 Open Access Article
Open Access ArticleMinimizing side reactions in chemoenzymatic dynamic kinetic resolution: organometallic and material strategies†
Ciara L.
Pollock
,
Kevin J.
Fox
,
Sophie D.
Lacroix
,
James
McDonagh
,
Patricia C.
Marr
*,
Alanna M.
Nethercott
,
Annie
Pennycook
,
Shimeng
Qian
,
Linda
Robinson
,
Graham C.
Saunders
* and
Andrew C.
Marr
*
School of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Belfast, BT9 5AG, UK. E-mail: a.marr@qub.ac.uk
First published on 7th September 2012
Abstract
Chemoenzymatic dynamic kinetic resolution (DKR) of rac-1-phenyl ethanol into R-1-phenylethanol acetate was investigated with emphasis on the minimization of side reactions. The organometallic hydrogen transfer (racemization) catalyst was varied, and this was observed to alter the rate and extent of oxidation of the alcohol to form ketone side products. The performance of highly active catalyst [(pentamethylcyclopentadienyl)IrCl2(1-benzyl,3-methyl-imidazol-2-ylidene)] was found to depend on the batch of lipase B used. The interaction between the bio- and chemo-catalysts was reduced by employing physical entrapment of the enzyme in silica using a sol–gel process. The nature of the gelation method was found to be important, with an alkaline method preferred, as an acidic method was found to initiate a further side reaction, the acid catalyzed dehydration of the secondary alcohol. The acidic gel was found to be a heterogeneous solid acid.
Introduction
Combining bio- and chemo-catalysis provides powerful methods for chemical synthesis.1 Chemoenzymatic dynamic kinetic resolution (DKR) of secondary alcohols has received considerable attention. In kinetic resolution an enzyme-catalyzed reaction is used to kinetically discriminate between two enantiomers in a racemic mixture and convert one but not the other into the product. In DKR a chemical catalyst is added in order to racemize the enantiomers, ensuring that all the substrate can be utilized. The combination of lipase B and an organometallic hydrogen transfer catalyst for the conversion of secondary alcohols into enantiopure acetates has been particularly effective, for example the DKR of rac-1-phenyl ethanol into R-1-phenylethanol acetate (Scheme 1).2,3 The success of this combination has led to commercialization.4 Lipase B is frequently added as an immobilized enzyme aggregate such as Novozym® 435, a macroporous acrylic resin containing CalB (Candida antarctica lipase B). Highly successful catalysts are often Ru(II) complexes,5,6 the most recognizable of which is Shvo's catalyst7 (Fig. 1), but examples of efficient Rh(III)8 and Ir(III)8,9 catalysts have also been reported. The metal complex catalyses racemization by a hydrogen transfer mechanism (Scheme 2).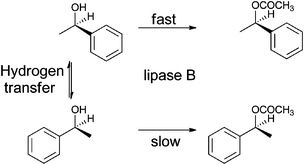 | ||
| Scheme 1 Dynamic kinetic resolution of rac-1-phenyl ethanol into R-1-phenylethanol acetate. | ||
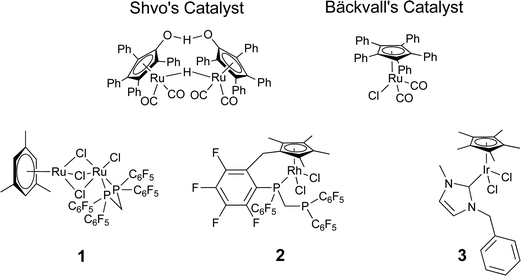 | ||
| Fig. 1 Chemocatalysts for chemoenzymatic DKR. | ||
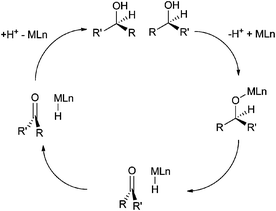 | ||
| Scheme 2 Hydrogen transfer mechanism for the racemization of a secondary alcohol by catalyst MLn. | ||
The chemoenzymatic DKR of secondary alcohols to yield chiral esters is susceptible to oxidation of the alcohol by dehydrogenation or Oppenauer oxidation10–12 to form ketone side products. This side reaction is expected to vary as the organometallic catalyst varies since the relative rate of oxidation versus racemization should be chemocatalyst dependent.
A further side reaction that may occur in chemoenzymatic DKR is the reaction between the chemical catalyst and biocatalyst. Enzymes such as CalB have a wide variety of ionic domains and regions rich in groups that may act as donors for the dissolved metal ion. In instances in which the chemocatalyst becomes associated with the protein structure racemization will be prevented, or at least, severely reduced; and the interaction may also disrupt the enzyme's activity by altering the protein structure. The prevalence of this type of interaction explains why many active hydrogen transfer catalysts exist which are inactive in the presence of lipase.5
Results and discussion
Varying the organometallic chemocatalyst
The hydrogen transfer activity of a range of piano stool complexes or Ru(II), Rh(III) and Ir(III) has been reported previously.13 Here we report the application of these catalysts to the chemoenzymatic dynamic kinetic resolution (DKR) of secondary alcohols. Activity was screened for DKR of secondary alcohols with Candida antarctica lipase B (CalB). Pentamethylcyclopentadienyl (Cp*) iridium N-heterocyclic carbene (NHC) complexes, have already been shown to a promote DKR.8In order to validate the work-up and analysis methods kinetic resolutions were first carried out in the absence of an organometallic complex. As expected the procedure gave 50% conversion to R-1-phenylethanol acetate, the percentage enantiomeric excess (% ee) was 94% at 20 °C. DKR results were obtained for a variety of Ru(II) (Table 1), Rh(III) and Ir(III) (Table 2) complexes. The structure of 1, 2 and 3 is given (Fig. 1).
| Catalyst | % Estera (% ee) | % Ketone |
|---|---|---|
| Toluene (2.4 mL), catalyst (0.0072 mmol), rac-1-phenyl ethanol (7.2 mmol), isopropenyl acetate (7.2 mmol), Novozym® 435 (40.5 mg), potassium carbonate (0.26 g), temperature 70 °C, reaction time 18 h. 1 is ½[(η6-mesitylene)Ru(μ-Cl)3RuCl(dfppm)].a Percentage conversion to 1-phenylethanol acetate by NMR.b 5 h reaction. Mesitylene is 1,3,5-trimethylbenzene, p-cymene is 1-methyl-4-(1-methylethyl)benzene, DPEN is 1,2-diphenyl-1,2-ethylenediamine (R,R), PHEN is 2-phenylglycinol (S), dppe is bis(diphenylphosphino)ethane, dppp is 1,3-bis(diphenylphosphino)propane, dfppm is (C6F5)2PCH2P(C6F5)2. | ||
| ½[(η6-Mesitylene)RuCl(μ-Cl)]2 + DPEN | 70 (97) | 3 |
| ½[(η6-p-Cymene)RuCl(μ-Cl)]2 + DPEN | 73 (97) | 2 |
| ½[(η6-Mesitylene)RuCl(μ-Cl)]2 + PHEN | 80 (97) | 1 |
| ½[(η6-p-Cymene)RuCl(μ-Cl)]2 + PHEN | 86 (94) | <1 |
| [(η6-Mesitylene)RuCl(dppe)]BF4 | 94 (85) | <1 |
| [(η6-Mesitylene)RuCl(dppp)]BF4 | 83 (98) | <1 |
| [(η6-Mesitylene)RuCl(dfppm)]BF4 | 89 (92) | — |
| [(η6-p-Cymene)RuCl(dfppm)]BF4 | 75 (91) | <1 |
| 1 | >99 (96) | <1 |
| 1 b | >99 (98) | <1 |
| Catalyst | % Ester (% ee) | % Ketone |
|---|---|---|
| Toluene (2.4 mL), catalyst (0.0072 mmol), rac-1-phenyl ethanol (7.2 mmol), isopropenyl acetate (7.2 mmol), Novozym® 435 (40.5 mg), potassium carbonate (0.26 g), temperature 70 °C, reaction time 18 h. 2 is [{η5,κP-C5Me4CH2C6F4-2-P(C6F5)CH2P(C6F5)2)}RhCl2]. 3 is [Cp*IrCl2(1-benzyl,3-methyl-imidazol-2-ylidene)].8a 48 h reaction. | ||
| ½[Cp*IrCl(μ-Cl)]2 + DPEN | 82 (84) | 2 |
| ½[Cp*IrCl(μ-Cl)]2 + PHEN | 86 (91) | 3 |
| [Cp*IrCl(dfppm)]BF4 | 84 (83) | 1 |
| ½[Cp*RhCl(μ-Cl)]2 + DPEN | 71 (96) | 12 |
| ½[Cp*RhCl(μ-Cl)]2 + PHEN | 88 (97) | 4 |
| [Cp*RhCl(dppe)]BF4 | 70 (83) | 2 |
| [Cp*RhCl(dfppm)]BF4 | 36 (86) | 5 |
| [Cp*RhCl2(PPh3)] | >99 (90) | <1 |
| [Cp*RhCl2(p-C6H4F)3] | 87 (98) | 2 |
| 2 | 63 (96) | — |
| 2 a | 97 (96) | <3 |
| 3 | >99 (97) | <1 |
Most of the organometallic complexes tested afforded reasonable conversion over 18 h. The enzyme was affected by the presence of the metal in many cases, affording lower enantioselectivity than obtained in the absence of the complex. Of the Ru(II) complexes tested bimetallic complex 113 gave optimum activity and fully conserved selectivity. The synthesis of similar complexes with non-fluorinated ligands has been reported.14 Comparing 1 and Shvo's catalyst (Fig. 1)7 both these active catalysts have hindered bimetallic cores and produce two different complexes on dissociation.
Loss of stereoselectivity was more common for Rh(III) and Ir(III) complexes (Table 2), and this points towards an increased interaction between the metal and lipase B. Complex 2 was selective, but exhibited lower activity, requiring longer reaction times; 97% conversion and good enantioselectivity could be obtained after 48 h. The slow rate of reaction could be attributed to steric crowding as the metal ion is surrounded by fluorinated groups. Such bulk is expected to retard interactions between the bio- and chemo-catalyst, but this must be balanced with the rate of racemization, as very bulky ligands will slow the approach of the organic substrate to the metal centre.
Levels of ketone formation were low for Ru(II) and Ir(III) catalysts and higher for Rh(III) catalysts. [Cp*RhCl(μ-Cl)]2 + DPEN was the most active system for the dehydrogenative oxidation of 1-phenyl ethanol under these conditions.
Overall the results suggest that the activity and selectivity can be tuned, we suggest that the key to a more active chemoenzymatic system is the positioning of enough steric bulk to prevent interaction between the enzyme and chemocatalyst, but not too much, as this will slow down the overall rate of DKR by retarding racemization.
The most active Ru(II) catalyst 1 was tested as a racemization catalyst on the aliphatic substrate rac-pinacolyl alcohol (3,3-dimethyl-2-butanol). Under similar conditions 81% ester at 82% ee was obtained with 3% ketone, Ir(III) catalyst 3 achieved 99% ester, 99% ee.8
The activity of 3 was compared to that of [(η5-Ph5C5)RuCl(CO)2], a highly active racemization catalyst (Fig. 1),15 both catalysts facilitated good conversion (>99%) and good enantioselectivity (>97% ee) in the presence of base, but only 3 was found to be active in the absence of base. Liquid sampling of a scaled up reaction employing 3 (containing base) revealed the reaction was complete after 2 h 30 min. Isolation of the ester by distillation from a repeated reaction gave a 70% isolated yield of R-1-phenylethanol acetate (97% ee).
Changing the enzyme and bio-chemo compatibility, entrapping the enzyme
Results obtained from a single batch of the enzyme exhibit excellent reproducibility, with results repeatable multiple times, with little variation, independent of the experimenter. However, in our investigations, we found that the results of organometallic chemoenzymatic DKR can vary markedly as the batch of lipase B is changed.1 These problems are assumed to arise from the interaction between the bio- and chemo-catalysts. If the chemocatalyst associates with the enzyme, this will prevent further racemization and can alter the selectivity of the enzyme by distorting the protein's structure. One way of retarding association, which has been very effectively applied by Bäckvall and co-workers, is to add bulk to the organometallic chemocatalyst, Shvo's catalyst and Bäckvall's catalyst contain bulky phenyl substituents on the cyclopentadienyl ring (Fig. 1).5 An alternative approach is to associate the catalyst with a solid material.A materials approach to the prevention of mutual poisoning of bio- and chemo-catalysts has been reported by Blum and Avnir.16 The catalysts were compartmentalized by entrapping them inside porous silica by sol–gel processes. This method physically imprisoned the catalyst preventing free diffusion through the solvent and therefore making processes that result from the interaction between the bio- and chemo-catalyst unlikely.17 Entrapping both catalysts in chemoenzymatic DKR is likely to lead to a highly diffusion limited process, therefore we investigated the entrapment of just the biocatalyst (CalB) in silica gels.
The activity of a batch (different from that above) of lipase acrylic resin from Candida Antarctica (Novozym® 435) and 3 was evaluated. Kinetic resolution gave the expected result of 50% 1-rac-phenyl ethanol conversion to the R-1-phenylethyl acetate. The reaction was repeated as a DKR in the presence of 3, after 48 h only 47% conversion to 1-rac-phenyl acetate was observed. This drop in performance is indicative of an interaction between the chemocatalyst and enzyme. In order to compartmentalize the catalyst and reduce poisoning the Novozym® 435 was entrapped in silica by sol–gel methods. This provides a porous silica coating around the lipase acrylic resin. Entrapment methods have been successfully applied by Hoyos et al.18 as an enzyme stabilising method in the dynamic DKR of benzoins by Pseudomonas stutzeri employing Shvo's catalyst. In order to entrap the enzyme a basic sol–gel method was applied catalyzed by benzylamine; this method was based on an organogel-templated method previously described.19 Upon moderate heating and cooling organogelator 2,5-di-O-methanesulfonyl-1,4:3,6-dianhydro-D-sorbitol gels a range of solvents, including alcohols. In the presence of a silica precursor and promoter the organogel formed acts as a template transcribing silica formation and dictating structure on the micro- and nano-scale. This method was capable of providing a stable matrix that entrapped the enzyme. The distribution of the enzyme throughout the matrix is important to the activity of the resultant material. A base catalysed method of gelation and transcription was used as base promotes the hydrogen transfer reaction. AOT (dioctyl sodium sulfosuccinate) and PEG (polyethylene glycol) 400 were used to stabilize the enzyme and create the correct environment within the matrix.20 The charged head group on AOT has the potential to interact with the enzyme, and the PEG forms stable microemulsions within a gel matrix.21
An acidic sol–gel method was also used to entrap lipase B employing PEG 400 to stabilise the enzyme and phosphoric acid to catalyse the gelation.
In order to investigate the effects of the silica support, the entrapped enzyme catalysts were tested in kinetic resolution. The activity of the doubly immobilized basic gel was approximately 1/10th that of the acrylic resin Novozym® 435 (Table 3). The activity of the acidic gel could not be measured as the resulting 1H NMR spectrum contained many additional resonances, indicating that new side reactions had occurred; these were further investigated and the results reported below.
| Catalyst | Active/g | % Ester |
|---|---|---|
| Toluene (2.4 mL), rac-1-phenyl ethanol (7.2 mmol), isopropenyl acetate (7.2 mmol), temperature 70 °C, reaction time 4 h, NA = not applicable due to side reactions. | ||
| Novozym® 435 | 0.02 | 36 |
| Novozym® 435 | 0.04 | 50 |
| Basic gel | 0.33 | 39 |
| Acidic gel | 0.35 | NA |
In order to assess the extent to which the lipase B in the basic gel and in Novozym® 435 was heterogeneous and recyclable, the following procedure was carried out. The enzyme-containing material was exposed to reaction conditions for 2 h, the solution was then filtered off and returned to reaction conditions for a further 2 h, the lipase-containing material was added to a fresh reactant solution and run for 4 h (Table 4).
| Catalyst | Initial conversion/% | Filtrate conversion/% | Recycled conversion/% |
|---|---|---|---|
| Toluene (2.4 mL), rac-1-phenyl ethanol (7.2 mmol), isopropenyl acetate (7.2 mmol), temperature 70 °C. | |||
| Novozym® 435 | 32 | 36 | 9 |
| Basic gel | 10 | 15 | 27 |
Under these conditions this batch of Novozym® 435 did not recycle well. A significant further conversion (from 32 to 36%) was observed when the filtrate was exposed to reaction conditions for a further 2 h, indicating leaching of active material. Furthermore the used enzyme aggregate gave only 9% conversion after 4 h, indicating little active material was left. The basic gel performed better but not ideally. Initial conversion was reduced relative to Novozym® 435, this was expected as the reactant must now diffuse through the silica gel to reach the entrapped lipase. Upon separation the filtrate was still active, and this reveals that active enzyme was leaching, the activity was 50% of that observed in the presence of the gel. The used and recycled entrapped enzyme gave 27% conversion after 4 h, consistent with conservation of enzyme activity.
Comparing the DKR activity of the entrapped enzyme with 3 and the activity of the parent Novozym® 435 with 3, the entrapped enzyme enabled a 5% improvement in conversion, affording 52% ester (Table 5).
| Catalyst | Active/g | Time/h | % Conversion | % Ester |
|---|---|---|---|---|
| Toluene (2.4 mL), rac-1-phenyl ethanol (7.2 mmol), isopropenyl acetate (7.2 mmol), Novozym® 435 (41 mg), potassium carbonate (0.26 g), temperature 70 °C. | ||||
| Novozym® 435 | 0.041 | 20 | 50 | 50 |
| Novozym® 435 + 3 | 0.020 + 0.0022 | 48 | 47 | 47 |
| Basic gel + 3 | 0.15 + 0.0022 | 48 | 52 | 52 |
We propose that the DKR reaction of the basic gel and 3 failed to achieve full conversion for two reasons. Firstly there is a reduction in the activity of the enzyme due to the additional diffusion processes necessary to approach the active site. Secondly there is measureable leaching from the gel, the active enzyme in solution is believed to interact with the chemocatalyst, retarding activity and preventing full conversion of the substrate. As the entrapment of lipase B was only partially successful and the basic gel exhibited some leaching, higher but not complete conversion was observed in the resultant DKR. We suggest that a better entrapment method would lead to a more robust and recyclable DKR.
In order to investigate the side-reactions occurring when the acidic gel was employed, a gel was prepared consisting of PEG 400, phosphoric acid and silica, with no entrapped lipase. The gel was heated to 105 °C (unstirred) in a toluene solution containing rac-1-phenylethanol. The products detected correlated with the side products during kinetic resolution and were identified as the products of dehydration [1-(1-phenylethoxylethyl)benzene] and styrene (Scheme 3). In order to test whether the catalyst behaved as a homogeneous or heterogeneous Brønsted acid catalyst, the following procedure was carried out. The dehydration was performed for 2 h and the solution and gel separated by filtration. The filtrate was returned to the reaction conditions for a further 2 h. For the final period of 2 h the gel was returned to the solution. The reaction was followed by 1H NMR and showed straightforward heterogeneous behaviour (Table 6, Chart 1).
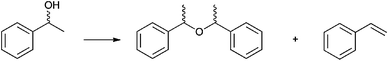 | ||
| Scheme 3 The dehydration of rac-1-phenyl ethanol. | ||
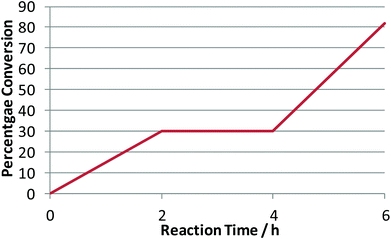 | ||
| Chart 1 Heterogeneous solid acid behaviour in the dehydration of rac-1-phenyl ethanol. | ||
Conclusion
There are a number of side reactions that can reduce the efficiency of the chemoenzymatic DKR of secondary alcohols to esters. Base-catalyzed DKR is susceptible to alcohol dehydrogenation to form ketones due to the Oppenauer oxidation activity of the chemocatalyst. As the relative amount of this side reaction depends on the organometallic catalyst, it can be largely avoided by picking a suitable catalyst precursor. In the absence of base an acid catalyzed dehydration is possible to form ethers and alkenes. These side reactions are catalyzed by acids and can be excluded through sensible choice of catalysts, reagents and conditions.The most detrimental and unpredictable side reaction in chemoenzymatic DKR is the interaction between the chemo-catalyst and bio-catalyst, this reduces the activity of the enzyme, can hamper enantioselectivity, and frequently stops the racemization catalyst working altogether. The application of bulky groups on the metal catalyst can help to avoid this interaction, but this can be at the detriment of the overall rate, as steric bulk will slow down racemization. In instances where considerable poisoning occurs it is difficult to prevent, and extremely active hydrogen transfer catalysts can be rendered inactive. We suggest that the further protection of the enzyme in a porous material, or alternatively, the adoption of a heterogeneous hydrogen transfer catalyst, can reduce poisoning. A sol–gel entrapped dehydrogenation catalyst was recently reported by Oded et al.22
Experimental
NMR spectra were recorded on 400 MHz and 300 MHz Bruker NMR spectrometers. HPLC was performed on an Agilent 1100 with a chiral AD-H column (25 × 0.46 cm), wavelength 190 nm, column flow 1 cm3 min−1, solvents hexane–propan-2-ol (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5), injection volume 5 μl.
:5), injection volume 5 μl.
Solvents were dried and degassed using standard methods.
Complexes [(η6-mesitylene)RuCl(μ-Cl)]2, [(η6-p-cymene)RuCl(μ-Cl)]223 [Cp*IrCl(μ-Cl)]2, [Cp*RhCl(μ-Cl)]224 and [(η5-Ph5C5)RuCl(CO)2]15 were prepared by literature methods. The synthesis of [(η6-mesitylene)RuCl(dfppm)]BF4, [(η6-p-cymene)RuCl(dfppm)]BF4, 1 [(η6-mesitylene)Ru(μ-Cl)3RuCl(dfppm)], [Cp*IrCl(dfppm)]BF4, [Cp*RhCl(dfppm)]BF4 and 2 [{η5,κP-C5Me4CH2C6F4-2-P(C6F5)CH2P(C6F5)2)}RhCl2] were conducted as previously reported.13 [(η6-Mesitylene)RuCl(dppe)]BF4, [(η6-mesitylene)RuCl(dppp)]BF4 were prepared by methods analogous to [(η6-mesitylene)RuCl(dfppm)]BF4 by substituting dfppm with the equivalent amount of dppe and dppp. [Cp*RhCl(dppe)]BF4 was prepared in a method analogous to [Cp*RhCl{(C6F5)2PCH2CH2(C6F5)2}]BF4 by substituting dfppm with the equivalent amount of dppe.25
[Cp*RhCl2(PPh3)] was prepared by a method analogous to [Cp*RhCl2P(C6H4F-4)].
[Cp*RhCl(μ-Cl)]2 (0.071 g, 0.11 mmol) and P(C6H4F-4)3 (0.073 g, 0.23 mmol) were dissolved in dichloromethane (20 mL). The resulting red solution was kept at ambient temperature overnight. Concentration by rotary evaporation afforded the product as a red solid, which was filtered and dried in vacuo. Yield 0.116 g (81%).
1H NMR (CDCl3): δ 7.80 (6H, m, Ho or Hm), 7.08 (6H, m, Ho or Hm), 1.38 [15H, d, 4J(P–H) 3.5, CH3]. 19F NMR (CDCl3): δ −108.43 (2F, br), −110.45 (1F, br). 31P NMR (CDCl3): δ 28.9 [d, 1J(Rh–P) 146]. EI-MS: m/z 589 [M − Cl]+. (Found: C, 53.4; H, 4.2. Calc. for C28H27Cl2F3PRh: C, 53.8; H, 4.35%).
3 [Cp*IrCl2(1-benzyl,3-methyl-imidazol-2-ylidene)] was prepared as previously reported.26,27 2,5-Di-O-methanesulfonyl-1,4:3,6-dianhydro-D-sorbitol was prepared as previously reported.19
The following reagents were used as supplied; Novozym® 435, isopropenyl acetate, DPEN, PHEN, benzylamine, Tetraethylorthosilicate (TEOS), PEG 400 and AOT (Sigma-Aldrich), Rac-1-phenyl ethanol (Fluka and Sigma-Aldrich), potassium carbonate (Lancaster), phosphoric acid (85% aqueous solution Lancaster).
KR/DKR to convert rac-1-phenyl ethanol to R-1-phenylethanol acetate
The catalyst precursor(s) (if required), isopropenyl acetate and rac-1-phenyl ethanol were dissolved in toluene and degassed in vacuo. Potassium carbonate was added and the suspension stirred at 70 °C under nitrogen for 15 min. Novozym® 435 was added and the reaction stirred gently at 70 °C under nitrogen for the reaction time. The product solution was filtered through a silica plug eluted with hexane–diethyl ether (10:1). The filtrate was reduced by rotary evaporation to yield a colourless liquid. The product was analysed by 1H NMR to determine conversion and HPLC to determine enantiopurity. 1H NMR was used to determine conversion as the spectrum can be recorded in the presence of the metal complex (the presence of the metal catalyst in solution would be damaging to the HPLC column). This enables the degree of conversion to be measured before and after work-up in order to check the validity of the work-up method.
Entrapping lipase in porous silica, basic method
Ethanol (1.0 mL) was added to ground Novozym® 435 (0.04 g) in a glass vial. AOT (0.0445 g, 0.100 mmol) was added and the mixture stirred until it dissolved. 2,5-Di-O-methanesulfonyl-1,4:3,6-dianhydro-D-sorbitol (0.10 g, 0.33 mmol), PEG 400 (2.5 mL), ethanol (1.25 mL) and water (0.25 mL) were heated in a separate vial in a water bath to 60 °C and stirred to obtain a homogeneous solution. The solutions were combined maintaining heating and stirring. Benzylamine (0.14 g, 1.3 mmol) and TEOS (0.475 g, 2.38 mmol) were added and the mixture heated to 60 °C and stirred for a further 10 min. The gel was aged for 2–3 weeks before use.Entrapping lipase in porous silica, acidic method
Ground Novozym® 435 (0.04 g) was weighed into a glass vial. PEG 400 (1.0 mL) and TEOS (1.0 mL) were added, followed by phosphoric acid (0.40 mL, 85%). The mixture was stirred until the liquids homogenized, then left to gel. The gel was aged for 2–3 weeks before use.Dehydration of rac-1-phenylethanol by an acidic gel
The dehydration was performed under 1 atm N2, filtrations and work-up were performed in air. Acidic gel (0.0433 g), prepared as above but omitting Novozym® 435, was heated with anhydrous toluene (2 mL) and rac-1-phenyl ethanol (0.67 mL, 5.6 mmol) without stirring, heating oil temperature 105 °C, for 2 h. The solution was filtered from the gel under gravity and a sample taken for 1H NMR analysis. The solution was returned to the heat for a further 2 h and another sample taken and analyzed by 1H NMR. The gel and solution were recombined and heated for a final 2 h and then a final 1H NMR sample was analysed.Acknowledgements
Thanks to Shifang Liu (CCM research), Peter Goodrich, QUILL, ASEP (QUB) and the European Social Fund.References
- A. C. Marr and S. Liu, Trends Biotechnol., 2011, 29, 199–204 CrossRef CAS.
- P. M. Dinh, J. A. Howarth, A. R. Hudnott and J. M. J. Williams, Tetrahedron Lett., 1996, 37, 7623–7626 CrossRef CAS.
- A. L. E. Larsson, B. A. Persson and J.-E. Bäckvall, Angew. Chem., Int. Ed., 1997, 36, 1211–1212 CrossRef CAS.
- G. K. M. Verzijl, J. G. De Vries and Q. B. Broxterman, DSM Fine Chemicals. ‘Process for the preparation of enantiomerically enriched esters and alcohols’, World Pat., WO0190396 A1 20011129, 2001 Search PubMed.
- B. Martín-Matute and J.-E. Bäckvall, Curr. Opin. Chem. Biol., 2007, 11, 226–232 CrossRef.
- Y. Ahn, S.-B. Ko, M.-J. Kim and J. Park, Coord. Chem. Rev., 2008, 252, 647–658 CrossRef CAS.
- N. Menashe and Y. Shvo, Organometallics, 1991, 10, 3885–3891 CrossRef CAS.
- A. C. Marr, C. L. Pollock and G. C. Saunders, Organometallics, 2007, 26, 3283–3285 CrossRef CAS.
- Y. Sato, Y. Kayaki and T. Ikariya, Chem. Commun., 2012, 48, 3635–3637 RSC.
- R. V. Oppenauer, Recl. Trav. Chim. Pays-Bas, 1937, 56, 137–144 CrossRef CAS.
- T. Suzuki, K. Morita, M. Tsuchida and K. Hiroi, J. Org. Chem., 2003, 68, 1601–1602 CrossRef CAS.
- F. Hanasaka, K. I. Fujita and R. Yamaguchi, Organometallics, 2006, 25, 826–831 CrossRef CAS.
- A. C. Marr, M. Nieuwenhuyzen, C. L. Pollock and G. C. Saunders, Organometallics, 2007, 26, 2659–2671 CrossRef CAS.
- A. C. da Silva, H. Piotrowski, P. Mayer, K. Polborn and K. Severin, Eur. J. Inorg. Chem., 2001, 685–691 CrossRef CAS.
- (a) B. Martín-Matude, M. Edin, K. Bogár and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2004, 43, 6535–6539 CrossRef; (b) B. Martín-Matude, M. Edin, K. Bogár, F. B. Kaynak and J.-E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 8817–8825 CrossRef.
- F. Gelman, J. Blum and D. Avnir, J. Am. Chem. Soc., 2002, 124, 14460–14463 CrossRef CAS.
- A. C. Marr and P. C. Marr, Dalton Trans., 2011, 40, 20–26 RSC.
- P. Hoyos, A. Buthe, M. B. Ansorge-Schumacher, J. V. Sinisterra and A. R. Alcántara, J. Mol. Catal. B: Enzym., 2008, 52–53, 133–139 CrossRef CAS.
- S. J. Craythorne, C. L. Pollock, A. J. Blake, M. Nieuwenhuyzen, A. C. Marr and P. C. Marr, New J. Chem., 2009, 33, 479–483 RSC.
- M. M. R. Talukder, M. M. Zaman, Y. Hayashi, J. C. Wu, T. Kawanishi, C. Ogino and N. Shimizu, J. Chem. Technol. Biotechnol., 2004, 79, 273–276 CrossRef CAS.
- K. Soni and D. Madamwar, Process Biochem., 2001, 36, 607–611 CrossRef CAS.
- K. Oded, S. Musa, D. Gelman and J. Blum, Catal. Commun., 2012, 20, 68–70 CrossRef CAS.
- (a) M. A. Bennett and A. K. Smith, J. Chem. Soc., Dalton Trans., 1974, 233–241 RSC; (b) D. A. Tocher, R. O. Gould, T. A. Stephenson, M. A. Bennett, J. P. Ennett, T. W. Matheson, L. Sawyer and V. K. Shah, J. Chem. Soc., Dalton Trans., 1983, 1571–1581 RSC.
- C. White, A. Yates and P. M. Maitlis, Inorg. Synth., 1992, 29, 228–234 CrossRef CAS.
- M. J. Atherton, J. Fawcett, J. H. Holloway, E. J. Hope, A. Karacüar, D. R. Russell and G. C. Saunders, J. Chem. Soc., Dalton Trans., 1996, 3215–3220 RSC.
- S. McGrandle and G. C. Saunders, J. Fluorine Chem., 2004, 126, 451–455 Search PubMed.
- R. Corberán, M. Sanaú and E. Peris, J. Am. Chem. Soc., 2006, 128, 3974–3979 CrossRef.
Footnote |
| † This paper is dedicated to Prof. David J. Cole-Hamilton on occasion of his retirement in celebration of his distinguished (and ongoing) contribution to transition metal catalysis, and his role in introducing ACM to organometallic chemistry for homogeneous catalysis, his enthusiasm was truly infectious. |
| This journal is © The Royal Society of Chemistry 2012 |