and D. Dini, in The Porphyrin Handbook, Academic Press, San Diego, CA, 2003 Search PubMed.
- M. Hanack, S. Deger and A. Lange, Coord. Chem. Rev., 1988, 83, 115–136 CrossRef CAS.
- M. Hanack, K. Dürr, A. Lange, J. Osío Barcina, J. Pohmer and E. Witke, Synth. Met., 1995, 71, 2275–2278 CrossRef CAS.
- S.-J. Kim, M. Matsumoto and K. Shigehara, Synth. Met., 1999, 107, 27–33 CrossRef CAS.
- S. Hayashida and M. Hanack, Synth. Met., 1992, 52, 241–255 CrossRef CAS.
- C. G. Claessens, U. Hahn and T. Torres, Chem. Rec., 2008, 8, 75–97 CrossRef CAS.
- M. Munakata, L. P. Wu, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga and K. Sugimoto, Inorg. Chem., 1997, 36, 4903–4905 CrossRef CAS.
- M. Munakata, L. P. Wu, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga, G. L. Ning and T. Kojima, J. Am. Chem. Soc., 1998, 120, 8610–8618 CrossRef CAS.
- G. L. Ning, L. P. Wu, K. Sugimoto, M. Munakata, T. Kuroda-Sowa and M. Maekawa, J. Chem. Soc., Dalton Trans., 1999, 2529–2536 RSC.
- M. Munakata, G. L. Ning, Y. Suenaga, T. Kuroda-Sowa, M. Maekawa and T. Ohta, Angew. Chem., Int. Ed., 2000, 39, 4555–4557 CrossRef CAS.
- J. C. Zhong, M. Munakata, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga and H. Konaka, Inorg. Chem., 2001, 40, 3191–3199 CrossRef CAS.
- S. Q. Liu, T. Kuroda-Sowa, H. Konaka, Y. Suenaga, M. Maekawa, T. Mizutani, G. L. Ning and M. Munakata, Inorg. Chem., 2005, 44, 1031–1036 CrossRef CAS.
- Y. Takazaki, Z. Yang, M. Ebihara, K. Inoue and T. Kawamura, Chem. Lett., 2003, 120–121 CrossRef CAS.
- Y. Fuma and M. Ebihara, Chem. Lett., 2006, 1298–1299 CrossRef CAS.
- P. Amo-Ochoa, L. Welte, R. González-Prieto, P. J. Sanz Miguel, C. J. Gómez-García, E. Mateo-Martí, S. Delgado, J. Gómez-Herrero and F. Zamora, Chem. Commun., 2010, 46, 3262–3264 RSC.
- C. N. R. Rao, A. Ranganathan, V. R. Pedireddi and A. R. Raju, Chem. Commun., 2000, 39–40 RSC.
- Z. T. Xu, Coord. Chem. Rev., 2006, 250, 2745–2757 CrossRef CAS.
- W. P. Su, M. C. Hong, J. B. Weng, R. Cao and S. F. Lu, Angew. Chem., Int. Ed., 2000, 39, 2911–2914 CrossRef CAS.
- K. H. Low, V. A. L. Roy, S. S. Y. Chui, S. L. F. Chan and C. M. Che, Chem. Commun., 2010, 46, 7328–7330 RSC.
- Y. Zhao, M. Hong, Y. Liang, R. Cao, J. Weng, S. Lu and W. Li, Chem. Commun., 2001, 1020–1021 RSC.
- W. Su, R. Cao, M. Hong, W.-T. Wong and J. Lu, Inorg. Chem. Commun., 1999, 2, 241–243 CrossRef CAS.
- Y. Fuma, M. Ebihara, S. Kutsumizu and T. Kawamura, J. Am. Chem. Soc., 2004, 126, 12238–12239 CrossRef CAS.
- A. Aumuller, P. Erk, G. Klebe, S. Hunig, J. U. Vonschutz and H. P. Werner, Angew. Chem., Int. Ed., 1986, 25, 740–741 CrossRef.
- D. F. Sun, R. Cao, J. B. Weng, M. C. Hong and Y. C. Liang, Dalton Trans., 2002, 291–292 RSC.
- R. B. Fu, S. Q. Xia, S. C. Xiang, S. M. Hu and X. T. Wu, J. Solid State Chem., 2004, 177, 4626–4631 CrossRef CAS.
- M. J. Kelly, Low-Dimensional Semiconductors, Clarendon Press, Oxford, 1995 Search PubMed.
- Several reviews of the Special issue devoted to Metal Organic Frameworks, Chem. Soc. Rev., 2009, 38.
- R. Mas-Balleste, G. Gomez-Navarro, J. Gomez-Herrero and F. Zamora, Nanoscale, 2011, 3, 20–30 RSC.
| This journal is © The Royal Society of Chemistry 2012 |