Photosensitized electron transfer processes of nanocarbons applicable to solar cells
Francis
D'Souza
*a and
Osamu
Ito
*b
aDepartment of Chemistry, University of North Texas, 1155 Union Circle, #305070, Denton, TX 76203-5017, USA. E-mail: Francis.DSouza@unt.edu; Fax: +1 940-565-4318; Tel: +1 940-369-8832
bPRESTO Adviser, Japan Science and Technology Agency and CarbonPhotoScience, Kita-Nakayama 2-1-6, Izum-kui, Sendai, 981-3215, Japan. E-mail: ito@tagen.tohoku.ac. jp; Fax: +81-22-376-5970
First published on 5th October 2011
Abstract
Photosensitized electron-transfer processes of nanocarbon materials hybridized with electron donating or electron accepting molecules have been surveyed in this tutorial review on the basis of the recent results reported mainly from our laboratories. As nano-carbon materials, fullerenes and single wall carbon nanotubes (SWCNTs) have been employed. Fullerenes act as photo-sensitizing electron acceptors with respect to a wide variety of electron donors; in addition, the fullerenes act as good ground state electron acceptors in the presence of light-absorbing electron donors such as porphyrins and phthalocyanines. In the case of SWCNTs, their ground states act as electron acceptor and electron donors, depending on the photosensitizers. For example, with respect to the photoexcited porphyrins and phthalocyanines, SWCNTs usually act as electron acceptors, whereas for the photoexcited fullerenes, SWCNTs act as electron donors. The diameter sorted semi-conductive SWCNTs have been used to verify the size-dependent electron transfer rates. For the confirmation of the electron transfer processes, the transient absorption methods have been widely used, in addition to the time-resolved fluorescence spectral measurements. The kinetic data thus obtained in solution are found to be quite useful to predict the efficiencies of photovoltaic cells constructed on semiconductor nanoparticle modified electrodes and their photocatalytic processes.
 Francis D'Souza | Francis D'Souza is a Professor of Chemistry, and Materials Science and Engineering at the University of North Texas (UNT), Denton, TX. Prior joining UNT in 2011, he was a Professor of Chemistry at Wichita State University, Wichita, KS. He received PhD (1992) from the Indian Institute of Science, Bangalore, India. His research covers wide areas of chemistry, nanophotonics and materials science. Principal research interests include chemistry and supramolecular chemistry of porphyrins and carbon nanomaterials, light energy harvesting, photoelectrochemistry and photovoltaics, electrochemical and photochemical sensors and catalysts, fluorescent chemosensors and biosensors, conducting nanocomposite hybrid materials for energy storage and conversion. Honors and Award include Excellence in Research Award, 2006, Japan Society for the Promotion of Science (JSPS) Fellow, 2008, Fellow of the Electrochemistry Society, 2010. |
 Osamu Ito | Osamu Ito is an Emeritus Professor at Tohoku University of Sendai, Japan. He received PhD (1973) from the Department of Chemistry of Tohoku University. He fulfilled his academic positions at Tohoku University as an Associate Researcher (1973–1983), an Associate Professor (1984–1992) and a Full Professor of the Institute of Multi-disciplinary Research for Advanced Materials (1993–2007). After retirement, he holds the positions of Adjunct Professor of Tohoku University, and Research Adviser at the National Institute for Materials Science (Tsukuba, Japan) and at Japan Science and Technology Agency. He was a recipient of the Award for Distinguished Achievements to Japanese Photochemistry Association (2009). His present research interests include the photo-physical studies using laser flash photolysis of various molecules and carbon nanomaterials. |
Introduction
Recently, supramolecular nanocarbon architectures involving fullerenes and single wall carbon-nanotubes (SWCNTs) linked to photosensitizers have received considerable attention due to their distinguishing electronic properties suitable for optoelectronic and light-induced applications.1–9The chemically functionalized fullerenes and SWCNTs with photoactive molecules such as light-harvesting donors have been recognized to be useful materials for photocatalytic and light-energy conversion applications, as shown in Fig. 1.1–9
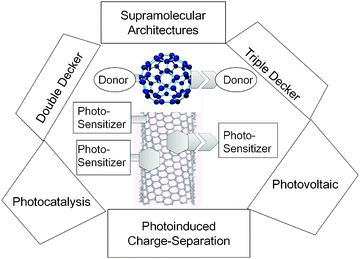 | ||
| Fig. 1 Functionalized fullerenes and SWCNTs linked with photosensitizers via various self-assembly methods and their potential light-induced applications via photoinduced charge-separation processes; hexangular shape is adsorbent with π–π stacking, bar is covalent bond, and other shape with space is non-covalent bonding. | ||
Covalent functionalization is structurally a simpler method; however, the procedures are intricate due to the multi-step synthetic processes.10–13 This problem especially comes up against nanocarbons which demonstrate poor solubility in organic solvents used for the syntheses. Furthermore, direct covalent functionalization converts the sp2 carbons of some of the double bonds of fullerenes and SWCNTs to sp3 carbons, cutting-off the π-conjugations. However, since these carbon materials contain a large number of π-bonds, loss of a few π-bonds would only slightly alter their electronic properties impacting their electronic conductivities and light absorption abilities.10–13
On the other hand, the noncovalent self-assembly methods using intermolecular interactions with the photosensitizers retain completely the π-networks of SWCNTs making them useful for the wide applications to various photodevices and optoelectronics.11 Among the non-covalent functionalization approaches, the simplest method is the direct π–π stacking of the sensitizers such as porphyrins (MP) and phthalocyanines (MPc) onto the SWCNT surface. However, such direct π–π interactions promote the closest contact between the entities, which is sometimes crucial for photoinduced electron transfer, although it alters their electronic properties of the corresponding individual characteristic features with charge-transfer interactions.10–13
Usage of adsorbent-connected sensitizer molecules can avoid such direct interaction making appropriate space, preserving the charge-separated states induced by the light illumination for longer times.14–17 For this purpose, appropriate linkages are necessary to connect the sensitizers to fullerenes and/or SWCNTs. In the case of fullerenes, the functional groups with nonbonding abilities are easily linked with covalent bonds. In the case of SWCNTs, the aromatic compounds such as pyrene appended with the desired sensitizers can be employed as adsorbents onto the SWCNTs surface via π–π stacking.15–17 When the covalent bonds are used to connect the photosensitizers to the pyrene unit, a “double-decker” architecture could be envisioned as seen in the left side of Fig. 1.18
When additional intermolecular binding motifs such as metal–ligand coordination, crown-ether inclusion complexation, and ion-paring and/or hydrogen bonding interactions are employed to connect the sensitizer via appropriately functionalised pyrene receptors, novel “triple-decker” architectures could be envisioned as seen in the right side of Fig. 1.15–17
Dendrimers possessing photosensitizers can also be used to construct photosensitizer–SWCNT nanohybrids, since the dendrons intertwine with the SWCNT and probably prevent direct interaction between the photosensitizer and SWCNT.19 Additionally, photosensitizers with long alkyl or alkyl-ether chains can form the hybrids by intertwining the SWCNT with these long chains.
Recent progresses on the SWCNTs include separation of the mixtures of SWCNTs into the metallic and semiconductor, which would exhibit quite different properties in photosensitizing electron transfer reactions. Furthermore, size-sorted SWCNTs are now available, which makes researchers to consider them as single molecular events.
This tutorial review documents the recent progresses on supramolecular constructions of nano-architectures using photosensitizing electron-donor and electron-acceptor molecules with fullerene and diameter sorted SWCNTs as one of the components of photovoltaic devices. The key findings in the areas of photoinduced charge separation (CS), photocatalysis, and photoelectrochemistry, mainly from our laboratories are discussed.
Fullerenes
Efficient photoinduced electron transfer (ET) can occur between the fullerene and electron donor (D) in solution in a wide range of polarity. As shown in Fig. 2(a), the light excitation of the D molecule elevates an electron on the HOMO(D) to the LUMO(D), from which the electron transfers to the LUMO(C60), giving D˙+ and C60˙−. The energy difference between the LUMO(D) and the LUMO(C60) (ΔG0ET(D*)) corresponds to the difference between ΔG0ET (LUMO(C60)–HOMO(D)) and EEX(D) (LUMO(D)–HOMO(D)).20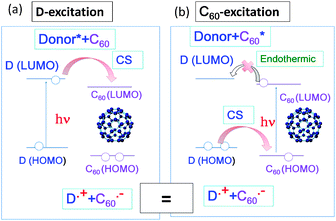 | ||
| Fig. 2 Molecular orbital (MO) diagrams showing photoinduced electron transfer for donor+C60 systems: (a) D-excitation and (b) C60-excitation. For a covalently bonded donor–C60 dyad, similar events would be expected to occur. | ||
On the other hand, light-excitation of C60 raises an electron from the HOMO(C60) to the LUMO(C60) (EEX(C60)); then, one of the electrons of the HOMO(D) transfers to the half-vacant HOMO(C60), forming C60˙− and D˙+ (Fig. 2(b)). Although the electron configuration of the radical ions is same as that of the D-excitation, the trail of electron transfer is different. That is, for the D-excitation, the extra electron of C60˙− on the LUMO(C60) comes from the HOMO(D), whereas for the C60-excitation, the extra electron of C60˙− on the HOMO(C60) comes from the HOMO(D).20
For example, in the case of a mixture of C60 and ZnPc, selective excitation of C60 generates the 3C60* (740 nm band), which decays by accepting an electron from ZnPc; then, ZnPc˙+ (860 nm) and C60˙− (1000 nm) are produced as intermolecular ET intermediates as shown in Fig. 3(a).21 The energy level diagram for this process is shown in Fig. 3(a′), in which the energy level of the radical ions (ZnPc˙+ and C60˙−) is lower than the 3C60* energy in polar solvents. In non-polar solvents, where the energy level of sum of ZnPc˙+ and C60˙− entities is higher than the 3C60* energy, only a slow decaying 3C60*-band could be expected.
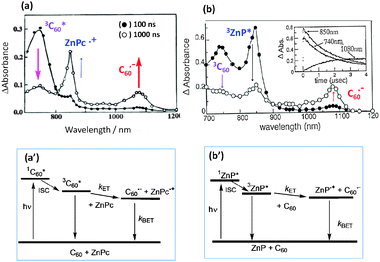 | ||
| Fig. 3 Nanosecond transient absorption spectral traces confirming intermolecular photoinduced ET under (a) C60-excitation in the presence of ZnPc, and (b) ZnP-excitation in the presence of C60 in benzonitrile. (a′) and (b′) The energy-level diagrams for the ET for these two donor–acceptor systems. | ||
On the other hand, the predominant excitation of ZnP produces the 3ZnP* (840 nm), which decays to produce C60˙− (1000 nm) and ZnP˙+ (620 nm), although the latter absorption band is not shown in Fig. 3(b). The energy level diagram for ZnP-excitation shown in Fig. 3(b′) is essentially the same as that of C60-excitation, in which the energy level of sum of ZnP˙+ and C60˙− is lower than the energy level of 3ZnP*, which is similar to 3C60*.
However, since the energy level of 1ZnP* is higher than the 1C60*, the excited singlet state energy-transfer (EnT) process occurs from 1ZnP* to C60, generating 3C60*, from which the ET is also initiated. In Fig. 3 (upper right), the 740 nm band of 3C60* appears due to this EnT process in addition to the direct C60-excitation; thus, the observed absorption intensity of C60˙− is a total of the ET processes via1ZnP* and 3C60*.22 The ET process via1ZnP* is also possible from the energy diagram; for this process, it is necessary to increase the C60-concentration to prevent the competitive unimolecular intersystem crossing to 3ZnP*. Therefore, for intermolecular ET, the appropriate selections of the solvent polarity, concentration of the individual reactants, and excitation light are all important factors.
Fullerene derivatives covalently linked to D molecules via spacers are also good candidates for light-controlling molecular devices.23 For about two decades, the C60∼spacer∼D molecules have successfully been synthesized, which gave usually long-living radical ion-pairs such as C60˙−∼D˙+ after the photoinduced charge separation (CS) process (∼ refers to a covalent bond with a suitable spacer).24,25 Although the lifetimes of the radical ion-pairs (RIPs) produced by the photoinduced CS process depend on the length and kind of spacer in addition to property of D molecules, some of them gave surprisingly long-persisting RIPs even in dyads with short spacers, mainly due to the small reorganization energy characteristic of the large spherical fullerene molecules.26–28 Therefore, various fullerene derivatives are incorporated into a number of molecular photovoltaic devices, which are denoted as photoinduced charge-separation type solar cells, to distinguish from the dye-sensitized solar cells (Grätzel cell).29,30
In the case of covalently linked C60 with MP and MPc, two types of dyads can be constructed: (i) C60 connected to the peripheral positions of MP and MPc in an in-plane arrangement, and (ii) C60 connected to the axial positions of MP and MPc, producing upright positioning with respect to the plane of the macrocycle.
In a simple in-plane connected C60∼ZnP dyad with an amide group as a spacer linking the meso-phenyl group of the porphyrin (Fig. 4(a)),31,32 the picosecond fluorescence emission and transient absorption measurements in the visible region revealed that the CS mainly takes place via1ZnP* and partly via1C60* in polar solvents, generating the C60˙−∼ZnP˙+ radical ion pair. Most of the radical ion absorption bands of the C60˙−∼ZnP˙+ moiety decayed within about 1 ns. Additionally, the C60˙−∼ZnP˙+ may retain the singlet spin character of the precursors (1ZnP* and 1C60*) wherein the CR rate of 1(C60˙−∼ZnP˙+) is expected to be much faster.31,32
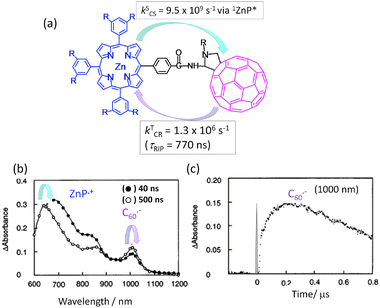 | ||
| Fig. 4 (a) Molecular structure of a covalently linked ZnP∼C60 dyad and experimentally determined rate parameters. (b) Transient absorption spectra of the ZnP∼C60 dyad and (c) time profile of the 1000 nm band corresponding to C60˙− in Ar-saturated PhCN (τRIP is lifetime of RIP). Adapted from ref. 31. | ||
In the nanosecond time region, the transient absorption bands of the C60˙−∼ZnP˙+ were clearly observed at 1000 and 640 nm after the decay of the 3C60* moiety as shown in Fig. 4(b).31,32 The time profile of the C60˙− moiety at 1000 nm shows the slow rise and slow decay (Fig. 4(c)), after the quick CS and quick CR processes via1ZnP* and/or 1C60*. Therefore, the slow rise of C60˙−∼ZnP˙+ can be attributed to the CS process via3ZnP* and 3C60*, generating 3(C60˙−∼ZnP˙+) with long τRIP. As origins of 3ZnP* and 3C60*, their ISC process can be considered. Triplet spin character of ZnP˙+∼C60˙− can be confirmed by the rapid decay in the presence of O2 due to the triplet EnT process. Such C60∼ZnP derivatives have been applied to the photoinduced CS-type solar cells, giving high light-to-electricity conversion efficiency.33
Compared with the above in-plane linked C60∼MP dyad, dyads with upright positioning of the fullerene entity with respect to the plane of the macrocycle at one side (MP∼C60, MPc∼C60) or both sides (MP∼C60∼MP, MPc∼C60∼MPc) and (C60∼MP∼C60, C60∼MPc∼C60) have recently been reported.8,9 An example for the one-side type dyad is shown in Fig. 5(a), in which the C60 unit is linked axially to the aluminium(III) porphyrin (AlPor) via a rigid benzoate spacer; thus, this dyad is represented as AlPor∼C60 in this review.34 The fluorescence quenching of AlPor with the C60 unit suggests that the CS process from the 1AlPor* to C60 entity occurs very fast, generating AlPor˙+∼C60˙−, which is confirmed by the transient absorption band of the C60˙− (1000 nm) as shown in Fig. 5(b).
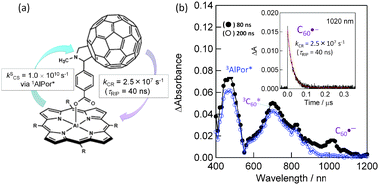 | ||
| Fig. 5 (a) Structure of the AlPor∼C60 dyad and the experimentally determined rate parameters. (b) Nanosecond transient absorption spectra observed with 532 nm laser irradiation in Ar-saturated o-DCB. Inset: absorption-time profile. Adapted from ref. 34. | ||
From the time profile at 1020 nm, the τRIP value was evaluated to be about 40 ns of AlPor˙+∼C60˙− in o-dichlorobenzene (o-DCB) at RT. This value is lesser than the τRIP value of the in-plane positioned dyad, ZnP˙+∼C60˙−. This may reflect a stronger electronic coupling of the vertically positioned entities, AlPor˙+ and C60˙−, compared with the in-plane positioned ZnP˙+ and C60˙− entities in Fig. 4. In the transient spectra (Fig. 5(b)), although the 3AlPor* and 3C60* entities are observed, the CS process via these triplet states may not occur since no decay of these bands until 500 ns, a time scale beyond the formation of AlPor˙+∼C60˙−, is observed. Thus, the short τRIP of AlPor˙+∼C60˙− may be caused by the less contribution of the triplet spin character.
A charge-separation photoelectrochemical solar cell was constructed by adsorbing the AlPor∼C60 dyad onto the SnO2 nanoparticle surface pasted on FTO (Fig. 6(a)). Here, the photogenerated electron on C60 of AlPor˙+∼C60˙− is expected to transfer to SnO2 generating photo-current. Photocurrent measurements showed good photovoltaic performance of the cell (Fig. 6(b)); maximal incident photon-to-current conversion efficiency (IPCE) of FTO/SnO2/AlPor∼C60 was accomplished to be 25% at 450 nm, which is higher than those of the individual components (FTO/SnO2/AlPor and FTO/SnO2/C60). In addition to the fast CS rate and high efficiency of AlPor˙+∼C60˙− formation, and its low τRIP, the morphology seems to be also an important factor. As shown by the TEM images, the AlPor∼C60 dyad formed chained nanoparticles as shown in Fig. 6(c-B) compared with the globular nanoparticles for the starting materials.34 The chained nanoparticle morphology of the dyads might facilitate charge transportation.
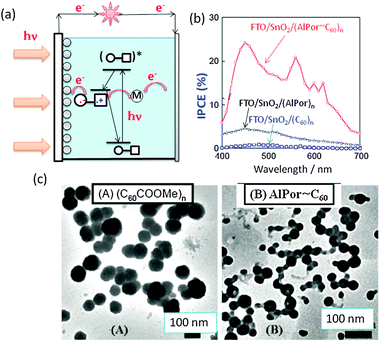 | ||
| Fig. 6 (a) Design of the photoelectrochemical solar cell (M: mediator (I−/I3−)). (b) The incident photon-to-current efficiency (IPCE) vs. wavelength of the illumination light of the FTO/SnO2 electrode coated with AlPor∼C60, AlPor and C60. (c) TEM images of (A) FTO/SnO2/RC60 and (B) FTO/SnO2/AlPor∼C60. Adapted from ref. 34. | ||
Similar to porphyrins and phthalocyanines, oligothiophenes (nTs; n = 4, 8, 12) are also known to be good electron donors especially for the C60 in the covalently linked systems.35 Otsubo's group synthesized C60∼nT type dyads shown in Fig. 7(a). By the selective excitation of the C60 moiety, a rapid CS process is shown to take place via1C60* in polar solvents; the CS efficiency increases with the length of the nT moiety. That is, even in longer nTs the CS process is observed.36,37 In Fig. 7(b), the nanosecond transient spectrum of C60∼4T is shown as an example; this spectrum is ascribed to the formation of C60˙−∼4T˙+ since the 680 and 1100 nm bands are associated to 4T˙+, whereas the 1000 nm band of C60˙− is hidden. The τRIP values of (C60˙−∼nT˙+) attain the microsecond range. The longest τRIP was obtained for the shortest C60∼4T (Fig. 7(b)) as a consequence of the closest energy level of C60˙−∼4T˙+ with that of 3C60*∼4T and C60∼34T*, increasing the triplet spin character in C60˙−∼4T˙+.36 This was supported by the rapid quenching of the C60˙−∼4T˙+ in the presence of O2 (inset of Fig. 7), as the individual radical ions are not reactive to O2. The excitation of the nT moiety in the C60∼nT dyads induces both the CS process via1nT* and the EnT process from 1nT* to 1C60*, from which the CS process subsequently occurs in polar solvent. However, in the presence of nanoparticles, such a high τRIP value was not observed in the relatively short nT, probably because of the extra intermolecular interactions among C60∼nT.38
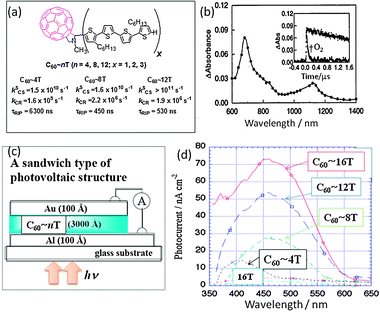 | ||
| Fig. 7 (a) Molecular structures of C60∼nT dyads (x = 4n) and observed rate parameters in PhCN at RT. (b) Nanosecond transient absorption spectrum of C60∼4T in PhCN at 100 ns after 532-nm laser irradiation. Inset: absorption time profile at 680 nm in the absence and presence of O2. Adapted from ref. 37 and 38. (c) Design of the organic thin-layer solar cells. (d) Photocurrent vs. wavelength of nT-C60 dyads in comparison with nT. Adapted from ref. 39. | ||
On the basis of these photochemical outcomes, organic thin-layer solar cells were constructed by Otsubo's group (Fig. 7(c)).39 The curves of photocurrent vs. wavelength for the C60∼nT dyads show similar spectra to the absorption spectra of nTs, indicating that the CS process takes place via1nT*. The peaks at 450 nm are extremely high compared to that observed in the pristine nTs, and they increase with the nT length as shown in Fig. 7(d).
Further connection of a porphyrin (ZnP) at the opposite end of C60 in the 8T structured system (Fig. 8) increases the visible light absorption ability.40 When the ZnP moiety is predominantly excited, the long distant CS process from the 1ZnP* to the C60 is attained through the nT chain, since the nT chain is π-conjugative linkage. The relative energy levels of the MOs of the LUMO and HOMO of the ZnP, nT and C60 moieties are important to understand the CS process, in which an electron jumps from the half-occupied LUMO of the 1ZnP* moiety to the LUMO of the nT chain (CS-1), forming ZnP˙+∼nT˙−∼C60 as an intermediate. Then, the electron of the LUMO of the nT chain transfers to the LUMO of C60 (CS-2), generating ZnP˙+∼nT∼C60˙− as a final stable CS state. After a while, the CR process is triggered by an electron-jump from the HOMO of the nT chain moiety up to the half-vacant HOMO of the MP˙+ moiety (CR-1).40 Then, the electron of C60˙− covers the electron loss of the HOMO of the nT chain, returning to the original neutral ZnP∼nT∼C60 molecule in the HOMO level.
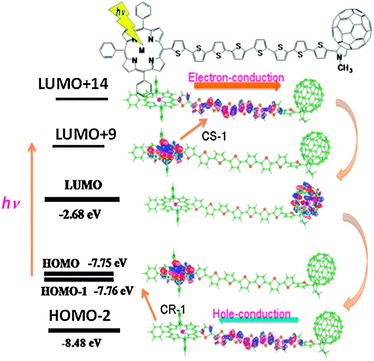 | ||
| Fig. 8 MO representation of the CS and CR processes of ZnP∼8T∼C60. The HOMO is localized on the ZnP moiety; the light excitation raises the electron to the LUMO+9 localized on the ZnP moiety; the electron of LUMO+9 jumps up to LUMO+14 localized on the 8T moiety; the electron falls down to the LUMO localized on the C60 moiety, generating the stable ZnP˙+∼8T∼C60˙−. As for the CR process, the electron of the HOMO−2 localized on the 8T moiety jumps up to the HOMO; then, the electron of the LUMO on the C60˙− moiety falls down to the HOMO−2, returning to the original neutral molecule. Adapted from ref. 20. | ||
With regard to noncovalent binding of fullerene to various electron donor molecules, coordination of either pyridine- or imidazole-appended fullerenes to the coordinatively unsaturated central metal of the porphyrins and phthalocyanines has been widely used to construct the photosensitizing donor–acceptor supramolecular hybrids.8,9 However, such coordination bonding is competitive with the solvent coordination; thus, such studies in polar solvents are limited. Hydrogen bonding (including complementary base-pairing) can also be used to build supramolecular hybrids between the fullerenes and donor molecules possessing hydrogen-bonding functionalities.41–43 In the subsequent studies such supramolecular hybrids have also been employed as components of photo-voltaic solar cell systems; high visible light-to-photocurrent efficiencies are reported by relative easy fabrication of the energy harvesting devices.4
SWCNTs
It is essential to employ size-sorted SWCNTs for both fundamental and application-oriented research in photosensitizer–SWCNT systems. One of the successful methods of separation of mixture of SWCNTs is the density gradient centrifugation with gel media reported by Hersam and Tanaka–Kataura group, as shown pictorially in Fig. 9.44,45 Semiconductive and metallic SWCNTs, which have quite different electronic properties,46 are now commercially available.47 Furthermore, diameter-sorted semiconductive (6,7)-SWCNT and (7,6)-SWCNT are also now commercially available.48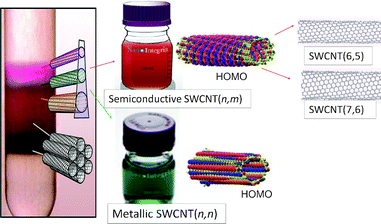 | ||
| Fig. 9 Separation of SWCNTs by a density gradient centrifugation method, commercially available semiconductive and metallic SWCNTs and their HOMOs; commercially available diameter sorted SWCNTs, SWCNT(6,5) and SWCNT(7,6). Adapted from ref. 44–48. | ||
Energy level diagrams depicting the possible photoinduced processes for the sensitizer molecule (M) and SWCNT are illustrated in Fig. 10, in which the narrow band gap of the SWCNT usually positions inside the wider HOMO–LUMO gap of M. This type of relative positioning of energy levels is quite different from that observed for the usual donor–acceptor molecular pair shown in Fig. 2. Therefore, when M is selectively excited (Fig. 10(a)), two types of photoinduced CS processes could be envisioned as shown by curved arrows, viz., CS-1 is a CS process from the LUMO of M to the conduction level of SWCNT, generating the M˙+/SWCNT˙− ion-pair and CS-2 is a CS process from the valence band of SWCNT to the half-filled HOMO level of the 1M*, generating the M˙−/SWCNT˙+ ion-pair.49 When M is a strong electron acceptor such as C60, even upon excitation of the C60 entity, the LUMO of 1C60* cannot transfer an electron to the conduction band of SWCNT, because the LUMO of C60 is lower than the conduction band, whereas an electron of the valence band of SWCNT can readily transfer to the half-vacant HOMO level of 1C60*, resulting in the C60˙−/SWCNT˙+ ion pair, illustrated as CS-2 in Fig. 10(a).49
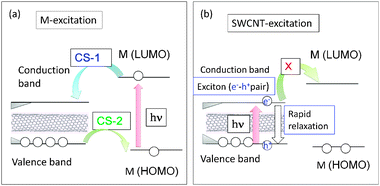 | ||
| Fig. 10 Schematic energy level diagrams for photoinduced CS processes of SWCNT(n,m) hybrids with M (molecule); (a) M-excitation, (b) SWCNT excitation. The curved arrows represent the CS paths and vertical arrows represent the excitation and relaxation processes. Modified from ref. 49. | ||
Alternatively, when the SWCNT is directly light-excited, the exciton (electron–hole pair) can be generated on the SWCNTs; however, the exciton rapidly relaxes to the ground state before the electron or hole transfers to M (Fig. 10(b)), since its lifetime is usually shorter than 10 ps.50 Furthermore, since the light absorptivity of the SWCNT in the visible region is quite low, it is not advantageous to employ such a process for an efficient light-energy conversion system, when SWCNTs are functionalized with light-harvesting molecules. Therefore, the photo-sensitizing processes of the type shown in Fig. 10a are widely studied and explained in this review.
The first example of the photo-sensitizing processes of SWCNTs is “double-decker” architectures via the π–π stacking and covalent-bonding. Among the non-covalent functionalization approaches, the simplest method is to utilize glue molecules such as aromatic π-systems, to which the desired photosensitizers are appended to adhere to the surface of the SWCNT. As indicated in Fig. 10, a wide variety of molecules are possible as photoinduced CS counter parts with respect to the SWCNT. For comparison, direct binding methodology and dendrimer methodology to functionalize the SWCNTs are also introduced here.
Many studied examples employed porphyrin as a photosensitizer with respect to the SWCNTs. The first example of a donor–acceptor hybrid using diameter sorted SWCNTs is a double-decker architecture held by π–π stacking interactions. As shown in Fig. 11b, this involved ZnP functionalized with four pyrene (Pyr) entities (ZnP(Pyr)4), in which ZnP is a typical visible-light sensitizer and the pyr entity is a π-stacking agent to the surface of SWCNT. The four pyr entities provided the necessary energy for π–π interaction.18 The ZnP(Pyr)4/SWCNT(n,m) hybrids are constructed by simple mixing of the individual components in DMF. The obtained homogeneous solution is stable and transparent for several days. TEM images of the dried ZnP(Pyr)4/SWCNT(n,m) samples are shown in Fig. 11(a), in which the treatment with ZnP(Pyr)4 unravelled the tangled SWCNT(n,m) in DMF; expanded images also show that each SWCNT(n,m) looks smeared and thicker, probably due to the attachments of the ZnP(Pyr)4 and its aggregates. Fig. 11(b) shows optical absorption spectra of the ZnP(Pyr)4/SWCNT(n,m) in DMF; the sharp peaks in the 600–1400 nm region are attributed to SWCNT(n,m) and the peak at 420 nm is due to the Soret band of the ZnP moiety. These results suggest the adduct formations; one possible structure is shown in the inset of Fig. 11(b).18
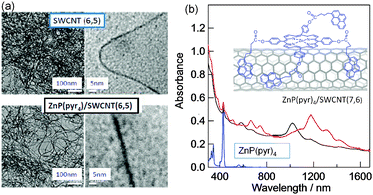 | ||
| Fig. 11 (a) TEM images of SWCNT(6,5) and ZnP(Pyr)4/SWCNT(6,5) at different magnification scales. (b) Absorption spectra of ZnP(Pyr)4/SWCNT(n,m) in DMF (black (6,5) and red (7,6)). Inset: presumed structure, in which the pyrene entities adsorb directly onto the surface of SWCNT, perhaps leaving an appropriate space between ZnP and SWCNT(n,m). Modified from ref. 18. | ||
From the transient absorption spectra showing the 600–700 nm band of ZnP˙+ and the 900–1300 nm band of SWCNT˙−, suggesting the formation of ZnP˙+(Pyr)4/SWCNT(n,m)˙−, lifetimes of ca. 100 ns are estimated. It is presumed that the electron generated near the ZnP moiety travels along the SWCNT resulting in the relatively long lifetime of ZnP˙+(Pyr)4/SWCNT(n,m)˙−. When the alkyl viologen dication (RV2+) is added as an electron accepting mediator with an appropriate sacrificial hole-shifter, one-electron reduced viologen, RV˙+, can be accumulated as blue solution by the visible light irradiation.18 Furthermore, the generated electron can be used for photovoltaic solar cell applications.
One example for H2-evolution is shown for a direct covalently bonded ZnP∼SWCNT hybrid with relatively short linkage in Fig. 12. In the transient absorption spectra, the CS radical ion pair with lifetime of 250 ns is observed by the transient absorption spectral studies.51 Further, the H2-evolution by the visible light irradiation of ZnP∼SWCNT in the presence of Pt-nanoparticles catalysts was observed as shown in Fig. 12. The observed pH dependence supports the photocatalytic H2 generation according to the mechanism shown in Fig. 12, in which the curved arrows indicate electron flow after the visible light illumination of the ZnP moiety.51 That is, after the photoinduced charge-separation, the electron generated on the SWCNT transfers to the fine Pt-particles, from which the electron is injected into H+, yielding H atoms; then, the H2 gas is generated by the coupling of the H atoms. The participation of H+ is supported by the pH dependence of the H2 gas evolution shown in Fig. 12.
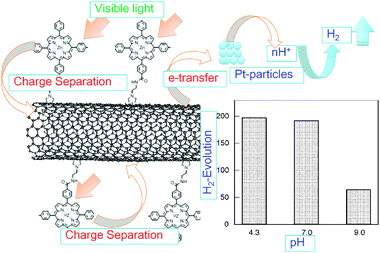 | ||
| Fig. 12 H2-evolution by the visible light irradiation of covalently bonded ZnP∼SWCNT in the presence of Pt-nanoparticles in aqueous solution; electron flows evaluated from fluorescence quenching, transient absorption and pH dependence on H2 evolution. Modified from ref. 51. | ||
Another example is a study with fullerene as a photosensitizer with respect to the SWCNT. When C60 is attached to SWCNTs via the Pyr moiety (C60Pyr/SWCNT(n,m)s in Fig. 13(a)), the CS-2 process in Fig. 10(a) would be expected to occur after the visible light-excitation of the C60 moiety in polar solvents.52 Fluorescence quenching of the C60 moiety with SWCNTs unequivocally indicates occurrence of the CS-2 process (Fig. 13(b)), since the LUMO of the C60 moiety is usually lower than the conduction band of SWCNTs. The rates of the CS-2 process are evaluated to be (2–3) × 109 s−1 from the shortening of the fluorescence lifetimes (see the inset of Fig. 13(b)); a faster CS-2 process was observed for C60Pyr/SWCNT(7,6) nanohybrids compared to C60Pyr/SWCNT(6,5) nanohybrids.
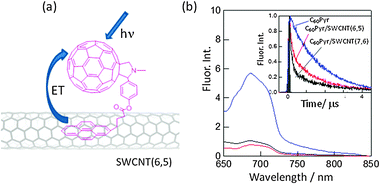 | ||
| Fig. 13 (a) Structure of C60Pyr/SWCNT(n,m)s, where (n,m) = (6,5) and (7,6), and (b) fluorescence spectra and time profiles of the C60 moiety in the 600–700 nm region in DMF; blue = C60Pyr, red = C60Pyr/SWCNT(6,5) and black = C60Pyr/SWCNT(7,6). Modified from ref. 52. | ||
The transient absorption spectra of C60Pyr/SWCNT(n,m)s show the 1000 nm band due to C60˙− and the 1300 nm band of SWCNT˙+ (Fig. 14(a)); from the decay time profile, the lifetime of C60˙−Pyr/SWCNT(7,6)˙+ is evaluated to be 70 ns, which is longer than that of C60˙−Pyr/SWCNT(6,5)˙+. Although the 700 nm band is similar to the 3C60*-absorption, the fast decay of this band with the same rate to that of C60˙− and SWCNT˙+ suggests the possible involvement of the radical ions.
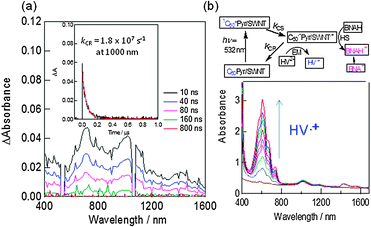 | ||
| Fig. 14 (a) Transient absorption spectra of C60Pyr/SWCNT(7,6) in DMF. Inset: time profile at 1000 nm. (b) HV˙+ accumulation by the selective illumination of the C60 moiety; an increase of the HV˙+ absorbance was observed with HV2+ and BNAH concentration, and also visible-light irradiation time. Inset: electron transfer mechanism. Modified from ref. 52. | ||
Upon addition of HV2+ (hexyl viologen dication), accumulation of HV˙+ is observed as a blue solution by the visible-light irradiation (Fig. 14(b)), suggesting that the electron on the C60˙− moiety transfers to HV2+ (EM process), while the cation site on SWCNT˙+ is faded away by accepting an electron from a sacrificial hole-shifter (HS process) BNAH (benzyl nicotinamide dihydride) as shown in the scheme in Fig. 14(b), in which the ET-processes (including CS, EM, HS processes) are illustrated (BNAH˙+ dissociates irreversibly into the benzyl nicotinamide cation (BNA+)).52 Furthermore, upon constructing the photoelectrochemical solar cell on the FTO/SnO2 electrode by coating C60Pyr/SWCNT(7,6) on the SnO2-nanoparticles, which is pasted on the fluorinated tin oxide (FTO), photocurrent is generated. Higher yields of HV˙+ formation and IPCE values are observed for C60Pyr/SWCNT(7,6) than that of C60Pyr/SWCNT(6,5).52
One example of dendrimer/SWCNT hybrids is reported; in this case, water-soluble dendrimers containing C60 as a central core are mixed with SWCNTs, producing the stable C60/SWCNT hybrids in aqueous solution.19 The C60-fluorescence quenching and the observation of C60˙− in the transient absorption spectra in addition to the 1400-nm band of SWCNT˙+ confirmed that C60˙−/SWCNT˙+ is formed via the CS-2 process, which was further confirmed by the photocatalytic MV˙+ accumulation in the presence of BNAH.
Next set of examples include triple-decker architectures via π–π stacking and coordination bond formation. Metal–ligand coordination is a widely used supramolecular approach to construct fullerene–porphyrin and fullerene–phthalocyanine dyads.8,9 This strategy is extended to the SWCNT hybrids (Fig. 15). Here, pyrene is functionalized to possess an imidazole coordinating ligand (PyrIm); thus, the appended Im can be used as a connector to ZnP, ZnPc, or ZnNc (zinc naphthalocyanine), as shown in Fig. 15.53,54
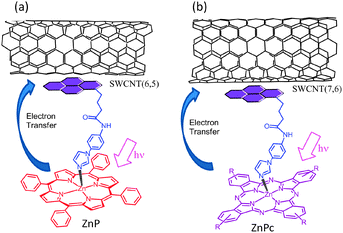 | ||
| Fig. 15 SWCNT/PyrIm→ZnP and SWCNT/PyrIm→ZnPc donor–acceptor hybrids via a metal–ligand axial coordination method. Modified from ref. 53 and 54. | ||
In the transient absorption spectra of SWCNT/PyrIm→ZnP, the peak at 640 nm is a diagnostic evidence of the ZnP˙+, while the 1300–1400 nm bands are assigned to SWCNT˙− (Fig. 16(a and b)), supporting the photoinduced CS-1 process via1ZnP*. Similarly, a set of bands in the 1300–1400 nm and 900 nm range supports the formation of SWCNT˙−/PyrIm→ZnPc˙+ (Fig. 16(c and d)). The absorption peaks of ZnP˙+ and ZnPc˙+ shift to the longer wavelength compared with the isolated cation radicals, probably due to the interaction of nearby SWCNTs. Compared with the spectra in Fig. 16(a and c) for SWCNT(6,5)˙− with the NIR peak at 1300 nm, the spectra in Fig. 16(b and d) for SWCNT(7,6)˙− show the NIR peak at 1400 nm; such spectroscopic changes can be observed only when the size-sorted SWCNT(n,m)s are employed.54
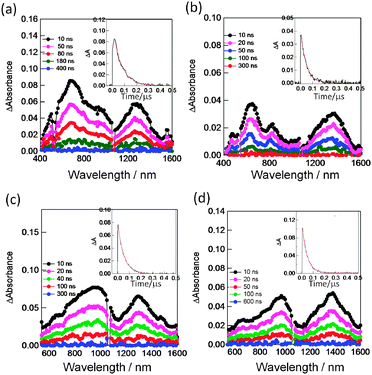 | ||
| Fig. 16 Nanosecond transient absorption spectra observed by the 532 nm (ca. 3 mJ per pulse) laser irradiation in Ar-saturated DCB. Inset: absorption-time profile. (a) SWCNT(6,5)˙−/PyrIm→ZnP˙+, (b) SWCNT(7,6)˙−/PyrIm→ZnP˙+, (c) SWCNT(6,5)˙−/PyrIm→ZnPc˙+ and (d) SWCNT(7,6)˙−/PyrIm→ZnPc˙+. Modified from ref. 54. | ||
Upon addition of HV2+ and BNAH, HV˙+ is accumulated as blue solution by the visible light irradiation of SWCNT(7,6)/PyrIm→ZnNc as shown in Fig. 17(a). It is important to note that the absorption intensity of ZnNc at 780 nm is remained constant during the formation of HV˙+, supporting that the ZnNc acts as a photocatalyst without being consumed in the photocatalytic HV˙+ accumulation process.53 Furthermore, the generated electron on SWCNT(n,m)s can be used for photovoltaic solar cells; the IPCE action spectrum of SWCNT(7,6)/PyrIm→ZnP on the FTO/SnO2 is shown in Fig. 17(b), in which the 420 nm peak and 500–650 nm peak correspond to the Soret band and Q bands of ZnP (inset of Fig. 17(b)), supporting that the photo-current is generated by the ZnP-excitation, but not SWCNT-excitation.54
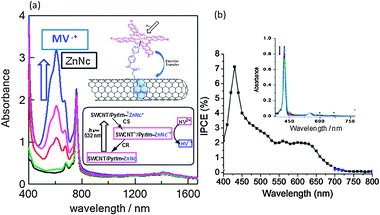 | ||
| Fig. 17 (a) Absorption spectra for the HV˙+-accumulation during the visible light illumination of the ZnNc part in SWCNT/PyrIm→ZnNc; ZnNc = 780 nm, HV˙+ = 620 nm. (b) IPCE action spectrum of SWCNT/PyrIm→ZnP on FTO/SnO2 in acetonitrile (I−/I3−). Inset: the ZnP absorption peaks at 420 nm and 550–600 nm. Modified from ref. 40 and 54. | ||
Light–current conversion efficiency based on the absorption intensity is higher for the Q-band region than that for the Soret band region, suggesting that the lowest excited state of ZnP is the origin of the CS process, while the second excited state energy may be consumed with some processes other than the CS process.
The next topic is ‘triple-decker’ architectures via π–π stacking and ion-pair interactions among the photosensitizer/glue/SWCNT nanohybrids. Here, charged tetrapyrroles as photo-sensitizers and the pyrene derivatives with opposite charges as glues are employed.52,53 Two examples are shown in Fig. 18; (a) negatively charged (TPPS−)M (tetrakis(4-sulfonatophenyl)porphyrin) is ion-paired with positively charged pyrNH3+, in which the Pyr moiety is π-stacked with the surface of SWCNT(n,m)s. (b) Positively charged (TMPyP+)M (tetrakis(4-N-methylpyridyl)porphyrin) is ion-paired with negatively charged PyrCOO−, in which the Pyr moiety is π-stacked with the surface of SWCNT(n,m)s. Formation of SWCNT(n,m)/PyrNH3+/(TPPS−)M and SWCNT(n,m)/PyrCOO−/(TMPyP+)M is confirmed by the TEM imaging and the steady-state absorption and fluorescence spectroscopic measurements.55,56
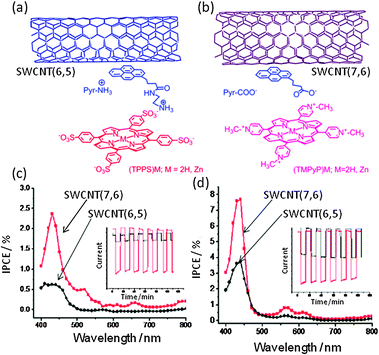 | ||
| Fig. 18 (a) SWCNT(6,5)/PyrNH3+/(TPPS−)M. (b) SWCNT(7,6)/PyrCOO−/(TMPyP+)Zn. (c) IPCE spectra of SWCNT(n,m)/PyrNH3+/(TPPS−)Zn. (d) IPCE spectra of SWCNT(n,m)/PyrCOO−/(TMPyP+)Zn. Inset: On–off current cycle. Modified from ref. 56. | ||
Occurrence of the photoinduced CS process via the excited singlet states of (TPPS−)M and (TMPyP+)M is confirmed by the transient absorption and time-resolved fluorescence measurements. The formation of the SWCNT˙−/PyrNH3+/(TPPS−)Zn˙+ ion-pair via1(TPPS−)Zn* in the CS-1 mechanism is anticipated, since (TPPS−)Zn has an electron rich porphyrin ring with a high LUMO level (see Fig. 10). As for SWCNT/pyrCOO−/(TMPyP+)M, the electron deficient (TMPyP+)M is capable of accepting an electron from the SWCNT, generating the SWCNT˙+/PyrNH3+/(TMPyP+)Zn˙− ion-pair via the CS-2 mechanism (see Fig. 10). The HV˙+-accumulation under visible light illumination of the porphyrin in the hybrid is observed, confirming the CS processes and also the photocatalytic cycle.
Studies of the photoelectrochemical solar cells using FTO/SnO2 electrodes modified with these donor–acceptor nanohybrids unanimously demonstrate the ability of these nanohybrids to harvest the visible light energy into electricity. Importantly, the photocurrent generation follows the trend observed for the CS rates and efficiency, in addition to the visible light-induced electron pooling in HV2+ as HV˙+. That is, the IPCE of a maximum of 8% at 420 nm was achieved for the photocell with SWCNT(7,6) and (TPPS−)Zn as sensitizers, while the IPCE value for the cell derived from SWCNT(6,5) and (TPPS−)Zn is smaller, reflecting the higher CS efficiency of the larger diameter SWCNT(7,6) with narrower band gap than that of the smaller diameter SWCNT(6,5) with wider band gap (see Fig. 18(d)).55,56 For SWCNT(n,m)/PyrCOO−/(TMPyP+)Zn, the IPCE values are smaller than the corresponding SWCNT(n,m)/PyrNH3+/(TPPS−)Zn, suggesting that the electronic properties and charges of the porphyrins play important roles. From these results, it is possible to achieve higher light-energy conversion efficiencies by the selection of the appropriate SWCNTs with the right band gap in combination with the given porphyrins.
The next topic is triple-decker architectures via π–π stacking and crown ether inclusion complex formation. Crown ether inclusion complexation has been widely adopted for the construction of the donor–acceptor conjugates using crown-ether appended porphyrins with alkyl ammonium cation functionalized fullerenes.57,58 This methodology can be also extended to SWCNT “Triple Decker” architectures using alkyl ammonium functionalized pyrene as an intermediate glue.
Cation–dipole interactions have also been utilized to assemble the SWCNT–donor hybrids as shown in Fig. 19,59,60 in which ZnP or ZnPc used as donors is functionalized to bear one or four benzo-18-crown-6 (crown) entities as ion–dipole interacting sites, while π-stacking pyrene is functionalized with an alkyl ammonium ion (PyrNH3+). Cation–crown ether inclusion complexation and pyrene–SWCNT π–π interactions result in SWCNT/PyrNH3+:(crown)nZnP (n = 1 or 4) or SWCNT/PyrNH3+:(crown)4ZnPc nanohybrids that are stable and soluble in DMF. After characterizing the SWCNT(n,m)/PyrNH3+:(crown)nMP conjugates using the TEM images and the spectroscopic methods, the CS process via the photoexcited ZnP and ZnPc can be confirmed using the time-resolved emission and transient absorption techniques.
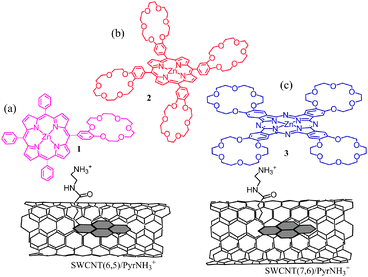 | ||
| Fig. 19 Structures of the nanohybrids of crown ether appended zinc porphyrin and zinc phthalocyanine derivatives with SWCNT(n,m) of different diameters viaPyrNH3+. (a) SWCNT(n,m)/PyrNH3+:(crown)ZnP, (b) SWCNT(n,m)/PyrNH3+:(crown)4ZnP, and (c) SWCNT(n,m)/PyrNH3+:(crown)4ZnPc, (: refers to the inclusion complex). Modified from ref. 59 and 60. | ||
For example, from the fluorescence quenching of ZnP and ZnPc upon complexing with the SWCNT via inclusion complexes, kSCS values in (3–5) × 109 s−1 are evaluated for SWCNT(n,m)/PyrNH3+:(crown)nMP in DMF. The transient absorption spectra showing the characteristic absorption bands of the radical ions and HV˙+-accumulation experiments support that the photoinduced CS process is certainly taking place in DMF. Furthermore, the photoelectrochemical solar cells constructed on the FTO/SnO2 show the IPCE action spectra with relatively high values. In Fig. 20, the IPCE action spectra for SWCNT(n,m)/PyrNH3+:(crown)nZnP are shown, in which the IPCE action spectra trace the absorption spectrum of ZnP, supporting the CS process via1ZnP*. The highest IPCE value is obtained to be 12% at 420 nm for SWCNT(7,6)/PyrNH3+:(crown)ZnP, which is higher than SWCNT(6,5)/PyrNH3+:(crown)ZnP. This trend is common for SWCNT(n,m)/PyrNH3+:(crown)4ZnP, although lower IPCE is obtained.59,60
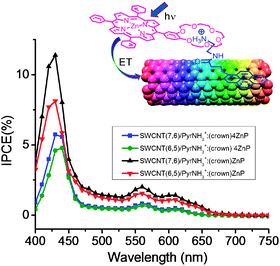 | ||
| Fig. 20 IPCE action spectra of SWCNT/PyrNH3+:(crown)ZnP/FTO/SnO2 and SWCNT/PyrNH3+:(crown)4ZnP/FTO/SnO2. Modified from ref. 60. | ||
Conclusions
As described in the present tutorial review, the photosensitizing nanocarbon materials are superior advanced materials to construct donor–acceptor hybrids, for achieving photoinduced charge separation processes, aiming at their applications to photovoltaics and photocatalysis. The successfully developed protocols involve the large curved nanocarbons in combination with the strongly light-absorbing dye molecules in the visible region. Although the electronic states of fullerenes belong to the usual molecules constructing donor–acceptor pairs, the band gaps of the SWCNTs are quite narrower than the HOMO–LUMO gaps of the photosensitizers. Therefore, the mechanisms of the photoinduced charge-separation processes are quite different, permitting construction of a variety of the donor–acceptor combinations.For fullerenes, the donor–acceptor hybrids are mostly constructed with the covalent bonding using the newly developed synthetic methods.61–65 In addition, non-covalent supramolecular constructions of the fullerene–donor hybrids have also been studied via the coordination and hydrogen-bonding self-assembly strategies. For SWCNTs, supramolecular approaches using π–π stacking, in addition to covalent bonding and alkyl chain intertwining, are used to adhere the glues, into which the photosensitizers are introduced via metal–ligand coordination, ion-pairing, and ion–dipole (with hydrogen bonding interactions) near the SWCNT surfaces. Although it is difficult to assess as to which self-assembly method is better for the formation of nanohybrids exhibiting efficient charge stabilization due to limited number of examples, it is clearly shown that the self-assembly approach with the π–π stacking largely preserves the electronic structures of the SWCNTs. Newer donor–acceptor hybrids are required to be built to probe distance and orientation aspects, by which through-bond and through-space electron transfer could be controlled. The role of host–guest chemistry adopted to create the nanohybrids in governing the efficiency of electron transfer and subsequent photochemical processes needs to be further clarified.
The photochemical behavior of donor–acceptor hybrids comprised of SWCNTs depends upon their electronic structures and their diameter (size) which controls their band gap and redox potentials. Thus, it is also possible to build donor–acceptor hybrids and demonstrate the diameter dependent photochemical properties. Studies performed to date have shown that it is possible to achieve higher efficiency of the photoinduced processes by the selection of the appropriately sized SWCNTs.
For future studies, in addition to fundamental studies involving photoinduced electron transfer originating in these supramolecular donor–acceptor conjugates of fullerenes and SWCNTs with different electronic properties and band gaps, practical applications especially in the areas of photovoltaics are promising, in addition to sensing, photocatalysis including hydrogen production, and building supercapacitors for energy storage. The robust donor–acceptor nanohybrids demonstrated here have revealed excellent photocatalytic behavior suggesting that improved catalytic schemes could be devised and tested for photolytic hydrogen production. The importance and practical results of photoelectrochemical solar cells of the nanohybrids point out that by choosing SWCNTs of appropriate diameter, it is possible to improve the light energy conversion efficiency. Much could be anticipated from these studies in the coming years.
Another important aspect is to extend the photosensitizing photochemistry of new donor–acceptor hybrids using endohedral fullerenes, since the redox potentials of the fullerene cages are much influenced by the entrapped metal or molecular ionic species, which would affect the scenario of the photoinduced events.65–67 More recently, 2D-carbon nanomaterials such as graphene and graphene oxide have attracted much attention, since these materials can be used to pair with photosensitizers as flat wide conductive electrode materials.68–70 Much more can be expected on these two- and three-dimensional materials in the coming years.
Acknowledgements
This work was financially supported by the National Science Foundation (Grant Nos. CHE-0804015 and CHE-1110942 to FD).Notes and references
- Introduction to Nanotechnology, ed. C. P. Poole and F. J. Owens, Wiley-Interscience, Weinheim, 2003 Search PubMed.
- Nanophysics and Nanotechnology: An Introduction to Modern Concepts in Nanoscience, ed. E. L. Wolf, John Wiley & Sons, New York, 2004 Search PubMed.
- J. L. Delgado, M. Á. Herranz and N. Martín, J. Mater. Chem., 2008, 18, 1417 RSC.
- S. Fukuzumi and T. Kojima, J. Mater. Chem., 2008, 18, 1427 RSC.
- F. D'Souza and O. Ito, Chem. Commun., 2009, 4913 RSC.
- A. S. D. Sandanayaka and O. Ito, J. Porphyrins Phthalocyanines, 2009, 13, 1017 CrossRef CAS.
- T. Hasobe, Phys. Chem. Chem. Phys., 2010, 12, 44 RSC.
- F. D'Souza and O. Ito, Electron Transfer in Self-assembled Supramolecular Fullerene Based Donor–Acceptor Conjugates, in Handbook of Organic Electronics and Photonics, ed. H. R. Nalwa, American Scientific Publishers, California, 2008, vol. 1, ch. 13, pp. 485–521 Search PubMed.
- F. D'Souza and O. Ito, Tetrapyrrole–Nanocarbon Hybrids: Self-Assembly and Photoinduced Electron Transfer, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing, New Jersey, 2010, vol. 1, pp. 307–438 Search PubMed.
- V. Sgobba, G. M. A. Rahman and D. M. Guldi, in Carbon Nanotubes in Electron Donor–Acceptor Nanocomposites, Chemistry of Carbon Nanotubes, ed. V. A. Basiuk, American Scientific Publishers, California, 2006 Search PubMed.
- T. Arai, S. Nobukuni, A. D. S. Sandanayaka and O. Ito, J. Phys. Chem. C, 2009, 113, 14493 CAS.
- V. Sgobba and D. M. Guldi, Chem. Soc. Rev., 2009, 38, 165 RSC.
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768 CrossRef CAS.
- C. Ehli, G. M. A. Rahman, N. Jux, D. Balbinot, D. M. Guldi, F. Paolucci, M. Marcaccio, D. Paolucci, M. Melle-Franco, F. Zerbeto, S. Campidelli and M. Prato, J. Am. Chem. Soc., 2006, 128, 11222 CrossRef CAS.
- R. Chitta, A. S. D. Sandanayaka, A. L. Schumacher, Y. Araki, O. Ito and F. D'Souza, J. Phys. Chem. C, 2007, 111, 6947 CAS.
- F. D'Souza, R. Chitta, A. S. D. Sandanayaka, N. K. Subbaiyan, L. D'Souza, Y. Araki and O. Ito, Chem.–Eur. J., 2007, 13, 8277 CrossRef CAS.
- A. S. D. Sandanayaka, R. Chitta, N. K. Subbaiyan, L. D'Souza, O. Ito and F. D'Souza, J. Phys. Chem. C, 2009, 113, 13425 CAS.
- E. Maligaspe, A. S. D. Sandanayaka, T. Hasobe, O. Ito and F. D'Souza, J. Am. Chem. Soc., 2010, 113, 81584 Search PubMed.
- A. S. D. Sandanayaka, Y. Takaguchi, Y. Sako, M. Tamura and O. Ito, Adv. Sci. Lett., 2010, 3, 353 CrossRef CAS.
- O. Ito and K. Yamanaka, Bull. Chem. Soc. Jpn., 2009, 82, 316 CrossRef CAS.
- T. Nojiri, M. M. Alam, H. Konami, A. Watanabe and O. Ito, J. Phys. Chem. A, 1997, 101, 7943 CrossRef CAS.
- T. Nojiri, A. Watanabe and O. Ito, J. Phys. Chem. A, 1998, 102, 5215 CrossRef CAS.
- L. Sanchez, M. A. Herranz and N. Martín, J. Mater. Chem., 2005, 15, 1409 RSC.
- J. W. Verhoeven, J. Photochem. Photobiol., C, 2006, 7, 40 CrossRef CAS.
- Y. Araki and O. Ito, J. Photochem. Photobiol., C, 2008, 9, 93 CrossRef CAS.
- S. Komamine, M. Fujitsuka, O. Ito, K. Moriwaki, T. Miyata and T. Ohno, J. Phys. Chem., 2000, 104, 11497 CAS.
- M. Yamazaki, Y. Araki, M. Fujitsuka and O. Ito, J. Phys. Chem. A, 2001, 105, 8615 CrossRef CAS.
- G. A. Rajkumar, A. S. D. Sandanayaka, K. Ikeshita, M. Itou, Y. Araki, Y. Furusho, N. Kihara, O. Ito and T. Takata, J. Phys. Chem. A, 2005, 109, 2428 CrossRef CAS.
- H. Imahori and S. Fukuzumi, Adv. Funct. Mater., 2004, 14, 525 CrossRef CAS.
- M. Ince, M. V. Martínez-Díaz, J. Barberá and T. Torres, J. Mater. Chem., 2011, 21, 1531 RSC.
- M. Fujitsuka, O. Ito, H. Imahori, K. Yamada, H. Yamada and Y. Sakata, Chem. Lett., 1999, 28, 721 CrossRef.
- H. Imahori, M. E. El-Khouly, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Phys. Chem. A, 2001, 105, 325 CrossRef CAS.
- H. Imahori, Y. Mori and Y. Matano, J. Photochem. Photobiol., C, 2003, 4, 51 CrossRef CAS.
- P. K. Poddutoori, A. S. D. Sandanayaka, T. Hasobe, O. Ito and A. van der Est, J. Phys. Chem. B, 2010, 114, 14348 CrossRef CAS.
- M. Fujitsuka, O. Ito, T. Yamashiro, Y. Aso and T. Otsubo, J. Phys. Chem. A, 2000, 104, 4876 CrossRef CAS.
- K. Matsumoto, M. Fujitsuka, T. Sato, S. Onodera and O. Ito, J. Phys. Chem. B, 2001, 104, 11632 CrossRef.
- M. Fujitsuka, K. Matsumoto, O. Ito, T. Yamashiro, Y. Aso and T. Otsubo, Res. Chem. Intermed., 2001, 27, 73 CrossRef CAS.
- M. Fujitsuka, A. Masuhara, H. Kasai, H. Oikawa, H. Nakanishi, O. Ito, T. Yamashiro, Y. Aso and T. Otsubo, J. Phys. Chem. B, 2001, 105, 9930 CrossRef CAS.
- Y. Kunugi, K. Takimiya, N. Negishi, T. Otsubo and Y. Aso, J. Mater. Chem., 2004, 14, 2840 RSC.
- T. Nakamura, M. Fujitsuka, Y. Araki, O. Ito, J. Ikemoto, K. Takimiya, Y. Aso and T. Otsubo, J. Phys. Chem. B, 2004, 108, 10700 CrossRef CAS.
- L. Sánchez, M. Sierra, N. Martín, A. J. Myles, T. J. Dale, J. Rebek Jr., W. Seitz and D. M. Guldi, Angew. Chem., Int. Ed., 2006, 45, 4637 CrossRef.
- S. Gadde, D.-M. S. Islam, C. A. Wijesinghe, N. K. Subbaiyan, M. E. Zandler, Y. Araki, O. Ito and F. D'Souza, J. Phys. Chem. C, 2007, 111, 12500 CAS.
- F. D'Souza, G. M. Venkadasula, K. Yamanaka, N. K. Subbaiyan, M. E. Zandler and O. Ito, Org. Biomol. Chem., 2009, 7, 1076 CAS.
- M. C. Hersam, Nat. Nanotechnol., 2008, 3, 387 CrossRef CAS.
- T. Tanaka, H. Jin, Y. Miyata and H. Kataura, Appl. Phys. Express, 2008, 1, 14001 CrossRef.
- S. Kilina, S. Tretiak, S. K. Doorn, Z. Luo, F. Papadimitrakopoulos, A. Piryatinski, A. Saxena and A. R. Bishop, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6797 CrossRef CAS.
- NanoIntegris (Skokie, IL, USA).
- CoMoCAT®, South West Nano Technologies, Inc. (Norman, OK, USA).
- F. D'Souza, A. S. D. Sandanayaka and O. Ito, J. Phys. Chem. Lett., 2010, 1, 2586 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Annu. Rev. Phys. Chem., 2007, 58, 71 CrossRef.
- T. Arai, S. Nobukuni, A. D. S. Sandanayaka and O. Ito, J. Phys. Chem. C, 2009, 113, 14493 CAS.
- A. S. D. Sandanayaka, E. Maligaspe, T. Hasobe, O. Ito and F. D'Souza, Chem. Commun., 2010, 46, 8749 RSC.
- R. Chitta, A. S. D. Sandanayaka, A. L Schumacher, L. D'Souza, Y. Araki, F. D'Souza and O. Ito, J. Phys. Chem. C, 2007, 111, 6947 CAS.
- S. K. Das, N. K. Subbaiyan, F. D'Souza, A. S. D. Sandanayaka, T. Wakahara and O. Ito, J. Porphyrins Phthalocyanines Search PubMed , in press.
- A. S. D. Sandanayaka, R. Chitta, N. K. Subbaiyan, L. D'Souza, O. Ito and F. D'Souza, J. Phys. Chem. C, 2009, 113, 13425 CAS.
- S. K. Das, N. K. Subbaiyan, F. D'Souza, A. S. D. Sandanayaka, T. Hasobe and O. Ito, Energy Environ. Sci., 2011, 4, 707 CAS.
- A. S. D. Sandanayaka, Y. Araki, O. Ito, R. Chitta, S. Gadde and F. D'Souza, Chem. Commun., 2006, 4327 RSC.
- O. Ito, A. S. D. Sandanayaka, R. Chitta and F. D'Souza, J. Indian Chem. Soc., 2009, 86, 1 Search PubMed.
- F. D'Souza, R. Chitta, A. S. D. Sandanayaka, N. K. Subbaiyan, L. D'Souza, Y. Araki and O. Ito, Chem.–Eur. J., 2007, 13, 8277 CrossRef CAS.
- A. S. D. Sandanayaka, N. K. Subbaiyan, R. Chitta, E. Maligaspe, T. Hasobe, O. Ito and F. D'Souza, ChemPhysChem, 2011, 12, 2266 CrossRef CAS.
- Handbook of Carbon NanoMaterials, ed. F. D'Souza and K. M. Kadish, World Scientific, Singapore, 2011, vol. 1 and 2 Search PubMed.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS.
- G. de la Torre, G. Bottari, U. Hahn and T. Torres, Struct. Bonding, 2010, 135, 1 CrossRef CAS.
- N. Martin, L. Sanchez, B. Illescas and I. Perez, Chem. Rev., 1998, 98, 2527 CrossRef CAS.
- Y. Takano, M. Á. Herranz, N. Martín, S. G. Radhakrishnan, D. M. Guldi, T. Tsuchiya, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2010, 132, 8048 CrossRef CAS.
- D. M. Guldi, L. Feng, S. G. Radhakrishnan, H. Nikawa, M. Yamada, N. Mizorogi, T. Tsuchiya, T. Akasaka, S. Nagase, M. Á. Herranz and N. Martín, J. Am. Chem. Soc., 2010, 132, 9078 CrossRef CAS.
- L. Feng, Z. Slanina, S. Sato, K. Yoza, T. Tsuchiya, N. Mizorogi, T. Akasaka, S. Nagase, N. Martín and D. M. Guldi, Angew. Chem., Int. Ed., 2011, 50, 5909 CrossRef CAS.
- N. Karousis, A. S. D. Sandanayaka, T. Hasobe, S. P. Economopoulos, E. Sarantopoulous and N. Tagmatrarchis, J. Mater. Chem., 2010, 21, 109 RSC.
- G. Bottari, D. Olea, V. López, C. Gomez-Navarro, F. Zamora, J. Gómez-Herrero and T. Torres, Nanoscale, 2011, 3, 20 RSC.
- X.-F. Zhang and Q. Xi, Carbon, 2011, 49, 3841 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |