 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Variable coordination of a chiral diphosphine containing an amidinium/NHC group within its backbone: μ-P,P′, κ2-P,P′ and κ3-P,C,P′ coordination modes‡†
Paul D.
Newman
*a,
Kingsley J.
Cavell
a and
Benson M.
Kariuki
b
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: newmanp1@cardiff.ac.uk; Fax: +44 (0)29 20874030; Tel: +44 (0)29 20870464
bSchool of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK
First published on 15th August 2012
Abstract
A diphosphine ligand (1·HPF66666), which is a potential precursor to a PCNHCP pincer, with a backbone containing two phenylene groups and a central bicyclic 4-aza-2-azoniabicyclo[3.2.1]oct-2-ene unit has been synthesised and coordinated to Pd(II) and Pt(II) to give trans-[M(κ2-P,P′-1·H)Cl2]PF6 where M = Pd (2) or Pt (3a). Single-crystal structure determinations of 2 and 3a show the complexes to be isostructural with the diphosphine coordinated in a trans-spanning fashion and the amidinium unit being protonated and non-coordinated. 2 and 3a react with CH3I to give the dimers trans-[Pd2(μ-1·H)2I4](PF6)2, 6, and trans-[Pt2(μ-1·H)2I4](PF6)2, 7, as the major products. This bridging mode of coordination of [1·H]+ is also seen in trans-[Rh2(μ-1·H)(1,5-COD)2Cl2]PF6, 4, and [Pt2(μ-κ2-1·H)(dvdms)]PF6, 5. Upon treatment with KOtBu complexes 2 and 3a undergo deprotonation at the amidinium carbon to give trans-[M(κ3-P,C,P′-1)Cl]PF6 where M = Pd (8), and Pt (9). The related trans-[Rh(κ3-P,C,P′-1)(CO)]PF6 (10) is prepared directly from 1·HPF66666 and Rh(acac)(CO)2: this and the palladium and platinum complexes 8 and 9 are isolated as isomeric mixtures as a consequence of a conformational isomerism. In situ deprotonation of 1·HPF66666 followed by addition of Ag(CF3SO3) gave SAg-[Ag(κ3-P,C,P′-1)(CF3SO3)], 11. Some preliminary studies of the reactivity of 2 and 8 in Suzuki-type reactions are reported and the Pt(0) system has been shown to be an active hydrosilylation catalyst.
Introduction
Wide angle diphosphines have been heavily exploited in homogeneous catalysis as demonstrated by the studies of van Leeuwen and others on the hydrocyanation and hydroformylation of alkenes.1 The success of such systems, e.g. the Xanthos series, resides in the ability of the two phosphine donors to adopt an obtuse bite angle enforced by the rigid nature of the ligand backbone. These relatively fixed bite angles, which typically lie between 110 and 130°, assist in promoting metal-based catalysis but preclude true trans coordination at a square planar metal. Other diphosphines are able to coordinate to give P–M–P angles of ∼180° and these are defined as trans-spanning ligands.1e,f,2 These are generally of two types: those with rigid backbones that strongly favour trans-coordination which can restrict catalysis through 2e redox processes;2a,3 and those that are flexible and able to promote catalysis through rearrangement to active cis- or monodentate forms.2a,4 Depending on the nature of the intervening ligand architecture, some trans-spanning diphosphines can undergo metallation at an atom in the ligand backbone to give PXP pincer complexes (where X is often carbon).5 Such complexes (which are not restricted to a PCP donor set) are finding increasing employment in homogeneous catalysis as they are usually robust by nature and able to withstand the high temperatures demanded by certain types of transformation notably alkane dehydrogenation.6 They are also proving useful in non-oxidative bond activation through metal–ligand cooperation.7The range of pincer ligands containing neutral NHC carbon donors includes those with a single central carbene donor8 and those with two carbenes in the terminal positions9 while homoleptic tricarbenes tend to be of the facial type.10 We are interested in expanded-ring NHC-based tridentates incorporating a central CNHC donor and have recently reported a dipyridyl-ER-NHC (Lpy2) containing a bicyclic core derived from camphor.11 These earlier investigations established some of the coordination chemistry of the ligand and highlighted its relative flexibility as a N,CNHC,N′ donor with both planar and non-planar species being isolated. In [Ni(κ3-N,C,N′-Lpy2)Cl]+ where the three donor atoms and the metal are essentially coplanar, twisting in the ligand backbone was observed giving rise to λ and δ conformational isomers.11a However the related Pd(II) and Pt(II) complexes of Lpy2 proved elusive. Isomerism of a different type was also observed in the [Rh(1,5-COD)(Lpy2)]+ and [Ir(1,5-COD)(Lpy2)]+. The ligand occupies one axial and two equatorial sites in these trigonal bipyramidal five-coordinate complexes and the source of the isomerism here lies in the relative orientation of the central bicyclic structure and the pyridyl groups leading to endo,endo or exo,exo species.11b
As coordination flexibility in multidentate ligands can have a profound effect on catalytic performance, we were interested in exploring the coordination behaviour of NHC based systems bearing donors other than pyridine groups. As part of this broader study we have opted to explore the coordination chemistry of a diphosphino-ER-NHC containing the bicyclic 6/7 membered NHC skeleton, the initial results of which are presented here.
Results and discussion
Pro-ligand synthesis
The synthesis of the pro-ligand 1·HPF66666 is highlighted in Scheme 1 and follows an already established procedure for the related dipyridyl analogue.11a The condensation of two mols of 2-(diphenylphosphino)benzaldehyde with 1R,3S-diamino-1,2,2-trimethylcyclopentane gives the diimine as an off-white solid. Subsequent reduction with NaBH4 produces the diamine which is readily condensed with triethylorthoformate in the presence of NH4PF6 to give 1·HPF66666 as a white solid. The salt is relatively air-stable in the solid state but does slowly oxidise when in solution. The two phosphine groups in 1·HPF66666 are different and, although the source of this distinction is remote (H versus CH3 at the ε position), two signals are observed in the 31P{1H} NMR spectrum at δP = −16.6 and −17.5 ppm. The 1H NMR spectrum shows the NCHN proton at δH = 7.79 ppm which compares to a value of 8.14 ppm for this hydrogen in the dipyridyl derivative Lpy2.11a The only other signals of any diagnostic value in the 1H spectrum are those for the benzylic hydrogens which occur as three doublets of doublets between 4.84 and 4.42 ppm and a doublet at 4.28 ppm. The pro-ligand is only modestly soluble in alcohol and (although it appears to slowly oxidise in CDCl3) can be crystallised from MeOH in air to give crystals suitable for structural analysis by single-crystal X-ray techniques. The molecular structure of the compound is shown in Fig. 1. The C–N bond lengths to the amidinium carbon in the two independent molecules of the unit cell average 1.315 Å and the C–N–C angles are 123.1° (average). These compare to values of 1.314(3) Å and 123.5(2)° and 1.307(6) Å and 123.0(4)° in the dibenzyl and dipyridyl derivatives respectively.11a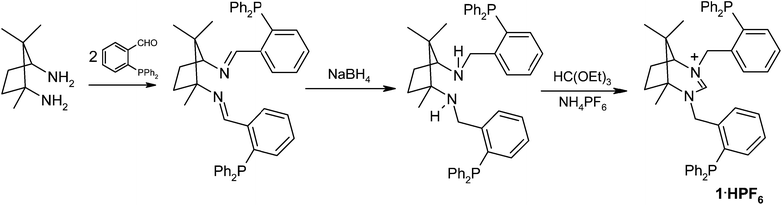 | ||
| Scheme 1 Synthesis of pro-ligand 1·HPF66666. | ||
![Ortep view of the molecular structure of [1·H]+. For clarity the PF6− anion is not shown. Thermal ellipsoids are drawn at 50% probability, hydrogens are omitted for clarity. Selected bond lengths (Å) and angles (°) for [1·H]+: C1–N1 1.334(7), C1–N2 1.316(8), N1–C1–N2 122.7(6).](/image/article/2012/DT/c2dt31475d/c2dt31475d-f1.gif) | ||
| Fig. 1 Ortep view of the molecular structure of [1·H]+. For clarity the PF6− anion is not shown. Thermal ellipsoids are drawn at 50% probability, hydrogens are omitted for clarity. Selected bond lengths (Å) and angles (°) for [1·H]+: C1–N1 1.334(7), C1–N2 1.316(8), N1–C1–N2 122.7(6). | ||
![(i) K2[PdCl4], EtOH, Δ; (ii) K2[PdCl4], EtOH, Δ; (iii) xs MeI, Δ; (iv) KOtBu; (v) 0.5 [Rh(1,5-COD)Cl]2, MeOH, Δ; (vi) Pt2(dvdms)3; (vii) [Rh(acac)(CO)2], THF, Δ; (viii) KOtBu, Ag(OTf).](/image/article/2012/DT/c2dt31475d/c2dt31475d-s2.gif) | ||
| Scheme 2 (i) K2[PdCl4], EtOH, Δ; (ii) K2[PdCl4], EtOH, Δ; (iii) xs MeI, Δ; (iv) KOtBu; (v) 0.5 [Rh(1,5-COD)Cl]2, MeOH, Δ; (vi) Pt2(dvdms)3; (vii) [Rh(acac)(CO)2], THF, Δ; (viii) KOtBu, Ag(OTf). | ||
(κ2-P,P′-1·H) complexes
The design of the ligand is such that coordination as a bidentate through both phosphorus atoms can lead to cis- and trans-isomers although the latter may be encouraged by the lengthy and semi-rigid backbone. Initial interest was to see whether κ2-P,P′ coordination could be achieved without activation of the amidinium group to investigate potential cis/trans isomerism in the bidentate systems. When one mol of 1·HPF66666 and K2[PdCl4] were heated in EtOH at 70 °C a bright yellow solid precipitated from the solution The isolated complex gives an AB pattern in the 31P{1H} NMR spectrum with δP values of 24.7 and 22.3 ppm respectively. The large magnitude (542 Hz) of the 2JP–P coupling constant suggests that the two inequivalent phosphorus donors are mutually trans.2a,b The compound is confirmed as trans-[Pd(κ2-P,P′-1·H)Cl2]PF6, 2, through determination of its molecular structure by single-crystal X-ray techniques (Fig. 2). The P–Pd–P bond angles average 173.6° for the two independent molecules in the unit cell and the average Pd–P bond length of 2.344 Å is slightly longer than the 2.322 Å and 2.316 Å observed in related trans-spanning diphosphine complexes of palladium.4a,12 The Pd–Cl bond lengths average 2.293 Å and compare with values of 2.292 and 2.312 Å for the related complexes.4a,12 The amidinium group has not undergone deprotonation during the preparation of 2 but it does appear pre-set for coordination upon treatment with base; this conclusion derives from the orientation of the NCN carbon which points towards the metal. This conformation is favoured by an intramolecular electrostatic interaction between the positively charged amidinium group and the chloride ligands that manifests itself as a noticeable bending of the two chlorides towards the amidinium group. Although the average Cl–Pd–Cl bond angle of 170.0° is not atypical, as similar or even greater distortions are observed in related trans-Pd(P2)Cl2 complexes,2a the nature of the distortion in 2 is distinct as the flexing in these other complexes is away from the backbone of the diphosphine as dictated by steric effects.2a,4a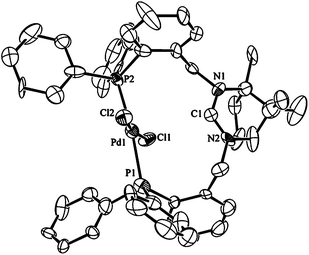 | ||
| Fig. 2 Ortep view of the molecular structure of 2. Thermal ellipsoids are drawn at 50% probability, hydrogens are omitted for clarity. Selected bond lengths (Å) and angles (°) for 2: Pd1–P1 2.315(5), Pd1–P2 2.356(5), Pd1–Cl1 2.297(4), Pd1–Cl2 2.306(5), C1–N1 1.34(2) C1–N2 1.38(2), P1–Pd1–P2 173.2(2), Cl1–Pd1–Cl2 170.0(2), P1–Pd1–Cl1 85.30(16), P1–Pd1–Cl2 93.52(16), N1–C1–N2 118.3(17). | ||
The 1H NMR spectrum of 2 shows the NCN hydrogen at δH 9.58 ppm, a shift of 1.79 ppm downfield of its position in the uncoordinated ligand. This may be a consequence of the electrostatic (possibly weak H-bonding) interaction with the Cl ligands or might be a result of its position along the z axis of the palladium. Downfield shifts for hydrogens orientated in relatively fixed positions over a palladium or platinum coordination plane have been noted previously.2i,j All four hydrogens of the methylene groups are inequivalent as evidenced by the presence of four doublets in the 1H NMR spectrum between 5.02 and 4.61 ppm with 2JH–H coupling constants of 14.5 and 15.2 Hz. The remainder of the 1H NMR spectrum is less informative providing little diagnostic information. The amidinium carbon is observed at δC = 154.5 ppm in the 13C{1H} NMR spectrum of 2. This is little shifted from its position in the spectrum for the free salt. The remainder of the 13C{1H} NMR spectrum mimics closely that for 1·HPF66666 except there is no observable coupling between the benzylic carbons and the phosphorus centre.
The complex [Pt(κ2-P,P′-1·H)Cl2]PF6 is isolated as a cream solid from the reaction of 1·HPF66666 with K2[PtCl4] in EtOH. Inspection of the 31P{1H} NMR spectrum of the isolated solid shows it to be a mixture of two complexes: a major species 3a described by an AB pattern in the 31P{1H} NMR spectrum and a minor species 3b that gives a broad singlet (see below). The two complexes were separated by simple treatment with CHCl3 which readily dissolved the major compound leaving the minor component as an insoluble solid.
The chloroform soluble complex 3a gives an AB pattern in the 31P{1H} NMR spectrum at δP = 20.0 (1JP–Pt = 2523 Hz) and 19.7 (1JP–Pt = 2526 Hz) ppm. The 1H NMR spectrum is closely similar to that for trans-[Pd(κ2-P,P′-1·H)Cl2]PF6 with a downfield shifted NCHN resonance at 9.90 ppm. The similarity in the spectra for this complex and 2 suggests that they are isostructural which is confirmed upon inspection of the solid state structure (Fig. 3). The Pt–P bond lengths average 2.332(5) Å which are close to the value of 2.304(5) Å seen for Pt(SPANphos)Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3b and 2.3204(12) Å in a related complex.4c The P–Pt–P angle however is significantly more obtuse at 178.9(2)° in 3a compared to the lower values of 171.9(4)° and 174.43(5)° seen with the reported complexes.
3b and 2.3204(12) Å in a related complex.4c The P–Pt–P angle however is significantly more obtuse at 178.9(2)° in 3a compared to the lower values of 171.9(4)° and 174.43(5)° seen with the reported complexes.
The broad resonance seen in the 31P{1H} NMR spectrum of the minor product 3b sharpens to a singlet (δP = 13.5 ppm, 1JP–Pt = 2556 Hz) upon heating to 80 °C in d6-DMSO. The 1H NMR spectrum recorded at this temperature showed the amidinium hydrogen at 8.31 ppm which is not significantly shifted from its position in the spectrum of the uncoordinated ligand. The benzylic CH2 hydrogens are inequivalent and appear as four separate resonances between δH = 5.80 and 4.77 ppm. One of the methyl groups is seen to resonate at an unusually high position (0.17 ppm) in the 1H NMR spectrum of 3b which might indicate a residence within the shielding region of one or more of the aromatic rings. Although the two phosphorus centres are inequivalent they clearly have the same chemical shift in the 31P{1H} NMR spectrum of 3b as only one resonance is seen. The magnitude of the 1JP–Pt coupling constant would suggest that it is not a cis-isomer as a larger J value would have been expected if the phosphines were trans to chloride.13 Thus the complex has likely trans-P donors but is seemingly not a monomer as the chloroform soluble compound 3a has been shown to be this species. The most likely structure of this complex is therefore the dimer trans-[Pt2(μ-1·H)2(Cl)4](PF6)2, although this could not be confirmed by mass spectrometry as only monomeric species were observed for the compound.
(μ-1·H) complexes
The 1:1 reaction of 1·HPF66666 and [Rh(1,5-COD)Cl]2 in MeOH at near reflux led to the formation of a light yellow solid. Inspection of the 1H NMR spectrum of the isolated solid revealed the presence of coordinated COD and [1·H]+ in a 2:1 ratio. Other characteristic features of the 1H NMR spectrum include the amidinium hydrogen at δH = 8.18 ppm and the four inequivalent benzylic methylene hydrogens which appear as four doublets between δH 6.5 and 5.6 ppm. The relative ratio of the [1·H]+ and COD resonances coupled with the chemical shift of the amidinium hydrogen would suggest that the isolated complex is [Rh2(1,5-COD)2(μ-1·H)Cl2]PF6, 4. The 31P{1H} NMR spectrum consists of two doublets at δP 21.7 and 21.4 with 1JP–Rh being 146 Hz. Although a coordination shift is evident and both resonances show coupling to rhodium, the lack of any 2JP–P coupling supports the above assignment where the amidinium diphosphine is coordinating in a bridging mode. The magnitude of the 1JP–Rh coupling is commensurate with that expected for a triarylphosphine trans to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C donor of COD as noted in several complexes of the type [Rh(1,5-COD)(PAr3)Cl].14
C donor of COD as noted in several complexes of the type [Rh(1,5-COD)(PAr3)Cl].14
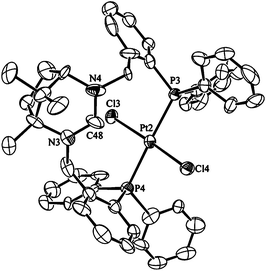 | ||
| Fig. 3 Ortep view of the molecular structure of 3a. Thermal ellipsoids are drawn at 50% probability, hydrogens are omitted for clarity. Selected bond lengths (Å) and angles (°) for 3a: Pt2–P3 2.329(5), Pt2–P4 2.315(4), Pt2–Cl3 2.310(5), Pt2–Cl4 2.303(4), C48–N3 1.31(2), C48–N4 1.32(2), P3–Pt2–P4 174.9(3), Cl3–Pt2–Cl4 174.73(19), P3–Pt2–Cl3 87.63(14), P3–Pt2–Cl4 94.15(14), N3–C48–N4 120.5(19). | ||
The reaction of 1·HPF66666 with Karstedt's complex Pt2(dvdms)3 in THF gave an air-stable cream coloured solid upon work-up. The 31P{1H} NMR spectrum consisted of two singlets at 20.7 and 18.7 ppm each with Pt-195 satellites (1JP–Pt = 3494 and 3506 Hz, respectively). As for the rhodium complex 4 above, the presence of both a coordination shift and P–Pt coupling confirms coordination of the phosphine donors and the absence of any 2JP–P coupling indicates a μ bonding mode. Inspection of the 1H NMR spectrum of the complex reveals the amidinium hydrogen at δH 8.18 ppm which is exactly as observed in the Rh dimer 4. Integration of selected resonances of the diphosphine and the dvdms reveal the ratio to be 1:2 and the most likely formulation of the product is [Pt2(dvdms)2(μ-1·H)]PF6. This accords with similar systems reported by Lappert where comparable values of Δδ and 1JP–Pt are noted.15 This assignment is further supported upon inspection of the 195Pt NMR spectrum which shows two doublets of multiplets at δPt = −5553 and −5563 ppm for the two platinum centres in the dimer. These chemical shift values again compare with related Pt(PR3)(dvdms)2 complexes.15,16
When 2 was reacted with excess MeI at 50 °C in CHCl3 an orange precipitate was formed. Inspection of the 31P{1H} NMR spectrum of the product showed a singlet at δP = 21.1 ppm. The 1H NMR spectrum was similar to that for 2 albeit with notable shifts in δH for the amidinium hydrogen and the benzylic CH2 hydrogens. The MS data showed a peak for the complex [Pd(1·H)I2]+ at 1063 amu confirming that the chloride ligands had been exchanged for iodide presumably though two successive oxidative addition (MeI)/reductive elimination (MeCl) cycles. The similar spectroscopic features of 2 and 6 suggest that 6 is a monomer containing trans-bound [1·H]+. The orange solid could be recrystallised from MeCN to give crystals suitable for analysis by single-crystal X-ray techniques (Fig. 4). As can be seen from the structure shown in Fig. 4 the complex has rearranged upon recrystallisation to give the [Pd2(μ-1·H)2I4]I0.27(PF6)1.73 species containing two bridging [1·H]+ ligands. The coordination about each of the Pd centres is the expected square planar arrangement with the P-donors (and by necessity the I− ligands) being mutually trans. The Pd–P bond lengths average 2.33 Å which is slightly shorter than in 2 indicating a small level of strain in the chelate. The P–Pd–P angles are unexpectedly acute at 166° compared to the 174° in 2 while the I–Pd–I angles at 169° are similar to the value of 170° seen for the Cl–Pd–Cl angle in 2. The nature of the bending is such that the palladium atoms point inwards with regard to the cavity defined by the dimetallo macrocycle (Fig. 4). When the crystals were redissolved in CD3CN for spectroscopic analysis, the resultant 31P{1H} and 1H NMR spectra were complex indicating the presence of a number of species in solution. Substitution of up to four iodides in the dimer by CD3CN is one possible source of solution complexity, however it seems likely that the μ-1·H coordination mode of the diphosphine is not so robust that bidentate modes are precluded in solution.
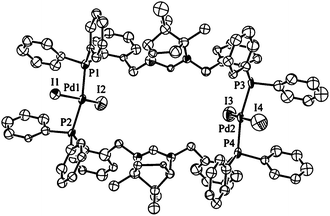 | ||
| Fig. 4 Ortep view of the molecular structure of 6. Thermal ellipsoids are drawn at 50% probability, hydrogens are omitted for clarity. Selected bond lengths (Å) and angles (°) for 6: Pd1–P1 2.321(7), Pd1–P2 2.330(6), Pd1–I1 2.632(2), Pd1–I2 2.592(3), Pd2–P3 2.362(7) Pd2–P4 2.295(6), Pd2–I3 2.565(2), Pd2–I4 2.531(3), P1–Pd1–P2 166.36(18), I1–Pd1–I2 168.06(8), P1–Pd1–I1 88.31(17), P1–Pd1–I2 91.13(18), P3–Pd2–P4 167.69(17), I3–Pd2–I4 168.95(9), P3–Pd2–I3 91.30(18), P3–Pd2–I4 87.23(17). | ||
The reaction of 3a or 3b (or a mixture of both) with MeI gave an orange solid 7 that showed a similar pattern to the 3a/b mixture in its 31P{1H} NMR spectrum. Significantly however the position of the resonances had shifted ∼10 ppm upfield commensurate with the replacement of the two chloride ligands in 3a/b with iodide. This was confirmed by mass spectrometry where a species correlating to [Pd(1·H)I2]+ was observed at 1151 amu. The magnitude of the 1JP–Pt coupling constants were comparable to those in 3a/b as expected for phosphorus donors situated mutually trans. No attempt was made to separate the mixture.
(κ3-P,C,P′-1) complexes
In the examples discussed above the diphosphine ligand coordinates solely through the phosphorus atoms and the third potential donor (the NHC carbon) is protonated and hence non-coordinating. The solid-state structures of the platinum and palladium complexes of κ2-[1·H]+ show this carbon to be poised for coordination upon removal of the proton so that tridentate forms should be accessible upon addition of base to 2 and 3a. When 2 was treated with 1.2 equivalents of KOtBu in THF the colour of the solution changed from yellow to a deep blood-red and the complex [Pd(κ3-P,C,P′-1)Cl]PF6, 8, was isolated as a red solid after work-up. Inspection of the 31P{1H} NMR spectrum of 8 revealed the presence of two sets of AB doublets around 5 ppm suggesting the presence of two isomers in solution. This was confirmed upon observation of the 1H NMR spectrum where a total of six CH3 resonances and eight separate doublets for the benzylic methylenes were seen. Some notable features of the 1H NMR spectrum include an unusual chemical shift for two of the methyl resonances to highfield of TMS and a large δH separation for the hydrogens of any given benzylic–CH2 group with one resonating over 2 ppm downfield of its geminal partner. It would appear that the binding of the ligand in the κ3-mode has placed one of the methyl groups of each isomer in a region that experiences a local shielding effect presumably from one or more of the aromatic rings. Similarly, one geminal hydrogen of each benzylic group in either isomer resides in a deshielded zone. Isomerisation in heterodonor terdentates containing a central NHC donor has been observed by us previously in square planar and five-coordinate trigonal bipyramidal metal systems.11 Conformational differences in the ligand backbone are the source of the isomerism which is dictated by the desire of the NHC donor to be canted with respect to the coordination plane as shown schematically in Fig. 5; in related square planar metal systems where the NHC does not have pendant donors the NCN link of the carbene donor is known to lie approximately perpendicular to the coordination plane.11a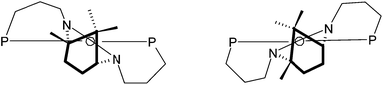 | ||
| Fig. 5 The two conformers for κ3-P,C,P′-1 at a square planar metal centre. | ||
As noted above the 31P{1H} NMR spectrum of 8 consists of two AB patterns for the two conformers with 2JP–P coupling appreciably smaller at 436 and 435 Hz than observed in 2. More surprising is the upfield shift in δP that accompanies the change from κ2 to κ3 binding as the reverse (downfield) trend is more often observed upon formation of smaller chelates. These more familiar downfield shifts often reflect the presence of shorter M–P bond lengths and it might be that the transition to the κ3 mode leads to longer Pd–P bond lengths compared to those seen in 2; unfortunately this could not be confirmed as crystals suitable for single-crystal structure determination were not forthcoming. There was no evidence of 2JC–P coupling to the NCN carbons in the 13C{1H} NMR spectrum of 8 where two singlets at 178.2 and 177.2 ppm occur for each isomer respectively. The remainder of the 13C{1H} spectrum is not of diagnostic value other than to confirm the presence of the two isomers.
The analogous platinum complex [Pt(κ3-P,C,P′-1)Cl]PF6, 9, was prepared by a similar procedure to that for 8 and isolated as a cream solid. The 1H NMR spectrum of 9 mimics closely that of 8 as does the 31P{1H} NMR spectrum although this does have the usual Pt-195 satellites with 1JP–Pt coupling constants of 2552 and 2570 Hz. These compare with those reported for 3a/b above confirming the retention of the trans-P arrangement in 9. The NCN carbons for each isomer are observed as triplets at 166.2 and 164.9 ppm with 2JC–P of 9.2 Hz in the 13C{1H} NMR spectrum of 9. Similar values of 9.2 and 9.4 Hz have been observed in the complexes trans-[Pd(BenzNHC)(PPh3)2Cl]BF417 where BenzNHC is a benzimidazole derived NHC and [Pt([16]ane–P2CNHC2)](PF6)2 containing a macrocyclic ligand.18 A larger value of 25 Hz has been reported for a related P,C,P complex containing a central NHC donor.8h
As a chelating complex was not obtained from the reaction of 1·HPF66666 with [Rh(1,5-COD)Cl]2, access to a κ3 complex through the addition of base to a preformed [Rh(κ2-P,P′-H·1)(L)2]n+/− species was precluded and a different means of synthesis was required. [Rh(acac)(CO)2] was considered an ideal starting material as it contains an internal base (acac−) for in situ deprotonation after coordination of one or both phosphines. The reaction of 1·HPF66666 with 1 equivalent of [Rh(acac)(CO)2] in THF at reflux gave, after removal of all volatiles, a yellow solid that was recrystallised from MeOH. The 1H NMR spectrum confirmed the loss of the amidinium proton and the coordinated acac ligand suggesting that the desired reaction had occurred to give [Rh(κ3-P,C,P′-1)(CO)]PF6, 10. The structure of the complex was confirmed upon closer inspection of the 1H NMR spectrum which showed the characteristic features for κ3-1 coordination, i.e. the presence of two isomers with two methyl resonances upfield of TMS and eight doublets for the benzylic hydrogens with each geminal pair being separated by >2 ppm. The 31P{1H} NMR spectrum consisted of two almost overlapping ABX multiplets with 2JP–P having a magnitude of 268 and 274 Hz and 1JP–Rh = 134 Hz. This value is typical of a trans-phosphine complex19 being only slightly smaller than the value of 142 Hz for the trans-PPh3 groups in Rh(PPh3)3Cl20 and is exactly the same as the values quoted for complexes of the type trans-[Rh(L)(PPh3)2(CO)]X21 where L is a saturated five-membered NHC. The infrared spectrum of 10 showed the CO stretch for the coordinated carbonyl ligand at 2008 cm−1 which is directly comparable to the values observed by Lappert and co-workers for trans-[Rh(5-NHC)(PPh3)2(CO)]X.21
The examples above are systems in which deprotonation of the amidinium group succeeds coordination of the phosphine donors (this is known for the preformed Pd and Pt complexes 2 and 3a/b and implied for the Rh). The common approach of in situ deprotonation of 1·HPF66666 followed by addition of the resultant free carbene to an appropriate metal precursor had, to this point, not been necessary. However, it was of interest to know if such an approach could be used with 1·HPF66666 as the N,N′-dibenzyl analogue of 1·HPF66666 does furnish complexes via this approach while the N,N′-di(2-pyridyl) derivative does not.11a Part of the problem with benzyl derived NHCs is the facility by which they undergo a 1,2-migration of the benzylic substituent in their unprotected form.22 Fortunately when this approach was attempted in THF with AgOTf the complex [Ag(κ3-P,C,P′-1)(OTf)], 11, was isolated as a grey solid. This was encouraging as the common method of preparing Ag–NHC complexes directly from the amidinium salt and Ag2O did not work with 1·HPF66666. The 31P{1H} NMR spectrum of the isolated solid appears relatively complex because of the presence of P–Ag(107/109) coupling but is essentially two superimposed ABX patterns, one for each of the two isotopomers. The value of the 1JP–Ag(107) and 1JP–Ag(109) coupling constants are 378 and 327 Hz respectively, reflecting the inherent difference in the gyromagnetic ratio for the two isotopes. The relatively small value of 65.5 Hz for 2JP–P is not consistent with the large values of 300–600 Hz expected for trans-orientated P-donors and it would appear that the silver complex does not contain a PCPM plane. This is largely to be expected as d10 Ag(I) does not tend to form octahedral or square planar complexes and, if 11 is monomeric (although bridging forms of PCNHCP ligands are known23 only monomeric species were observed in the mass spectrum of 11), then the likely geometry would approximate to tetrahedral as noted for the related [Cu(κ3-P,C,P′-1)X] species.24 The absolute magnitude of the 2JP–P coupling constant compares well with known tetrahedral phosphine containing complexes of silver(I) such as [AgBr(κ2-P,P′-dppp)(κ1-P-dpppO)] where a value of 58 Hz is observed.25 The 1JP–Ag coupling constants are somewhat larger than those of 253 and 220 Hz observed in [AgBr(κ2-P,P′-dppp)(κ1-P-dpppO)].25 Even though the coordination in the Ag(I) complex does not contain a planar PCPAg unit, there are still two possible isomers as shown in Fig. 6. The related copper(I) systems [Cu(κ3-P,C,P′-1)X] show a halide-dependant diastereomerism with the RCu configuration being favoured when X = I− and the SCu configuration when X = Cl−.24 The 1H NMR spectrum of 11 shows no evidence of the presence of more than one isomer although some of the signals in the 1H NMR spectrum are broadened slightly, most notably for one set of benzylic CH2 resonances (AB pattern) and one of the methyl signals. The absence of a CH3 resonance upfield of TMS in the 1H NMR spectrum would suggest that the preferred isomer of 11 has the S configuration at the metal and that this isomer is formed preferentially. Some very slight broadening is also evident for the benzyl methylenes in the 13C{1H} NMR spectrum of 11 although the spectrum is otherwise well resolved and there is some evidence of a second species in solution as each of the major peaks is associated with a minor peak. The NCN resonance however was not clearly resolved in the spectrum and was seen as a broad unstructured peak at 211 ppm. This is not surprising as this carbon is expected to be coupled to both phosphorus atoms and the silver for each of the isotopomers of the complex. The chemical shift of this carbon is downfield that of the dibenzyl analogue in a complex of the type [Ag(L)2]OTf11a but compares to other silver complexes of 6- and 7-membered NHCs.26
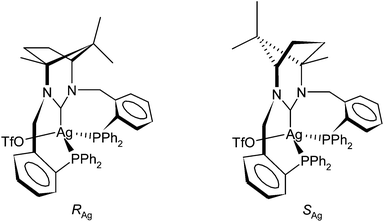 | ||
| Fig. 6 Diastereomeric possibilities for κ3-1 at pseudo tetrahedral silver(I) in complex 11. | ||
There have been very few studies of PCP ligands where the central donor is an NHC carbon.8b,c,e,22 Of these, none contain a ring-expanded NHC and only one consists of two 7-membered chelates.8e The Cu(I) complex of the latter ligand does not show a normal coordination mode but rather the NCN carbon acts as a bridging ligand in a dimeric Cu(I)–Cu(I) complex.8e Such a motif is more common for monovalent silver27 but is not considered to apply to complex 11 as both phosphines are clearly attached to a single Ag(I) centre as evidenced by the presence of 2JP–P coupling. It is also noteworthy that the related Cu(I) complex is a tetrahedral monomer.24
Catalysis
Complexes of staunchly trans-P2 chelates are not anticipated to catalyse transformations which involve reductive elimination. This is especially so when the catalytic cycle requires a transition from a square planar d8 complex to a tetrahedral or lower coordinate d10 system upon reduction, e.g. Pd(II) → Pd(0). Such inactivity is associated with the relative position of the two potential coupling partners: being mutually trans they cannot eliminate. Less rigid P2 donors may show a preference for trans-coordination but their increased flexiblity enables cis-bidentate binding or monodentate coordination (either through bridging or transient loss of one P-donor) and thus facilitate catalysis by cycles that do involve reductive elimination. Both [1·H]+ and 1 have been shown to be relatively promiscuous in their mode of metal binding and selected palladium and/or platinum complexes were anticipated to be active for catalysis.In order to test this assumption some preliminary C–C coupling and hydrosilylation catalytic tests were performed with 2, 8 and 5. The results of the Suzuki coupling of various aryl halides and phenylboronic acid are shown in Table 1. Good conversions were seen for the aryl bromides after 18 h at 80 °C but no activity was observed with the aryl chlorides. The palladium complexes 2 and 8 show little difference in reactivity which may suggest a common catalytic cycle. The fact that κ3-P,C,P′-1 has been shown to support d8 and d10 metal systems coupled with the basic conditions of the catalysis tends to suggest the presence of κ3-P,C,P′ complexes in the catalytic cycle for both 2 and 8.
| Catalyst | Substrate | Conversionb (%) |
|---|---|---|
| a Catalytic conditions: 1 × 10−3 mol substrate, 1.2 × 10−3 mol PhB(OH)2, 2 × 10−3 mol K2CO3, 0.5 mol% catalyst, 3 ml 1,4-dioxane, 80 °C, 18 h. b Total GC-MS yield based upon consumption of substrate using n-decane as internal standard. | ||
| 2 | Bromobenzene | 84 |
| 2 | 4-Bromotoluene | 100 |
| 2 | 4-Bromoanisole | 72 |
| 2 | 4-Bromoacetophenone | 100 |
| 2 | 2-Bromotoluene | 59 |
| 2 | 2-Bromopyridine | 54 |
| 2 | 4-Chlorotoluene | 0 |
| 2 | 4-Chloroanisole | 0 |
| 2 | 2-Chlorotoluene | 0 |
| 8 | 4-Bromotoluene | 100 |
| 8 | 4-Bromoanisole | 60 |
| 8 | 4-Bromoacetophenone | 100 |
| 8 | 2-Bromotoluene | 84 |
| 8 | 4-Chlorotoluene | 0 |
| 8 | 4-Chloroanisole | 0 |
| 8 | 2-Chlorotoluene | 0 |
Platinum complexes have long been known as catalysts for the hydrosilyation of unsaturated hydrocarbons. Zerovalent platinum complexes containing divinyldisiloxane feature prominently in the literature28 usually with a secondary controlling ligand as Karstedt's complex Pt2(dvdms)3, although active, is poorly selective.29 Given this history it is not surprising that 5 acts as a catalyst for the hydrosilylation of alkynes with Et3SiH (Table 2). Complete conversion is observed for all the alkynes examined after 18 h at 70 °C with a catalyst loading of 0.01 mol%. Regio-control is an important consideration in hydrosilylation as alkynes can give a number of products upon addition, e.g. with Et3SiH and terminal alkynes as substrates, E and Z 1-triethylsilyl and 2-triethylsilyl products can be formed. Although the thermodynamically more stable 1-substituted E-alkene is usually the major product, the observed selectivity for this isomer is very high for phenylacetylene and oct-1-yne with 5 compared with the best of the reported highly selective Pt(0) catalysts.28g,h,30 This selectivity is reduced for trimethylsilylacetylene where the 2-substituted isomer is produced as 25% of the product: this is not surprising as the alkyne substrate is electronically distinct and tends to form more 2-substituted product in other catalyst systems.28e The internal hex-2-yne gives two products with the 2-isomer predominating (Table 2). Although the selectivity here is modest it is better than the essentially 1:1 mixtures observed with complexes of the type Pt(ER–NHC)(dvdms)2.28h The reactivity towards alkenes is poor as exemplified by the two entries in the table. Oct-1-ene was chosen as a typical terminal alkene and styrene as a somewhat activated alkene; only the latter showed any conversion under the conditions employed for the reaction.
| Substrate | Conversionb (%) | Selectivityc |
|---|---|---|
|
a Catalytic conditions: 1 × 10−3 mol substrate, 3 ml toluene, 70 °C, substrate:Et3SiH ratio 1:1, 0.01 mol% catalyst.
b Total GC-MS yield based upon consumption of substrate using n-decane as internal standard.
c Determined by 1H NMR of the distilled products.
d 1-(E)-Triethylsilyloct-1-ene: 1-(Z)-triethylsilyloct-1-ene: 2-triethylsilyloct-1-ene.
e 2-(E)-Triethylsilylhex-2-ene: 3-(E)-triethylsilylhex-2-ene.
f 2-(E)-Triethylsilylstyrene: 2-triethylsilylstyrene.
g 1-(E)-Triethylsilyl-2-trimethylsilylethene: 1-triethylsilyl-1-trimethylsilylethene.
h 2-Triethylsilylethylbezene: 2-(E)-triethylsilylstyrene.
|
||
| 1-Octyne | 100 | 98.5:1:0.5d |
| 2-Hexyne | 100 | 63:37e |
| Phenylacetylene | 100 | >99:1f |
| Diphenylacetylene | 100 | — |
| Trimethylsilylacetylene | 100 | 75:25g |
| Styrene | 29 | 89:11h |
| Cyclohexenone | 100 | — |
| Octene | 0 | — |
Experimental
Methods and materials
All synthetic procedures and manipulations were performed under dry argon or nitrogen using standard Schlenk line techniques. All solvents were freshly distilled from sodium (toluene), sodium/benzophenone (THF) or calcium hydride (acetonitrile, methanol and dichloromethane) under nitrogen before use. All other chemicals were obtained commercially and used as received. The 1H and 13C NMR spectra were recorded on a Jeol Eclipse 300 MHz or Bruker 400 MHz spectrometer and referenced to tetramethylsilane (δ = 0 ppm). All 31P NMR spectra were recorded on a Jeol Eclipse 300 spectrometer operating in the proton-decoupled mode at 121.7 MHz. Mass spectra were obtained using a Waters LCT Premier XE mass spectrometer. Elemental analyses were performed by Stephen Boyer at London Metropolitan University, UK.31Syntheses
1·HPF66666. N,N′-Bis{2-(diphenylphosphanyl)benzyl}-1,2,2-trimethylcyclopentane-1,3-diamine (2 g, 2.89 mmol) and NH4PF6 (0.52 g, 3.2 mmol) were heated in triethylorthoformate (15 ml) at 100 °C under N2 for 2 h. After cooling, the white precipitate was filtered, washed with cold EtOH (2 × 3 ml), then diethyl ether and air-dried. Yield = 2.3 g (94%). 1H NMR (CD3CN, 400 MHz) δ 7.79 (1H, br), 7.52–7.04 (25H, m), 6.95 (1H, m), 4.84 (1H, dd, J 14.5, 3.1 Hz), 4.73 (1H, dd, J 14.5, 1.7 Hz), 4.42 (1H, dd, J 14.6, 1.3 Hz), 4.28 (1H, d, J 14.6 Hz), 3.20 (1H, d, J 4.0 Hz), 2.36 (1H, m), 1.89 (2H, m), 1.68 (1H, m), 1.31 (3H, s), 0.95 (3H, s), 0.84 (3H, s) ppm. 13C{1H} DEPT NMR (CDCl3, 75.6 MHz) δ 151.7 (C, d, J 10.4 Hz), 137–127 (aromatics), 71.3 (CH, s), 65.1 (C, s), 54.7 (CH2, d, J 17.3 Hz), 50.7 (CH2, d, J 25.4 Hz), 40.2 (C, s), 38.5 (CH2, s), 30.5 (CH2, s), 20.6 (CH3, s), 15.9 (CH3, s), 13.5 (CH3, s) ppm. 31P (CD3CN, 121.6 MHz): δ −16.6, −17.5 ppm. MS: 701 ([1·H]+, 100%).
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O). Anal.: Calc. for C48H46N2P3OF6Rh: C, 59.02; H, 4.76; N, 2.87%. Found: C, 58.9; H, 4.8; N, 3.0%.
O). Anal.: Calc. for C48H46N2P3OF6Rh: C, 59.02; H, 4.76; N, 2.87%. Found: C, 58.9; H, 4.8; N, 3.0%.
Catalysis
Suzuki coupling
A mixture of substrate (1 mmol), PhB(OH)2 (1.2 mmol), K2CO3 (2 mmol), 0.5 mol% catalyst in 1,4-dioxane (3 ml) with decane as internal GC standard (1 mmol) was heated at 80 °C for 18 h. A sample of the solution was diluted with CH2Cl2 and passed through a short column of silica. Total GC-MS yields based upon consumption of substrate using n-decane as internal standard were determined via an internal standard method of quantification on an Agilent Technologies 6890 N GC system equipped with an Agilent Technologies 5973 inert MS detector with MSD. Column: Agilent 190915-433 capillary, 0.25 mm × 30 m × 0.25 mm. Program for analysis: initial temperature of 40 °C, held for 2.5 min, ramp 5 °C min−1 next 150 °C, ramp 10 °C min−1 next 220 °C, held for 10 min. The temperature of both the injector and detector were maintained at 240 °C.Hydrosilylation
A mixture of substrate (1 mmol), Et3SiH (1 mmol), and 0.05 mol% catalyst in toluene (3 ml) with decane as internal GC standard (1 mmol) was heated at 70 °C for 12 h. A sample of the solution was diluted with CH2Cl2 and passed through a short column of silica. Total GC-MS yields based upon consumption of substrate using n-decane as internal standard were determined via an internal standard method of quantification on an Agilent Technologies 6890 N GC system equipped with an Agilent Technologies 5973 inert MS detector with MSD. Column: Agilent 190915-433 capillary, 0.25 mm × 30 m × 0.25 mm. Program for analysis: initial temperature of 40 °C, held for 2.5 min, ramp 5 °C min−1 next 150 °C, ramp 10 °C min−1 next 220 °C, held for 10 min. The temperature of both the injector and detector were maintained at 240 °C. Selectivities were determined by 1H NMR on the isolated materials.Crystallography
Data collection was carried out on a Bruker–Nonius Kappa CCD diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). The instrument was equipped with an Oxford Cryosystems cooling apparatus. Data collection and cell refinement were carried out using COLLECT32 and HKL SCALEPACK.33 Data reduction was applied using HKL DENZO and SCALEPACK.33 The structures were solved and refined with SHELX-97.34 Absorption corrections were performed using SORTAV.35 Generally, all non-hydrogen atoms were generally refined anisotropically, while the hydrogen atoms were inserted in idealised positions with Uiso set at 1.2 or 1.5 times the Ueq of the parent atom. For 2, the large asymmetric unit is partially accountable for the weak high angle data and two anion sites show two-fold disorder. Geometric restraints were applied to the PF6− during refinement to stop distortion of the anions and restraints were also applied in order to maintain similar atomic displacement parameters for atoms in close proximity. For 3a, geometric restraints were applied to acetonitrile solvent molecules as well as to the atomic displacement parameters to stop distortion. For 6, difference Fourier synthesis showed large spaces occupied by diffuse electron density, interpreted as disordered solvent, and hence SQUEEZE was used.36 The large asymmetric unit combined with the disordered solvent are accountable for the weak high angle data which were omitted from the refinement. Geometric restraints were applied to the PF6− during refinement to stop distortion of the anions. Initial examination of the data for 1·HPF66666 and 2 suggested higher crystal symmetry but lower symmetry was necessary for the final ordered complex structures. Both anion sites show two-fold disorder. Geometric restraints were applied to the PF6− during refinement to stop distortion of the anions. Restraints were also applied in order to maintain similar atomic displacement parameters for atoms in close proximity. The details of the data collection and structure solution are collected in Table 3.| 1·HPF66666 | 2 | 3a | 6 | |
|---|---|---|---|---|
| Empirical formula | C47H47F6N2P3 | C48H48Cl5N2P3F6Pd | C51H53Cl2N4P3F6Pt | C94H94I4N4P4Pd2, I0.27(PF6)1.73 |
| Formula weight | 846.78 | 1143.44 | 1194.87 | 2409.12 |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Orthorhomic |
| Space group | P1 | C2 | P21 | P2(1)2(1)2 |
| a/Å | 11.9727(5) | 26.7720(9) | 11.9538(5) | 27.8377(9) |
| b/Å | 13.6776(6) | 14.6590(6) | 14.4075(4) | 19.8686(12) |
| c/Å | 13.8349(5) | 28.2434(13) | 29.2856(14) | 21.6459(13) |
| β/° | 75.759(2) | 117.031(2) | 93.809(2) | |
| U/Å3 | 2112.19(15) | 9873.3(7) | 5032.6(3) | 11972.3(11) |
| Z | 2 | 8 | 4 | 4 |
| D c/Mg m−3 | 1.331 | 1.498 | — | 1.337 |
| F(000) | 884 | 4524 | 2392 | 4735 |
| θ range/° | 3.04 to 27.50 | 2.25 to 24.71 | 1.80 to 27.54 | 1.19 to 20.82 |
| Index ranges | −14 ≤ h ≤ 15, −17 ≤ k ≤ 17, −17 ≤ l ≤ 17 | −31 ≤ h ≤ 31, −17 ≤ k ≤ 15, −33 ≤ l ≤ 33 | −15 ≤ h ≤ 15, −18 ≤ k ≤ 18, −38 ≤ l ≤ 37 | −25 ≤ h ≤ 24, −16 ≤ k ≤ 19, −21 ≤ l ≤ 14 |
| Reflections collected | 14999 |
15228 |
19515 |
12167 |
| Independent reflections | 10801 |
9398 | 11357 |
8498 |
| Data/restraints/parameters | 14999/606/1179 |
15228/265/1218 |
19515/173/1217 |
12167/40/1145 |
| Goodness of fit on F2 | 1.047 | 1.016 | 1.026 | 1.061 |
| Final R1, wR2 [I > 2σ(I)] (all data) | 0.0631, 0.1297 | 0.0853, 0.1835 | 0.0976, 0.1597 | 0.1023, 0.2721 |
| 0.1009, 0.1500 | 0.1530, 0.2224 | 0.1889, 0.1947 | 0.1281, 0.2961 | |
| Largest difference peak | ||||
| And hole/e Å−3 | 0.284 and −0.321 | 1.346, −0.911 | 1.514, −1.057 | 3.294, −1.076 |
| Flack parameter | 0.03(12) | 0.09(6) | 0.049(14) | 0.20(5) |
Conclusions
A diphosphino(pro-NHC) ligand has been prepared containing a central bicyclic core derived from camphor and two closely similar but formally unique RPPh2 arms. In its pro-ligand form (protonated NHC) the ligand binds solely through the phosphine donors and, depending on the metal ion, forms trans-chelating monomeric or bridging dimeric complexes. Addition of base to these preformed complexes liberates the NHC for coordination to give κ3-P,C,P′ complexes which, for square planar metals, show conformational isomerism. Prior deprotonation of the pro-ligand and subsequent addition of AgOTf gives the pseudo-tetrahedral complex SAg-[Ag(κ3-P,C,P′-1)(OTf)] which is isolated as a single isomer. Selected platinum and palladium complexes have been shown to be effective for the regioselective hydrosilylation of alkynes and for Suzuki coupling with aryl bromides. Studies of this and related ligands are continuing with a view to extending their application in catalysis.Acknowledgements
The authors would like to thank Cardiff University for providing support for this work.Notes and references
- (a) P. W. N. M. van Leeuwen and Z. Freixa, in Modern Carbonylation Methods, ed. L. Kollár, Wiley-VCH, 2008, p. 1 Search PubMed; (b) P. C. J. Kamer, P. W. N. M. Van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895 CrossRef CAS; (c) Z. Freixa and P. W. N. M. Van Leeuwen, J. Chem. Soc., Dalton Trans., 2003, 1890 RSC; (d) P. C. J. Kamer, P. W. N. M. van Leeuwen, J. F. Kees Goubitz, H. Schenk and C. Bo, J. Am. Chem. Soc., 1998, 120, 11616 CrossRef; (e) B. P. Morgan and R. C. Smith, J. Organomet. Chem., 2008, 693, 11 CrossRef CAS; (f) E. E. Marlier, S. J. Tereniak, K. Ding, J. E. Mulliken and C. C. Lu, Inorg. Chem., 2011, 50, 9290 CrossRef CAS.
- (a) Z. Freixa and P. W. N. M. van Leeuwen, Coord. Chem. Rev., 2008, 252, 1755 CrossRef CAS; (b) N. Naser, D. J. Eisler and R. J. Puddephat, Chem. Commun., 2010, 46, 1953 RSC; (c) C. P. Butts, E. Filali, G. C. Lloyd-Jones, P.-O. Norrby, D. A. Sale and Y. Schramm, J. Am. Chem. Soc., 2009, 131, 9945 CrossRef CAS; (d) C. P. Butts, J. Crosby, G. C. Lloyd-Jones and S. C. Stephen, Chem. Commun., 1999, 1707 RSC; (e) I. J. S. Fairlamb and G. C. Lloyd-Jones, Chem. Commun., 2000, 2447 RSC; (f) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1999, 121, 4545 CrossRef CAS; (g) L. Kaganovsky, D. Gelman and K. Rueck-Braun, J. Organomet. Chem., 2010, 695, 260 CrossRef CAS; (h) A. Velian, S. Lin, A. J. M. Miller, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2010, 132, 6296 CrossRef CAS; (i) C. Azerraf, O. Grossman and D. Gelman, J. Organomet. Chem., 2007, 692, 761 CrossRef CAS; (j) C. Azerraf and D. Gelman, Organometallics, 2009, 28, 6578 CrossRef CAS; (k) I. S. Mikhel, M. Garland, J. Hopewell, S. Mastroianni, C. L. McMullin, A. G. Orpen and P. G. Pringle, Organometallics, 2011, 30, 974 CrossRef CAS.
- (a) N. J. Destefano, D. K. Johnson, R. M. Lane and L. M. Venanzi, Helv. Chim. Acta, 1976, 59, 2674 CrossRef CAS; (b) Z. Freixa, M. S. Beentjes, G. D. Batema, C. B. Dieleman, G. P. F. van Strijdonck, J. N. H. Reek, P. C. J. Kamer, J. Fraanje, K. Goubitz and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2003, 42, 1284 CrossRef CAS; (c) F. B. Xu, Q. S. Li, X. S. Zeng, X. B. Leng and Z. Z. Zhang, Organometallics, 2002, 21, 4894 CrossRef CAS; (d) M. Sawamura, H. Hamashima and Y. Ito, Tetrahedron: Asymmetry, 1991, 2, 593 CrossRef CAS.
- (a) L. Chahen, B. Therrien and G. Süss-Fink, J. Organomet. Chem., 2006, 691, 4257 CrossRef CAS; (b) G. Lamb, M. Clarke, A. M. Z. Slawin, B. Williams and L. Key, Dalton Trans., 2007, 5582 RSC; (c) E. K. van den Beuken, A. Meetsma, H. Kooijman, A. L. Spek and B. L. Feringa, Inorg. Chim. Acta, 1997, 264, 171 CrossRef CASB. M. Trost, D. L. van Vranken and C. Brigel, J. Am. Chem. Soc., 1992, 114, 9327 CrossRef CAS.
- (a) A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257 CrossRef CAS; (b) E. M. Schuster, M. Botoshansky and M. Gandelman, Angew. Chem., Int. Ed., 2008, 47, 4555 CrossRef CAS; (c) E. Poverenov, I. Efremenko, A. I. Frenkel, Y. Ben-David, L. J. W. Shimon, G. Leitus, L. Konstantinovski, J. M. L. Martin and D. Milstein, Nature, 2008, 455, 1093 CrossRef CAS; (d) R. Ahuja, B. Punji, M. Findlater, C. Supplee, W. Schinski, M. Brookhart and A. S. Goldman, Nat. Chem., 2011, 3, 167 CrossRef CAS; (e) M. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS; (f) S. Musa, R. Romm, C. Azerraf, S. Kozhuz and D. Gelman, Dalton Trans., 2011, 40, 8760 RSC.
- (a) M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750 CrossRef CAS; (b) W. Leis, H. A. Mayer and W. C. Kaska, Coord. Chem. Rev., 2008, 252, 1787 CrossRef CAS; (c) R. H. Crabtree, J. Organomet. Chem., 2004, 689, 4083 CrossRef CAS; (d) D. F. Maclean, R. McDonald, M. J. Ferguson, A. J. Caddell and L. Turculet, Chem. Commun., 2008, 5146 RSC; (e) M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471 CrossRef CAS; (f) M. Findlater, A. S. Goldman and M. S. Brookhart, Alkane C–H activation by single-site metal catalysis, in Catalysis by Metal Complexes, ed. P. J. Perez, Springer, New York, 2011, ch. 4, vol. 38 Search PubMed; (g) J. Choi, A. MacArthur, M. S. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761 CrossRef CAS; (h) B. Punji, T. J. Emge and A. S. Goldman, Organometallics, 2010, 29, 2702 CrossRef CAS; (i) C. M. Jensen, Chem. Commun., 1999, 2443 RSC.
- (a) M. D. Fryzuk and C. D. Montgomery, Coord. Chem. Rev., 1989, 95, 1 CrossRef CAS; (b) G. van Koten, Pure Appl. Chem., 1989, 61, 1681 CrossRef CAS; (c) A. Choualeb, A. J. Lough and D. G. Gusev, Organometallics, 2007, 26, 5224 CrossRef CAS; (d) C. Gunanathan, B. Gnanaprakasam, M. A. Iron, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 14763 CrossRef CAS; (e) R. Langer, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 1947 CrossRef; (f) M. J. Sgro and D. W. Stephan, Dalton Trans., 2011, 40, 2419 RSC; (g) A. A. Koridze, A. V. Polezhaev, S. V. Safronov, A. M. Sheloumov, F. M. Dolgushin, M. G. Ezernitskaya, B. V. Lokshin, P. V. Petrovskii and A. S. Peregudov, Organometallics, 2010, 29, 4360 CrossRef CAS.
- (a) A. M. Magill, D. S. McGuinness, K. J. Cavell, G. J. P. Britovsek, V. C. Gibson, A. J. P. White, D. J. Williams, A. H. White and B. W. Skelton, J. Organomet. Chem., 2001, 617–618, 546 CrossRef CAS; (b) H. M. Lee, J. Y. Zeng, C. H. Hu and M. T. Lee, Inorg. Chem., 2004, 43, 6822 CrossRef CAS; (c) F. E. Hahn, M. C. Jahnke and T. Pape, Organometallics, 2006, 25, 5927 CrossRef CAS; (d) T. Yagyu, S. Oya, M. Maeda and K. Jitsukawa, Chem. Lett., 2006, 35, 154 CrossRef CAS; (e) S. Gischig and A. Togni, Eur. J. Inorg. Chem., 2005, 4745 CrossRef CAS; (f) J. C. C. Chen and I. J. B. Lin, Organometallics, 2000, 19, 5113 CrossRef CAS; (g) J. Y. Zeng, M.-H. Hsieh and H. M. Lee, J. Organomet. Chem., 2005, 690, 5662 CrossRef CAS; (h) T. Steinke, B. K. Shaw, H. Jong, B. O. Patrick and M. D. Fryzuk, Organometallics, 2009, 28, 2830 CrossRef CAS; (i) L. P. Spencer, C. Beddie, M. B. Hall and M. D. Fryzuk, J. Am. Chem. Soc., 2006, 128, 12531 CrossRef CAS; (j) L. P. Spencer and M. D. Fryzuk, J. Organomet. Chem., 2005, 690, 5788 CrossRef CAS; (k) M. C. Jahnke, T. Pape and F. E. Hahn, Eur. J. Inorg. Chem., 2009, 1960 CrossRef CAS; (l) C. Michon, A. Ellern and R. J. Angelici, Inorg. Chim. Acta, 2006, 359, 4549 CrossRef CAS; (m) S. J. Roseblade, A. Ros, D. Monge, M. Alcarazo, E. Alvarez, J. M. Lassaletta and R. Fernandez, Organometallics, 2007, 26, 2570 CrossRef CAS.
- (a) D. Pugh, H. J. Wells, D. J. Evans and A. A. Danopoulos, Dalton Trans., 2009, 7189 RSC; (b) A. A. Danopoulos, D. Pugh and J. A. Wright, Angew. Chem., Int. Ed., 2008, 47, 9765 CrossRef; (c) D. Pugh, A. Boyle and A. A. Danopoulos, Dalton Trans., 2008, 1087 RSC; (d) A. A. D. Tulloch, A. A. Danopoulos, G. J. Tizzard, S. J. Coles, M. B. Hursthouse, R. S. Hay-Motherwell and W. B. Motherwell, Chem. Commun., 2001, 1270 RSC; (e) E. Peris, J. Mata, J. A. Loch and R. H. Crabtree, Chem. Commun., 2001, 201 RSC; (f) S. Grundemann, M. Albrecht, J. A. Loch, J. W. Faller and R. H. Crabtree, Organometallics, 2001, 20, 5485 CrossRef; (g) J. A. Loch, M. Albrecht, E. Peris, J. Mata, J. W. Faller and R. H. Crabtree, Organometallics, 2002, 21, 700 CrossRef CAS; (h) D. J. Nielsen, K. J. Cavell, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2002, 327, 116 CrossRef CAS; (i) F. E. Hahn, M. C. Jahnke, V. Gomez-Benitez, D. Morales-Morales and T. Pape, Organometallics, 2005, 24, 6458 CrossRef CAS; (j) E. Mas-Marza, M. Poyatos, M. Sanau and E. Peris, Organometallics, 2004, 23, 323 CrossRef CAS; (k) G. T. S. Andavan, E. B. Bauer, C. S. Letko, T. K. Hollis and F. S. Tham, J. Organomet. Chem., 2005, 690, 5938 CrossRef CAS; (l) H. V. Huynh, D. Yuan and Y. Han, Dalton Trans., 2009, 7262 RSC; (m) D. S. McGuinness, J. A. Suttil, M. G. Gardiner and N. W. Davies, Organometallics, 2008, 27, 4238 CrossRef CAS; (n) D. J. Nielsen, K. J. Cavell, M. S. Viciu, S. P. Nolan, B. W. Skelton and A. H. White, J. Organomet. Chem., 2005, 690, 6133 CrossRef CAS; (o) M. Raynal, C. S. J. Cazin, C. Vallée, H. Olivier-Bourbigou and P. Braunstein, Chem. Commun., 2008, 3983 RSC; (p) A. R. Chianese, A. Mo, N. L. Lampland, R. L. Swartz and P. T. Bremer, Organometallics, 2010, 29, 3019 CrossRef CAS; (q) K. M. Schultz, K. I. Goldberg, D. G. Gusev and D. M. Heinekey, Organometallics, 2011, 30, 1429 CrossRef CAS.
- (a) R. Fränkel, U. Kernbach, M. Bakola-Christianopoulou, U. Plaia, M. Suter, W. Ponikwar, H. Nöth, C. Moinet and W. P. Fehlhammer, J. Organomet. Chem., 2001, 617–618, 530 CrossRef; (b) H. V. Rasika Dias and W. Jin, Tetrahedron Lett., 1994, 35, 1365 CrossRef; (c) H. Nakai, Y. Tang, P. Gantzel and K. Meyer, Chem. Commun., 2003, 24 RSC; (d) X. Hu, I. Castro-Rodriguez and K. Meyer, Organometallics, 2003, 22, 3016 CrossRef CAS; (e) X. Hu and K. Meyer, J. Organomet. Chem., 2005, 690, 5474 CrossRef CAS.
- (a) P. D. Newman, K. J. Cavell and B. M. Kariuki, Organometallics, 2010, 29, 2724 CrossRef CAS; (b) P. D. Newman, K. J. Cavell, A. J. Hallett and B. M. Kariuki, Dalton Trans., 2011, 40, 8807 RSC.
- R. C. Smith and J. D. Protasiewicz, Organometallics, 2004, 23, 4215 CrossRef CAS.
- L. Rigamonti, A. Forni, M. Manassero, C. Manassero and A. Pasini, Inorg. Chem., 2010, 49, 123 CrossRef CAS.
- (a) C. J. Elsevier, B. Kowall and H. Kragten, Inorg. Chem., 1995, 34, 4836 CrossRef CAS; (b) O. Niyomura, T. Iwasawa, N. Sawada, M. Tokunaga, Y. Obora and Y. Tsuji, Organometallics, 2005, 24, 3468 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert, C. MacBeath, F. P. E. Scott and N. J. W. Warhurst, J. Organomet. Chem., 1997, 534, 139 CrossRef CAS.
- G. Berthon-Gelloz, O. Buisine, J.-F. Brière, G. Michaud, S. Stérin, G. Mignani, B. Tinant, J.-P. Declercq, D. Chapon and I. E. Marko, J. Organomet. Chem., 2005, 690, 6156 CrossRef CAS.
- T. Kösterke, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2011, 133, 2112 CrossRef.
- A. Flores-Figueroa, T. Pape, K.-O. Feldmann and F. E. Hahn, Chem. Commun., 2010, 46, 324 RSC.
- P. E. Garrou, Chem. Rev., 1981, 81, 229 CrossRef CAS.
- T. H. Brown and P. J. Green, J. Am. Chem. Soc., 1970, 92, 2359 CrossRef CAS.
- M. J. Doyle, M. F. Lappert, P. L. Pye and P. Terreros, J. Chem. Soc., Dalton Trans., 1984, 2355 RSC.
- (a) P. L. Arnold, M. Rodden and C. Wilson, Chem. Commun., 2005, 1743 RSC; (b) C. Boehler, D. Stein, N. Donati and H. Grützmacher, New J. Chem., 2002, 26, 1291 RSC; (c) B. Cetinkaya, E. Cetinkaya, J. A. Chamizo, P. B. Hitchcock, H. A. Jasim, H. Küçükbay and M. E. Lappert, J. Chem. Soc., Perkin Trans. 1, 1998, 2047 RSC; (d) S. Solé, H. Gornitzka, O. Guerret and G. Bertrand, J. Am. Chem. Soc., 1998, 120, 9100 CrossRef.
- P. L. Chiu and H. M. Lee, Organometallics, 2005, 24, 1692 CrossRef CAS.
- P. D. Newman, K. J. Cavell and B. M. Kariuki, Chem. Commun., 2012, 48, 6511 RSC.
- D. Affandi, S. J. Berners-Price, Effendy, P. J. Harvey, P. C. Healy, B. E. Ruch and A. H. White, J. Chem. Soc., Dalton Trans., 1991, 1411 Search PubMed.
- (a) M. Iglesias, D. J. Beetstra, J. C. Knight, L.-L. Ooi, A. Stasch, S. J. Coles, L. Male, M. B. Hursthouse, K. J. Cavell, A. Dervisi and I. A. Fallis, Organometallics, 2008, 27, 3279 CrossRef CAS; (b) E. L. Kolychev, I. A. Portnyagin, V. V. Shuntikov, V. N. Khrustalev and M. S. Nechaev, J. Organomet. Chem., 2009, 694, 2454 CrossRef CAS; (c) W. A. Herrmann, S. K. Schneider, K. Öfele, M. Sakamoto and E. Herdtweck, J. Organomet. Chem., 2004, 689, 2441 CrossRef CAS.
- (a) J. C. Garrison, R. S. Simons, W. G. Kofron, C. A. Tessier and W. J. Youngs, Chem. Commun., 2001, 1780 RSC; (b) J. C. Garrison, R. S. Simons, W. G. Kofron, C. A. Tessier and W. J. Youngs, J. Organomet. Chem., 2003, 673, 1 CrossRef CAS; (c) V. J. Catalano and M. A. Malwitz, Inorg. Chem., 2003, 42, 5483 CrossRef CAS; (d) V. J. Catalano, M. A. Malwitz and A. O. Etogo, Inorg. Chem., 2004, 43, 5714 CrossRef CAS.
- (a) I. E. Markó, S. Stérin, O. Busine, G. Mignani, P. Branlard, B. Tinant and J. P. Declercq, Science, 2002, 298, 204 CrossRef; (b) I. E. Markó, S. Stérin, O. Busine, G. Berthon, G. Michaud, B. Tinant and J. P. Declercq, Adv. Synth. Catal., 2004, 346, 1429 CrossRef; (c) O. Buisine, G. Berthon-Gelloz, J. F. Brière, S. Stérin, G. Mignani, P. Branlard, B. Tinant, J. P. Declerq and I. E. Markó, Chem. Commun., 2005, 3856 RSC; (d) J. J. Hu, F. Li and T. S. A. Hor, Organometallics, 2009, 28, 1212 CrossRef CAS; (e) G. De Bo, G. Berthon-Gelloz, B. Tinant and I. E. Markó, Organometallics, 2006, 25, 1881 CrossRef CAS; (f) M. G. McLaughlin and M. J. Cook, Chem. Commun., 2011, 47, 11104 RSC; (g) M. Blug, X.-F. Le Goff, N. Mézailles and P. Le Floch, Organometallics, 2009, 28, 2360 CrossRef CAS; (h) J. J. Dunsford, B. M. Kariuki and K. J. Cavell, J. Organomet. Chem., 2011, 696, 188 CrossRef CAS.
- B. D. Karstedt, U.S. Patent, 3,775,452, 1973 Search PubMed.
- G. Berthon-Gelloz, J.-M. Schumers, G. De Bo and I. E. Markó, J. Org. Chem., 2008, 73, 4190 CrossRef CAS.
- S. Boyer, Elemental Analysis Service, London Metropolitan University, London, N7 7DD Search PubMed.
- COLLECT, Nonius BV, 1998, Delft, The Netherlands.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112 CrossRef CAS.
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33 CrossRef.
- P. v. d. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
Footnotes |
| † We would like to acknowledge the outstanding contribution made by David J. Cole-Hamilton to chemistry in general, and to catalysis in particular. His work has been widely recognised over many years for its imagination and thoroughness in implementation. |
| ‡ CCDC 891175–891178. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2dt31475d |
| This journal is © The Royal Society of Chemistry 2012 |