Design and development of catalysts for Biomass-To-Liquid-Fischer–Tropsch (BTL-FT) processes for biofuels production
Rafael
Luque
*a,
Ana Raquel
de la Osa
*b,
Juan Manuel
Campelo
a,
Antonio Angel
Romero
a,
Jose Luis
Valverde
b and
Paula
Sanchez
b
aDepartamento de Química Orgánica, Edificio Marie Curie (C-3), Ctra Nnal IVa, Km 396, E-14014, Córdoba, Spain. E-mail: q62alsor@uco.es
bDepartamento de Ingeniería Química, Universidad de Castilla la Mancha, Ciudad Real, España, E13071, Spain. E-mail: AnaRaquel.Osa@uclm.es; anraqel@gmail.com
First published on 2nd December 2011
Abstract
BTL-FT processes for hydrocarbon production from syngas obtained from biomass gasification are becoming increasingly trendy as suitable alternatives to produce various high quality fuels for different applications. Many investigations are ongoing to test the suitability of biomass syngas for FTS using the traditionally employed catalysts. The choice of catalysts for these processes is normally restricted to Fe and Co-based materials as they provide the best compromise between performance and price. In this perspective, we aim to provide a comparative overview of the performance between Fe and Co-based high performance FTS catalysts for hydrocarbon production via BTL-FTS processes.
Broader contextFTS processes from biomass derived syngas have received a great deal of interest in recent years as a suitable alternative to produce various high quality (free of sulfur, nitrogen and aromatics) fuels for different applications, since they are compatible and blendable with conventional fuels, thus offering the possibility to work with the existing fuel infrastructure. Part of current FTS research is actively devoted to test the suitability of biomass syngas for FTS using the traditionally employed catalysts. However, few studies have investigated in detail the suitability of different catalysts in the BTL-FT process. Hence, in this perspective, we aim to compile the latest developments in the preparation of high performance FTS catalysts for hydrocarbon production from different feedstocks, mostly gasified biomass, including a comparative overview of the Fe- and Co-based catalysts performance. In addition, the integration of biomass gasification processes and catalyst design is also a crucial step to be optimized for the successful implementation of BTL processes for biomass valorization and biofuels production. Other promising strategies such as the production of hydrocarbons from CO2 and the gasification of coal-biomass blends have been also highlighted. |
Introduction
The Fischer–Tropsch synthesis (FTS) process was originally developed in the early 1920s as an alternative way to convert various gaseous products (mainly CO/H2 mixtures denoted as syngas) into a wide variety of hydrocarbon products, from gases to waxes, including liquid hydrocarbons of commercial interest (different chain lengths), via elongation of the hydrocarbon chain in a reaction generally catalyzed by a supported metal (e.g.Fe, Co).1| CO + 2H2 → –CH2– + H2O |
The process became fairly popular over the years and currently the term FTS applies to a wide variety of processes that deal with the production of hydrocarbons from syngas originating from any carbon-containing feedstock including coal and natural gas (most widely employed feedstocks) and more recently biomass.1,2 This versatility concerning both feedstock and products is precisely one of the most important advantages of FTS, which is expected to become the world's fastest growing energy segment. Depending on the feedstock, the process is referred to as CTL (Coal-To-Liquids), GTL (Gas-To-Liquids) or BTL (Biomass-To-Liquids).
Synthetic fuels have also distinct environmental advantages over conventional crude-refined fuels since they are virtually free of sulfur, nitrogen and aromatics as well as being compatible and blendable with conventional fuels, thus offering the possibility to work with the existing fuel infrastructure. Nevertheless, the economic viability of the FT process largely depends on the price of crude oil due to the high price of synthetic FT fuels owing to its energy demanding nature and the large capital cost requirements of FT plants.
Technical advances in the FT process and the increasing crude oil prices (in combination with the depletion of the crude reserves) have led to a renewed worldwide interest in the FT process in the last decade, especially since high quality clean biofuels (compatible with the existing infrastructure and vehicle technology) can be produced from a wide variety of biomass resources. Biomass to be employed in the BTL process include wood (most attractive feedstock), forest (wood chips), food (almond shells, grape pulp, exhausted olive husk, olive pits) and agricultural (pruning vines) residues and waste by-products including bagasse, lignocellulosic feedstocks from processing residues (e.g. paper slurry, black liquor, etc.) and others.2–4
Commercialization of the FT technology began in 1936 (Germany) using coal as syngas source and low-temperature FT (LTFT) technology. The Carthage Hydrocol plant was an example of an earlier iron-based HTFT process developed in U.S.A. (1940–1950s) to maximize gasoline production from natural gas. Another three coal-based Sasol FT plants were constructed in South Africa beginning with Sasol 1 (1952) that combines the two LTFT and high-temperature FT (HTFT) variants. However, the design changed in 2004 substituting coal gasification by natural gas reforming to yield waxes and chemicals. Sasol 2 and Sasol 3 (1970–1980s) mainly produce motor gasoline and diesel although chemicals are also included. Another FT facility based on the Sasol iron-catalyzed HTFT Synthol technology is operated by PetroSA at Mossel Bay, South Africa, since 1993, whose process scheme is focused on producing gasoline and smaller amounts of distillates.5
Currently, outside South Africa, there are few FT plants in full industrial-scale operation producing fuels, lubricants and chemical feedstocks from coal and gas, let alone biomass. The vast majority of BTL schemes are in the pilot or demonstration phase.6 One example is Shell's first commercial GTL facility commissioned in Bintulu, Sarawak, Malaysia in 1993. Shell developed its LTFT technology based on the Shell middle distillate synthesis (SMDS) technology (recommissioned in 2000) and proprietary Co LTFT catalyst producing a synthesis gas conversion between 80 and 95% with C5+ selectivity in the range of 85–95%.5,7 The 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 bpd Oryx GTL plant at Las Raffan (Qatar), very similar to that of the Shell Bintulu refinery, uses LTFT technology based on the Sasol Slurry Phase Distillate (SPD) process to produce mainly diesel fuel and naphtha as a byproduct. It uses a Co-based Sasol proprietary catalyst typically operating at 230 °C and 2.5 MPa and the product is distilled to produce LPG (3–7%), naphtha (20–30%), and distillate (65–75%), with the unconverted >360 °C waxy product being recycled back to the hydrocracker.5,8
000 bpd Oryx GTL plant at Las Raffan (Qatar), very similar to that of the Shell Bintulu refinery, uses LTFT technology based on the Sasol Slurry Phase Distillate (SPD) process to produce mainly diesel fuel and naphtha as a byproduct. It uses a Co-based Sasol proprietary catalyst typically operating at 230 °C and 2.5 MPa and the product is distilled to produce LPG (3–7%), naphtha (20–30%), and distillate (65–75%), with the unconverted >360 °C waxy product being recycled back to the hydrocracker.5,8
The biggest FT project (Shell's Pearl GTL in Qatar—capacity of 260000 bbl of oil equivalent/day of fuels, lubricants and naphtha-will produce enough fuel to fill over 160000 cars a day and enough synthetic base oil each year to make lubricants for more than 225 million cars) received its first flow of dedicated gas in March 2011 and is expected to start operation in 2012. Its proprietary Co-based catalyst system has taken around 30 years to develop, and prototypes of it have been in commercial-scale application in a much smaller GTL facility at Bintulu, Malaysia, since 1993. The Pearl GTL scheme employs a multi-tubular fixed-bed technology (24 reactors).9
Furthermore, as BTL R&D in Europe is gathering momentum, the world's first commercial BTL Plant was inaugurated in 2008 in Frieberg Saxony (Germany), utilising the Choren Carbo-V®Process. The plant, owned by Choren (VW, Daimler and Shell as partners), produces BTL fuel from any kind of biomass, such as wood chips, straw, weeds or leftover milk rejected by the agrofood industry. Unfortunately, as the manufacturing costs and the technology appeared to be uncontrollable, Shell opted out in 2009, followed by VW and Daimler, and so insolvency was unavoidable.10 However, there are some other BTL projects such as the BioTfuel BTL demonstration one, which includes the construction and operation of two pilot plants (scheduled to go into operation in 2012) in France to produce biodiesel and biokerosene based on biomass gasification. Forschungszentrum Karlsruhe GmbH in partnership with LURGI GmbH is constructing a pilot plant (due 2016) for production of BTL and “gasoline type fuels” and CEA (Atomic and Alternative Energy Commission) France announced the construction of a pilot BTL plant in Bure Saudron 75000 tonnes per year of forestry and agricultural residues to produce ∼23000 tonnes per year of biofuel (diesel, kerosene and naphtha). On the other hand, NSE Biofuels Oy (a joint venture between Neste Oil and Stora Enso) has opened a BTL demonstration plant at Stora Enso's Varkaus Mill in Finland (656 t/a from a 12 Mw gasifier) and, in partnership with Foster Wheeler and VTT, is planning to develop a commercial production plant at one of Stora Enso's mills with a projected output capacity of 100000 t/a and a potential launch date of 2016. Table 1 takes inventory of some of the most important world FTS plants.11
| Company | Country | Capacity/barrels per day | Raw material | Status | Catalyst type |
|---|---|---|---|---|---|
| Sasol | South Africa | 150.000 | Coal | In operation | Fe/K |
| China | 2 × 80.000 | Coal | Abandoned | — | |
| Australia | 30.000 | Natural gas | Study | — | |
| Nigeria | 34.000 | Natural gas | Under construction | — | |
| Qatar | 34.000 | Natural gas | In operation | Co/Al2O3 | |
| Shell | Malaysia | 14.700 | Natural gas | In operation | Co/SiO2 |
| Qatar | 140.000 | In operation | Proprietary Co-based | ||
| Indonesia | 75.000 | Study | — | ||
| Iran | 70.000 | Abandoned | — | ||
| Egypt | 75.000 | Study | — | ||
| Argentina | 75.000 | Study | — | ||
| Australia | 75.000 | Study | — | ||
| Shell Choren | Germany | 300 | Biomass | In operation | — |
| Mossgas | South Africa | 22.500 | Natural gas | In operation | Fe/K |
| EniTechnologie | Italy | 20 | Natural gas | In operation | — |
| BP | USA | 300 | Natural gas | In operation | Proprietary Co-based |
| Rentech | USA | 1.000 | Natural gas | In operation | Proprietary Fe-based |
| South Africa | 10.000 | Study | — | ||
| Bolivia | 10.000 | Under Construction | — | ||
| Rentech pertamina | Indonesia | 15.000 | Natural gas | Study | — |
| Syntroleum | USA | 70 | Natural gas | Closed | — |
| Australia | 11.500 | Natural gas | Under construction | — | |
| Chile | 10.000 | Natural gas | Study | — | |
| Peru | 5.000 | Natural gas | Study | — | |
| Syntrol.-Tyson Foods | USA | 5.000 | Biomass | In operation | Proprietary catalyst |
| Gazprom syntroleum | Russia | 13.500 | Natural gas | Study | — |
| Repsol-YPF | Bolivia | 13.500 | Natural gas | Study | — |
| Syntroleum | Bolivia | 90.000 | Study | — | |
| ExxonMobil | Qatar | 90.000 | Natural gas | Abandoned | — |
| Conoco | Qatar | 60.000 | Natural gas | In operation | Proprietary catalyst |
| USA | 400 | In operation | |||
| Bioliq | Germany | — | Biomass | Under construction | — |
The production of synthetic fuels from biomass comprises of the four basic steps of all FT processes: (1) biomass pre-treatment, (2) gasification of the biomass feedstock to synthesis gas (syngas, CO + H2) followed by gas cleaning/conditioning; (3) FTS production and (4) upgrading of the FT liquids to high quality fuels (Fig. 1). However, as commented before, a fully scaled-up commercial BTL process has not been completely established to date. Biomass gasification can be in fact pretty different as compared to coal or natural gas gasification.2–5,12,13 Although the gasification of natural gas is well known not to be associated with the production of high amounts of contaminants, coal and biomass gasification (especially coal) involves some additional cleaning and conditioning steps to reach FTS syngas composition requirements. The non-homogeneous character of most biomass resources (inconsistent moisture-required level: from <10% (Lurgi) to <70% (Foster Wheeler), density, size and thermal energy content of most biomass feeds) poses difficulties in maintaining constant feed rates to gasification units. Moreover, the high oxygen and moisture content results in a low heating value for the product syngas, which results in a higher system energy requirement, a dirtier syngas and problems for downstream combustors designed for a consistent medium-to-high heating value fuel. High moisture content fuels generally decrease reactor-operating temperature and, therefore, may increase methane content and lower hydrogen content. However, its use for energy production can significantly contribute to the reduction of net CO2 emissions. Table 2 summarizes some physico-chemical parameters determined for different syngas raw materials.
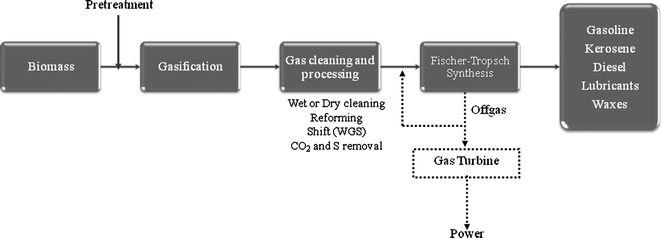 | ||
| Fig. 1 Schematic step process for converting biomass to FT-liquids (combined with gas turbine-IGCC power generation). Adapted from ref. 127. | ||
| Ref. | Sample | HHV/kWh kg−1 | wt% dry basis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Moisture | Ash | V.M | S | C | H | N | O | F.C | |||
| a F.C: fixed carbon; N.D.: non-detected; (p): pellets; V.M: volatile matter. | |||||||||||
| G. Chen, 2011 (ref. 137a) | Coal | 7.64 | 15.01 | 7.64 | 43.32 | 0.54 | 69.02 | 4.72 | 0.80 | 17.28 | — |
| Biomass | 5.48 | 6.57 | 0.56 | 80.27 | 0.01 | 46.65 | 5.90 | ND | 46.89 | — | |
| A. Smolinski, 2011 (ref. 137b) | Lignite | 5.27 | 14.52 | 8.62 | 42.81 | 1.91 | 50.68 | 3.9 | 1.31 | 20.37 | 34.05 |
| Hardcoal | 6.91 | 11.49 | 5.42 | 32.31 | 1.32 | 64.9 | 3.63 | 0.96 | 13.24 | 50.78 | |
| Biomass | 4.15 | 6.78 | 1.6 | 76 | 0.05 | 53.71 | 6.59 | 0 | 31.27 | 15.62 | |
| A. Lickrastina, 2011 (ref. 137c) | Wood (p) | 4.96 | 7.9 | 2.1 | 81.3 | 0.01 | 45.0 | 6.0 | 0.1 | — | — |
| Wheat straw (p) | 4.6 | 9.4 | 3.7 | 71.3 | 0.13 | 49.7 | 5.3 | 1.1 | — | — | |
| S.I. Ngo, 2011 (ref. 137d) | Pine woodchips | — | — | 0.01 | 50.8 | 5.4 | ND | 43.6 | — | ||
| W.T. Tsai, 2007 (ref. 137e) | Rice husks | 4.7 | 6.8 | 12.5 | — | 0.31 | 48.36 | 5.13 | 0.72 | 32.79 | — |
| J.M. Encinar, 2000 (ref. 137f) | Cardoon | 5.1 | — | 8.4 | 0.09 | 42.78 | 4.4 | 0.64 | 43.69 | — | |
| F.A. Agblevor, 1995 (ref. 137g) | Hybrid p. wood | 5.5 | 5.0 | 1.2 | 0.05 | 49.4 | 6.0 | 0.23 | 43.1 | — | |
| Switchgras | 5.4 | 5.0 | 4.6 | 0.11 | 46.9 | 5.8 | 0.58 | 42.0 | — | ||
| Corn stover | 5.2 | 5.4 | 5.0 | 0.12 | 46.0 | 5.9 | 0.88 | 41.4 | — | ||
| E. Schröder, 2004 (ref. 137h) | Beech wood | 5.4 | 7.8 | 0.47 | 0.02 | 49.47 | 5.6 | 0.16 | 44.39 | — | |
| A. Demirbas, 2006 (ref. 137i) | Sunflower shell | 5.0 | — | 4.0 | 0.05 | 47.4 | 5.8 | 1.4 | 41.4 | — | |
| I. Aigner, 2011 (ref. 132) | Hard coal | 8.1 (LHV) | — | 7.41 | 34.66 | 0.46 | 76.49 | 3.87 | 1.34 | 10.29 | 65.34 |
| Wood (p) | 5.2 (LHV) | — | 0.29 | 86.45 | 0.01 | 50.23 | 6.04 | 0.05 | 43.38 | 13.55 | |
| J. Fermoso, 2009 (ref. 137j) | Bituminous coal | 5.2 | — | 37.0 | 24.3 | 1.60 | 75.7 | 5.3 | 1.4 | 16.0 | — |
| Petcoke | 9.7 | 0.3 | 9.6 | 6.20 | 87.6 | 3.8 | 1.5 | 0.9 | |||
| Almond shells | 5.5 | 1.2 | 79.3 | 0.00 | 49.8 | 6.1 | 0.2 | 43.9 | |||
| Olive stones | 5.6 | 0.8 | 83.8 | 0.00 | 52.0 | 6.2 | 0.1 | 41.7 | |||
| Eucaliptus | 5.4 | 0.7 | 83.6 | 0.00 | 50.6 | 6.4 | 0.1 | 42.9 | |||
| M. Lapuerta, 2008 (ref. 137k) | Pinus pruning | 5.6 (LHV) | — | 2.67 | 82.10 | <0.01 | 50.55 | 6.12 | 0.45 | 40.20 | 15.13 |
| Olive pruning | 5.6 (LHV) | 3.67 | 82.35 | 0.04 | 47.50 | 6.00 | 1.06 | 43.66 | 13.98 | ||
| Grapevine-pruning | 5.0 (LHV) | 2.06 | 78.16 | 0.01 | 46.97 | 5.80 | 0.67 | 44.49 | 19.78 | ||
| Sawdust wastes | 5.7 (LHV) | 1.28 | 82.45 | 0.05 | 50.26 | 6.14 | 0.07 | 42.20 | 16.27 | ||
| Marc of grape | 5.4 (LHV) | 7.83 | 65.77 | 0.14 | 49.66 | 5.56 | 2.23 | 34.42 | 26.40 | ||
| Coal-coke blend | 6.3 (LHV) | 25.70 | 14.81 | 2.97 | 61.51 | 3.13 | 1.50 | 5.23 | 59.49 | ||
A great deal of research is actively ongoing on all four steps of the process in an effort to improve the overall efficiency, with special focus on the biomass gasification step and subsequent gas-conditioning and cleaning prior to the FTS in order to meet the strict FT gas purification requirements. For more details of these processes, readers are kindly referred to the recent available reports in the literature by many authors.2–5,12–20
The FT process is generally operated at fairly high temperatures (in the 150–350 °C range) to avoid the formation of high quantities of methane as a by-product. Increased pressures lead to higher conversion rates and also favour the formation of desired long-chain alkanes. Typical pressures are in the range of one to several tens of atmospheres, although it is usual to operate at 20 bars. The FT hydrogenation reaction is normally catalyzed by metal catalysts, while the size and distribution of the hydrocarbon products of the reaction are generally governed by the Anderson–Schulz–Flory (ASF) chain polymerization kinetics model.21 However, it has also been proposed that total product distribution for the LTFT process could be a combination of two separate superimposed ASF distributions that are on two types of sites for the chain growth on the catalyst surface and, therefore, each site might individually yield the ideal ASF distribution. For iron-based catalyst, a carbide surface is proposed for the production of hydrocarbons (including n- and iso-paraffins and internal olefins) and an oxide surface is suggested for the production of light hydrocarbons (including n-paraffins, 1-olefins, and oxygenates) and the water–gas shift (WGS) reaction.15 For cobalt-based catalysts, Patzlaff et al.23 indicated that chain length distributions were slightly modified by secondary chain growth of readsorbed alkenes and hydrogenolysis of hydrocarbons. They advanced that the degree of hydrogenation of monomers for the products described with α1 is higher than those with α2. Therefore, CH2 was assumed as C1 intermediate and attributed to the distribution of growth probability α1, whereas hydrocarbon chain growth probability α2 was built up due to monomers except CH2 species with low degree of hydrogenation.
Since the publication of one of the most relevant reviews dealing with the development of FTS catalysts,24 a plethora of FTS catalysts have been designed, developed, synthesized and utilized for various FT processes. These have mainly been Co- and Fe-based catalysts, with some minor reports of Ru and Ni-based catalysts. Co-based catalysts were originally reported to provide an optimum compromise between performance and cost for the synthesis of hydrocarbons from syngas (CO/H2) mixtures,24 but Fe-based materials have been attracting an increasing amount of attention due to their possibility to work under low and high temperature FTS conditions for both the synthesis of short chain alkanes (High T) and waxes or long chain hydrocarbons (low T).2,12,25 Typically, fixed-bed and slurry-phase reactors are employed for LTFT processes with either Co or Fe catalysts for the production of linear long-chain alkanes, whereas fluidized bed reactors are used for HTLT processes with Fe catalysts for the production of C1–C15hydrocarbons and a-alkenes.26 New types of reactors, such as monolith structured, microstructured, and membrane reactors, have also been reported for FT synthesis.27
In this perspective, we aim to compile the latest developments in the preparation of high performance FTS catalysts for hydrocarbon production from different feedstocks, mostly gasified biomass, following the preceding overviews in the field by Davis2 and van Steen and Claeys28 A promising methodology has also been recently developed to produce hydrocarbons from CO2 using similar FT processes and will be briefly discussed in the prospects and future section of this manuscript. For an overview of the heterogeneous catalytic CO2 conversion to hydrocarbons, readers are kindly referred to the recent publication by Dorner et al.29
Development of FTS catalysts: properties and types
An optimum FT catalyst mainly requires a high hydrogenation activity to catalyze the hydrogenation of CO to higher hydrocarbons. In this regard, there are only four transition metals which possess a sufficiently high hydrogenation activity to be employed in FTS processes. These are Fe, Co, Ni and Ru. The Ru catalyst has been reported as the most active for the Fischer–Tropsch synthesis.30 As compared with Co, Ru catalysts possess higher intrinsic activity and can work under higher partial pressures of water or other oxygenate-containing atmospheres,31 which becomes particularly important for the conversion of syngas produced from biomass.31b However, its use in large scale FTS processes is restricted due to its low availability and high price.31a,32 Therefore, it has been also used as a promoter of the Co-based catalyst or even as a bimetallic one since it seems to favor reduction of cobalt oxide particles,33 a lower fraction of barely reducible mixed oxides, cobalt dispersion34 and inhibition of catalyst deactivation.35Nickel has a comparable hydrogenation efficiency to Ru but is essentially a methanation catalyst (promotes the reaction of CO to produce large quantities of methane). Fe and Co are consequently employed as FT catalysts at the industrial scale. The choice of catalyst principally depends on the FT operating mode (high or low temperature) and the utilized feedstock (natural gas, coal or biomass). The lifetime of these catalysts is influenced at least by two factors: the physical properties (catalyst attrition, wax accumulation within the catalyst pellet, pressure drop across the bed, etc.) and the loss of catalyst sites through poisoning and/or fouling. Therefore, lifetime data for catalysts in commercial operations are difficult to find. As an example, the lifetime of the Fe based catalysts is short and in commercial installations is generally limited to 8 weeks.36 Sasol has indicated that iron catalysts are replaced because of physical changes in the catalyst or to attrition and not due to the loss of catalyst sites due to poisoning and/or fouling. On the other hand, Shell declared that they expect their supported cobalt catalyst to have a lifetime of five or more years in their fixed-bed reactor operations.2 Hence, the biggest problem with FT catalysts is their vulnerability to deactivation. The proposed mechanisms of inherent catalyst deactivation include sintering, surface carbon formation, carbidization, re-oxidation, metal–support mixed compound formation, surface reconstruction and mechanical deactivation through attrition. However, the loss of activity is also related to operational catalyst deactivation (poisoning, process conditions and the type of reactor—fixed-bed or slurry). A short description is summarized in Table 3. Thus, considerable progress is required in developing new support structures, mesoporous systems and promoters for enhancing activation in order to raise the efficiency of the catalysts. In the next sections, these topics will be discussed in detail.| Inherent catalyst deactivation | Operational catalyst deactivation | ||||||
|---|---|---|---|---|---|---|---|
| Effect | Cobalt catalyst | Iron catalyst | Effect | Cobalt catalyst | Iron catalyst | ||
| Sintering | It leads to a reduction of the active surface area. Crystallite transformations (coalescence) may increase surface migration and enhance the possibility of agglomeration. Highly depended on the support. Generally enhanced by water vapor. | It can cause 30% decrease in activity. Alumina stabilizes cobalt crystallites and makes the catalyst more resistant against sintering. Presence of water may accelerate this effect. | Deposition of catalytic species inside the pores is the physical encapsulation of catalytic particles, which can reduce the site sintering | Poisoning | Sulfur compounds lead to permanent catalyst deactivation by physical blocking of the sites and electronic modification of neighboring atoms. It does not affect the primary FTS product distribution but increases secondary hydrogenation of olefins. | It appears to be more a geometric blockage of sites than an electronic modification. It can be removed from syngas | Iron catalysts are more sulfur resistant than cobalt catalysts (S levels should be <0.02 mg m−3), especially those promoted with B or MnO. |
| Halogenated compounds (HCl) may result in the coverage of the catalytically active surface with support material. | Commonly in cobalt based-catalyst due to the larger amount of high surface area irreducible oxides present compared to precipitated iron catalysts. It can be removed from syngas. | ||||||
| Nitrogen compounds (NH3, HCN, NOx) appear to have a reversible kinetic inhibition effect caused by even ppb levels of them. | It results in a decrease in CO conversion for cobalt-based catalysts, but not for iron ones. It can be removed from syngas. | ||||||
| Alkali/alkali-earth metals, carbon or metal carbonyls may be also responsible for catalyst poisoning. | Small amounts of alkali/alkali earth metals usually increase chain growth probability. However, the activity is negatively influenced depending on the concentration level. | ||||||
| Possible deactivation due to iron carbonyl decomposition (minimized by operating at high temperatures and large crystallites). | Iron carbonyls do not lead to catalyst deactivation and may limit transport of the catalytically active compound out of the reactor. | ||||||
| Surface carbon formation | Reversible pore plugging and diffusion inhibition due to carbonaceous species formation that blocks the surface or chemisorb on the catalytically active sites. Thermodynamically favored under FTS conditions. The exact nature of the species is still unclear. Increased by higher crystallite size, but contradicting results reported. | Gradual deposition of largely polymeric carbon with TOS. | Weaker hydrogenation ability of iron-based catalysts does not enhance carbon formation. Additives such as Ru retard this effect. | Type of reactor: fixed-bed or slurry | The mechanical stability of the catalyst is important for slurry phase reactor and seems to highly depend on porosity, preparation, pre-treatment. | Alumina support resulted in better attrition resistance (slurry phase) than silica and titania for cobalt-based catalysts (12–20 wt% Co). | Mechanical stability (slurry phase) seems to be improved by the addition of moderate amounts of silica prior to spray-drying the catalyst, whereas alumina seems to decrease the mechanical stability. |
| Carbidization | Diffusion of carbon into the crystal of the catalytically active component results in the formation of carbide compounds. Thermodynamically not feasible. Reversible upon mild H2 treatments. | Rarely been taken into consideration | Rapid formation of iron carbide commonly observed | ||||
| Re-oxidation | Contradictory reports. Usually ascribed to the presence of H2O but thermodynamically unfavorable. Strongly dependent on experimental pretreatments and conditions, catalyst micro- and macroscopic properties and instrumentation. Minimized upon utilization of large active crystallites. | Only possible for cobalt crystallites <5 nm diameter. Some reports indicated further reduction under realistic conditions. | Large extent of oxidation (up to 40–50%). Iron carbide more resistant than metallic iron and stabilized by addition of Mn or alumina. | ||||
| Metal–support solid state reactions | Thermodynamically feasible, but kinetically restricted. Highly depended on the support, enhanced by the existence of surface re-oxidation. Water may promote this side effect. | Usual for SiO2, Al2O3 or TiO2 either as support or as a binder. Minimized by large crystallites. Not important for Co catalyst (Formed from unreduced CoO). | |||||
| Surface Reconstruction | It is induced by carbon. A link between reconstruction and deactivation has been reported but the phenomenon needs experimental evidences. | ||||||
| Attrition | Important for the industrial process (fluidized or slurry phase). Defined as the breakdown of solid particles (abrasion/erosion and fracture) that interferes with operation and causes catalyst loss. The attrition resistance of cobalt-supported catalysts decreased in the sequence Co/Al2O3 > Co/SiO2 > Co/TiO2(rutile) > Co/TiO2(anatase). It could be also affected to a much lesser extent by promoters (Ru, Cu or Zr, Ru–La, K, Cr oxides) but really suppressed by doping of the support with various bi-valent metals (e.g. Ni) and subsequent rise in calcination temperature. | ||||||
Iron catalysts
Iron-based catalysts are suitable for both High Temperature FT (HTFT, 300–350 °C) and Low Temperature FT (LTFT, 200–240 °C) processes. HTFT is generally utilized for the production of gasoline and linear low molecular mass olefins, while LTFT normally generates high molecular mass linear waxes.21 A list of industrially employed Fe materials in FT processes has been included in Table 4.Numerous research efforts have been performed on several aspects of the Fe catalysts, including investigations on the effect of promoters, supports, additives, pretreatments, preparation (metal nanoparticle size, morphology, phase transformations, etc.) and generally all chemical and physical properties of the materials in order to increase catalyst activity, enhance selectivity to the desired products, inhibit formation of unwanted products (especially methane), and improve resistance to sulfur poisoning. A summary of improved modified Fe catalysts employed in industry for the FT process is presented in Table 1.22
The most commonly employed iron materials for LTFT (waxes production) are prepared viaprecipitation and generally have high surface areas (>200 m2 g−1) upon calcination at 300–350 °C.22 For HTFT processes, catalysts are prepared by fusing magnetite with small amounts of promoters via microemulsion or spry drying methods.39Iron has also been supported on a range of supports (e.g.silica, alumina, titania, etc.) to stabilise the metal (structural promoters) and prevent its sintering under FTS conditions.27,40–42Carbon nanotubes (CNTs) have become an increasingly trendy support for the preparation of Fe FT catalysts, depositing Fe both on the outer walls43 or investigating the effect of Fe confination within the CNT.44
| Premium product | Catalysts | Reactors | Processes |
|---|---|---|---|
| C2–C4olefins | Fe/K, Fe/Mn, Fe/Mn/Ce Fe/K/S, Fe2O3Cx, Fe/C | Slurry, fluid-bed | Synthol, Koelbel, Rheinpreussen-Koppers, DowLPG |
| Gasoline | Fused Fe/K, Fe/K/ZSM-5, Fe/Cu/K and ZSM-5 | Fluid-bed, Fixed-bed Slurry/fixed-bed | Synthol, Gulf-Badger, Mobil One-Stage, Mobil Two-Stage |
| Diesel fuel | Fe/K | Fixed-bed (low T) | Sasol-Arge |
| Waxes | Fe/K, Fe/Cu/K | Slurry-bed (low T) | Mobil (First Stage) |
The phase compositions of the reduced iron catalysts and the phase transformations during FTS have great influence on the activity, selectivity and stability.45 It is well known that the activation with CO or syngas typically results in the reduction of Fe2O3 to Fe3O4 and finally to carbides. Generally, the activation process is a crucial step on iron catalysts since the differences in catalyst composition,45a,e pretreatment conditions45d and the type of reactor46 can result in different bulk phase compositions of the reduced catalyst. On the other hand, sequential phase-transformation of these iron catalysts during FTS also takes place and plays an important role in determining the structural integrity or attrition resistance of the catalyst particles.47Iron carbides can be re-oxidized to Fe3O4 during FTS if the ratios H2O/H2 or CO2/CO are high enough.45d In addition, the complexity of phase compositions and the transformations between these phases in syngas atmosphere results in controversies on the nature of active phases for FTS.46b,48 In fact, several types of iron carbides with different structures can be classified on the basis of the sites occupied by the carbon atoms: carbon atoms in trigonal prismatic interstitial sites leads to θ-Fe3C (cementite; after carburizing the reduced iron in syngas at >300 °C), χ-Fe5C2 (Hägg carbide; formed at higher carburization temperature) and Fe7C3 (Eckstrom and Adcock carbide), whereas the presence of carbon atoms in octahedral interstitial sites results in ε-Fe2C (formed by CO carburizing of iron oxides at low temperatures) and έ-Fe2.2C (hexagonal carbides). FexC (or FexCy) usually denotes iron carbides with poorly defined structures. Therefore, under working conditions, several iron species (iron oxides, α-iron, …) may coexist with iron carbides whose functions in the FTS process are still ambiguous. Some researchers suggested that carbide formation was necessary before the catalyst became active48,49 whereas others indicated that the mixture of χ-, έ-iron carbides and a small amount of α-iron was the active phase for hydrogenation.50
Regarding the reaction mechanism of iron catalysts, it is worth mentioning that although many reports do not make a distinction between “low and high” temperature operations, it is essential that this should be done. Under low-temperature conditions it was established following the discovery of FT processes and has interestingly changed in time through various hypotheses from a carbide mechanism (early years) to the intermediate oxygenated mechanism (currently the most supported option by research studies). A detailed account of this interesting and suggestive story of the changing mechanism was covered by Davis in a recent review.38 It is worth mentioning though that the structure of the stable low-temperature iron catalyst is considered to be comprised of a Fe3O4 core that is covered by a layer of iron carbide. It was considered that the catalyst is in a dynamic pseudo-steady-state whose stability depends on the catalyst composition, the state after the activation step for CO reduction (at low-temperature metallic iron is stable only in essentially a pure hydrogen atmosphere) and the reaction conditions that allow the layer of iron carbide to be maintained.51 On the other hand, as reported by Dry and Shroff et al.,52HTFT catalytic reaction over low surface area catalysts begins predominantly with iron in the metallic form. As the reaction time increases, the fraction of metallic iron decreases as it is converted to Fe3O4 and iron carbides. However, even after a long time-on-stream there is still metallic iron present in the catalyst. The reaction mechanism of iron supported catalysts for both HTFT and LTFT is schematized in Fig. 2. It has been recently demonstrated that among different mechanisms proposed, H-assisted pathways occur concurrently with unassisted CO dissociation on Fe-based catalysts.53 At higher temperatures, direct dissociation pathways contribute significantly, especially on alkali-promoted Fe catalysts. O* species are formed and ultimately rejected as CO2. However, at lower temperatures, H-assisted CO dissociation is more facile than direct CO dissociation on model Fe5C2 (100) surfaces.54
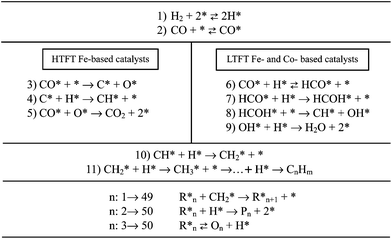
It is well known that agglomeration, sintering and reduction of iron based catalyst associated with particle size may lead to low catalytic activity.55 However, particle sizes do not have a significant influence on iron-based catalysts.56 It was reported that for supported iron catalysts with lower loading than 10%, it is very difficult to completely reduce the small-sized iron species due to strong metal–support interaction.57 In contrast, bigger particle sizes can be easily reduced due to weak interaction with the support but suppress the dispersion of iron oxide. The average crystallite size of the magnetite phase in precipitated Fe-based catalysts is usually larger (50–150 nm) than those based on simple oxidation of iron carbide (7–15 nm).58 Mabaso et al. reported the effect of crystal sizes on carbon supported iron catalysts. Smaller particles than 7–9 nm showed lower activity and higher selectivity of methane compared to the bigger-sized catalysts.56 Likewise, CO conversion in the HTFT process and hydrocarbon selectivities were strongly affected by the presence of metal particle size in the catalysts in the size range of 2.0–12.0 nm, with 6 nm selected to be optimal for HTFT on the Fe/Al2O3 catalyst.
In addition, Eliason and Bartholomew59 provided evidence that sintering is not responsible for the activity lost in iron catalysts. Therefore, observed losses in the activity of the alkali promoted iron catalysts could be attributed to the transformation of the active carbons into inactive carbons and active carbides into lower active carbides as detailed by Nakhaei Pour et al.60 Moreover, the external addition of water seems to have a positive effect on CO conversion at high temperatures (270 °C) whereas a decrease of CO conversion and catalyst deactivation occurs at lower reaction temperatures (230 °C) due to catalyst oxidation that transforms the iron carbide to the Fe3O4 phase. When the reaction was carried out at 270 °C, severe oxidation did not take place and a carbide phase was retained.61 Blocking of active sites by carbon deposition is also common at HTFT and/or in the presence of CO2 hydrogenation.62
Regardless of the preparation of the iron-based catalyst, its surface basicity has been proven to be significantly important to direct the FT process to the formation of certain hydrocarbons. Several studies have addressed the effect of alkali promoters in iron-based catalysts. Alkalization of iron catalysts has two different effects: it promotes the selectivity of 1-alkene formation and increases the growth probability. Dry showed the promotion of iron catalysts with Group I alkali metals (Li, Na, K, Rb) to have a remarkable effect on the formation of longer chain alkanes.37 The probability of chain growth was found to increase in the order Rb > K > Na > Li, as alkalis tend to enhance CO decomposition (into C and O atoms) due to the stronger chemisorption of CO in these promoters. Luo and Davis investigated the role of Group II alkali-earth metals (barium, beryllium, calcium and magnesium) as promoters in Fe FT catalysts and demonstrated that these metals possessed lower overall FTS activities and lower alpha values as compared to a K promoted iron catalyst but improved activities compared to those of unpromoted catalysts.63 In agreement, Dry and Oosthuizen63 found that the basicity increased in the order of Ba, Li, Ca, Na and K for the reduced catalyst. The amount of methane in FTS decreased as the surface basicity increased. In addition, the combination of sodium (Na/Fe = 0.1) with either aluminium (Al/Fe = 0.9) or manganese (Mn/Fe = 0.4) resulted in stable FTS catalysts with high selectivity for light olefins and concurrently methane selectivity suppression.64 However, it was important to note that residual sodium negatively influences physicochemical properties of the iron-based catalyst, as well as the catalytic activity and selectivity during FTS performance. The residual sodium was found to increase particle sizes (textural inhibitor), restraint and carburization of catalysts in pure H2 and/or CO environment (H2/CO = 0.67).
The appropriate amount of K, i.e., <1.5 wt%, could not only significantly increase both FTS activity and the activity for the WGS reaction but also improve catalyst stability.65 When alkali loading is high, CO dissociation proceeds faster than carbon hydrogenation, which leads to an excessive carbon deposition that eventually deactivates the catalyst surface.66 As an example, potassium is assumed to donate electrons to the vacant d orbital of the transition metal as evidenced by the lowering of the metalwork function. Hence, its presence would facilitate the dissociation adsorption of CO, which tends to accept electrons from iron.67Hydrogen at higher surface coverage is however prone to act as an electron donor to iron. Thus, the presence of potassium may weaken the strength of the Fe–H bond68 and influence its adsorption rate in same manner. Liet al.39c,65c,69 concluded that potassium promotes the formation of an increased number of active sites during reduction and carburization of iron oxides by facilitating the rapid formation of nucleation sites, resulting in the formation of smaller iron carbide crystallites.
Concerning the role of structural promoters, basic potassium can interact with the acid sites on structural promoters or be fixed in pores or holes of structural promoters decreasing or inhibiting the promotional effect of potassium. McVicker and Vannice70 reported that a high surface area support tends to reduce the direct contact between iron and potassium, which leads to a less effective potassium promotion. Comparatively, Bukur et al.65a interpreted a hydrocarbon selectivity improvement with the addition of support in terms of interactions between potassium and/or iron with supports. On the other hand, the surface diffusion of reactants and intermediates (spillover) between the metal sites and the support or structural promoters can apparently enhance the catalytic activity and vary the selectivity.71 Therefore, the improvement of the performance of alkali-promoted supported catalysts is believed to be due to the presence of a synergistic interaction between the alkali promoter and the structural promoter (e.g.silica, alumina, etc.). Thus, the catalyst is more stable when both promoters are simultaneously present.38 This fact supports the proposed theory that involves a slow continuous replacement of carbon in the iron carbide layer. Therefore a combination of catalyst composition and reaction conditions can be controlled to ensure a slow catalyst deactivation.
Copper (<3 wt%) is also typically added to enhance the reduction of Fe2O3 to Fe3O4 during the catalyst pre-treatment step72 increasing both FTS and WGS activities. However, there is some discrepancy about the Cu effects in terms of hydrocarbon selectivities that could be a consequence of both the specific catalyst and the different process conditions used.65c,73 It was found that 0.8–2 wt% Cu promotion over Fe/K/Activated Carbon (AC) does not benefit the catalyst activity due to high Cu loadings that lead to the suppression of iron carbonization and increase of the deposition of carbon on the catalyst surface. It was also reported that selectivity was also changed. Cu increases oxygenate selectivity (molecular CO adsorption was favored whereas CO dissociation was inhibited) and methanol formation. The olefin content was also decreased due to promotion of both hydrogen adsorption and hydrogenation/isomerization.74
Other widely utilized promoters include Pt (0.01–1.0 wt%),75Rh or Pd,76Mo (i.e. 0.5–1 wt% over CNTs, since higher contents of Mo (5–12 wt%) diminished catalytic activity due to catalytic site coverage and lower extent of reduction although the selectivity toward lighter hydrocarbons was increased),77Mn,78Ru,66c,78–80Zr (1–15 wt%)81 and Zn,39c but K (0.05–5 wt%)81 is practically used as the only promoter for iron based catalysts.
Iron-based Fischer–Tropsch catalysts have a high sensitivity towards sulfur compounds that can cause a permanent poisoning of the catalyst.34 Another important feature of iron based catalysts is that they show high catalytic activity towards water gas shift (WGS: CO + H2O → CO2 + H2) reactions.2 These catalysts are generally employed for hydrogen-poor synthesis gas FTS, most especially from coal (H2/CO molar ratio ranging from 0.67 to 1 or even lower). However, this is not always the case as other different authors have reported the use of such catalysts for various ratios35 to increase the hydrogen content of syngas (via WGS) to an optimum value of the H2/CO ratio for the FT reaction (about 2).
Cobalt catalysts
Cobalt-based catalysts are especially interesting from the commercial point of view due to their rather high activity (up to 60–70% conversion per single pass), selectivity and stability in the synthesis of linear hydrocarbons from CO/H2 mixtures,16,36 resistance to attrition in slurry bubble column reactors and the relatively lower negative effect of water on CO conversion. However, as compared to Fe-based catalysts (that can be employed in both HTFT and LTFT modes), Co-catalysts are only used under LTFT conditions, as an excess of methane is produced at higher temperatures.21 A list of industrially employed Co materials in FT processes has been included in Table 5.| Premium product | Catalysts | Reactors | Processes |
|---|---|---|---|
| Gasolines | Co/ThO2/Al2O3/Silicalite, Co/ZSM-5, Fe/Cu/K and ZSM-5 | Fixed-bed Slurry/fixed-bed | Gulf-Badger, Mobil One-Stage, Mobil Two-Stage |
| Diesel fuel | Co/Zr, Ti or Cr/Al2O3, Co/Zr/TiO2, Co–Ru/Al2O3 | Fixed-bed (low T), Slurry-bed (low T) | Gulf-Badger, Sasol-Two Stage, Shell-Middle Distillate, Eisenlohr/Gaensslen |
| Waxes | Co/Zr, Ti or Cr/Al2O3 Co/R/Al2O3, Prom. Fe/Ru | Fixed-bed (low T) | Shell-Middle Distillate (First Stage) |
Regarding Co catalysts, it is generally accepted that the metallic nanoparticles are the active phases.26 They can exist in different crystalline forms, including α-Co (hcp) and β-Co (fcc), the former being more stable at lower temperatures than the latter. However, cobalt crystallites with a particle size less than 20 nm are stable as pure fcc phase.82 Typically, the preparation of Co-based FTS catalysts involves the impregnation of a cobalt precursor salt over a porous inorganic solid, followed by calcination and reduction. The last step is particularly important but it increases the cost and complexity of the process. Unreduced Co species typically give lower CO conversions and higher CH4 selectivities. Recent studies have proved that under realistic FTS conditions, the transformation of Co0 to cobalt oxides, cobalt carbides, or mixed oxide compounds (e.g., cobalt aluminate or cobalt silicate) may occur as a result of a reaction with the supports, which can be assigned as the major deactivation mechanism.83
Regarding the reaction mechanism, it is generally accepted that FT synthesis proceeds through a surface-catalyzed polymerization mechanism, which uses CHx monomers formed by hydrogenation of CO. On the basis of the H-assisted CO dissociation and of the CH2* insertion alkyl mechanisms, a complete mechanistic model for FTS over the supported cobalt catalyst is schematized in Fig. 2.53 In such pathways, H2 chemisorbs reversibly on two adjacent free catalytic sites in the dissociated state. First, CO is reversibly chemisorbed in the molecular state. Then, it is hydrogenated two times resulting in the formyl intermediate and the hydroxymethylene species. Adsorbed O can be efficiently removed by H to form water, whereas adsorbed C can be recombined with H to yield various CHx intermediates (x = 0–3). Then, the chain growth through C–C coupling starts although competition with chain termination through hydrogenation, hydrogen abstraction, or insertion of non-dissociative adsorbed CO to produce alkanes, alkenes, or alcohols, respectively, is established.
The main deactivation mechanisms of cobalt catalysts from the literature can be classified as: (i) poisoning by sulfur and/or nitrogen compounds in the syngas feed (particularly important for CTL applications), (ii) oxidation of the active cobalt metal to an inactive cobalt oxide, (iii) cobalt-support compound formation (cobalt silicates and cobalt aluminates), (iv) sintering of small cobalt crystallites, (v) surface reconstruction and (vi) carbon formation (Table 3).
During the past two decades, the research on cobalt catalyst deactivation has been focused on oxidation despite it being recently found not to be the deactivation mechanism for supported Co catalysts with crystallite size in excess of 2 nm.84 It has been demonstrated that although smaller metallic crystallites should display a higher activity per unit mass of catalytically active material, cobalt-time-yield was improved by an increase of cobalt particle size up to 30–40 nm. For larger cobalt particles supported on high area (50–400 m2 g−1) Al2O3, SiO2 and TiO2, the FTS reaction rate was proportional to the number of cobalt surface sites85 suggesting that both hydrocarbon selectivity (attributed to the rate of olefin readsorption)86 and cobalt site time yield slightly depend on the cobalt particle size. Hence, a minimum crystallite size was required (6–8 nm)87 since lower particle sizes resulted in the lowest FT turnover frequency and highest methane selectivity and olefinic products. It might be attributed to both their re-oxidation under realistic FT conditions (by changes in thermodynamic properties) and modification of the electronic structure because of the quantum size effect. Moreover, as oxidation of Co at proper particle size may not take place, it can be concluded that the source of the cobalt aluminate is the unreduced CoO so that cobalt aluminate formation may not contribute to deactivation.
Sintering (contributing to up to 30% of deactivation), carbon deposition (responsible for the long-term deactivation due to surface blockage) and surface reconstruction (causing deactivation due to shape changes in the cobalt particle) are intrinsic to cobalt so they will be present on most Co based FTS catalysts. Therefore, a three-step process ((1) dewaxing, (2) oxidation and (3) reduction) was found to be useful for catalyst regeneration.84
It is generally accepted that several parameters affect the performance of cobalt catalyst for FTS, i.e., the catalyst support, the nature and amount of added promoters as well as the cobalt dispersion that in turn is influenced by not only the former parameters but also the preparation method applied. An industrial FT cobalt-based catalyst usually contains between 15 and 30 wt% of cobalt.37Cobalt is usually incorporated by incipient or slurry impregnation (most commonly with Co nitrate)88 although a homogeneous deposition–precipitation method has been also considered.89 Distribution of Co surface sites could be also critical for reaching a high yield of hydrocarbons. No intraparticle diffusion limitations have been commonly reported for slurry reactors operating with catalyst grains of about 50–80 mm whereas in fixed bed reactors (particles of 1–3 mm), diffusion of reagents, intermediates and final products could influence both the FT reaction rate and product selectivity.90
The influence of cobalt precursor on the catalytic performance and physical properties of the final catalyst has been intensively examined. It could result in different sizes of Co3O4 particles and affect the crystallinity of the supported cobalt species. It can be observed from Table 6 that the selection of a proper precursor leads to important differences in the cobalt species particle size. For alumina or silica supported catalysts, the cobalt particles are about ten times smaller when alternative precursors like cobalt carbonyls or EDTA compounds in comparison to cobalt nitrate (typical clusters diameter of ca. 100–400 nm) are applied. This change in cluster size was found to greatly modify both the carbon monoxide conversion and the hydrocarbon product distribution. Therefore, cobalt acetate seems to lead to inactive FT catalysts, while catalysts prepared from cobalt nitrate show higher activity in FT reaction and selectivity to high-boiling hydrocarbons.
| Cobalt catalyst | Precursor | Particle size (Co3O4) (XRD) (nm)b | Dispersion (%)b | Degree reduction (%) | T/°C | P/bar | GHSV | H2/CO | CO conversion (%) | SC1–4 (%) | SC5+ (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a n.m: not measurable; IN: iron nitrate; CuN: copper nitrate; KC: potassium carbonate; AlN; aluminium nitrate; A: ammonium; IWI: Incipient Wetness Impregnation; P: precipitation; S: Spreading. b Calculated by: D (%) = 96/dCo0(nm) and dCo0(nm) = 0.75dCo3O4(nm). | ||||||||||||
| Co/TiO2 | Nitrate (IWI) | 21.0 | 6.1 | 21.6 | 200 °C | 20 | 1200 h−1 | 2.0 | 14.7 | — | 79.4 | M. Kraum, 1999 (ref. 139a) |
| Acetate (IWI) | 16.4 | 7.8 | 31.4 | 26.2 | 68.2 | |||||||
| (II) Acetylacetonate (IWI) | 25.6 | 5.0 | 23.8 | 7.6 | 75.3 | |||||||
| Hydroxide (P) | 14.1 | 9.1 | 14.3 | 1.2 | 81.2 | |||||||
| EDTA (P) | 20.3 | 6.3 | 10.6 | 14.2 | — | |||||||
| Oxalate (S) | 16.6 | 7.7 | 12.9 | 32.0 | 76.3 | |||||||
| Co/ITQ-2 | Nitrate | 17.3 | 7.4 | 89 | 220 | 20 | 13500 Nml gcat−1 h−1 |
2.0 | 37.5 | 21.7 | 78.3 | P. Concepción, 2004 (ref. 139b) |
| Co/ITQ-6 | 20.9 | 6.1 | 88 | 220 | 20 | 13500 Nml gcat−1 h−1 |
2.0 | 21.9 | 27.4 | 72.6 | ||
| Co/MCM-41 | 5.5 | 23.3 | 38 | 220 | 20 | 13500 Nml gcat−1 h−1 |
2.0 | 24.3 | 55.3 | 44.7 | ||
| Co/SiO2 | 15.6 | 8.2 | 96 | 220 | 20 | 13500 Nml gcat−1 h−1 |
2.0 | 20.2 | 34.5 | 65.5 | ||
| 11Co/Al2O3 | 30.7 | 4.2 | — | 242 | 20 | 6000 Nml gcat−1 h−1 | 2.0 | 30.8 | 19.1 | 78.0 | A.R. de la Osa, 2011 (ref. 102) | |
| 10Co/CNTs | 9.1 | 14.1 | — | 230 | — | — | 2.0 | 30.0–35.0 | 20.0 | 60.0 | M. Trépanier, 2009 (ref. 139c) | |
| 11Co/CeO2 | — | n.m | n.m | 210 | 20 | 1500 h−1 | 1.2 | 5.0 | 13 (CH4) | 34 | W.J. Wang, 1991 (ref. 139d) | |
| 8.8Co/Mordenite | 7.1 | 18.0 | 52 | 240 | 20 | 1000 h−1 | 2.0 | 59.6 | 26 (CH4) | — | S. Bessel, 1993 (ref. 139e) | |
| 7.8Co/ZSM-5 | 21.3 | 6.0 | 100 | 240 | 20 | 1000 h−1 | 2.0 | 33.9 | 21 (CH4) | — | S. Bessel, 1993 (ref. 139e) | |
| 10Co/SiC | 45.0 | 2.8 | 79 | 242 | 20 | 6000 Nml gcat−1 h−1 | 2.0 | 72.1 | 6.3 | 92.8 | A.R. de la Osa, 2011 (ref. 102) | |
| Co/Al2O3 | Nitrate | 28.8 | 1.2 | 29.8 | 220 | 20 | 6000 Nml gcat h−1 | 2.0 | 25.6 | 4.3 | 95.5 | A.R. de la Osa, 2011 (ref. 119c) |
| Co–Ca/Al2O3 | 55.6 | 2.0 | 44.8 | 33.0 | 4.4 | 95.4 | ||||||
| Co–Mg/Al2O3 | 31.8 | 2.4 | 53.5 | 27.5 | 5.5 | 94.3 | ||||||
| Co–K/Al2O3 | 76.6 | 2.2 | 35.5 | 13.0 | 10.3 | 89.2 | ||||||
| Co–Na/Al2O3 | 40.0 | 1.9 | 47.0 | 30.3 | 1.8 | 98.0 |
| Iron catalyst (wt%) | Preparation method | Precursor | Reactor | T/°C | P/bar | GHSV | H2/CO | CO conversion (%) | SC1–4 (%) | SC5+ (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fe/K-free | Combination method of continuous co-precipitation and spray-drying | — | Fixed bed reactor | 250 | 15 | 4000 h−1 | 2.0 | 34.3 | 36.3 | 63.7 | G. Zhao, 2008 (ref. 67b) |
| Fe/K-ZSM-5 | 53.7 | 32.0 | 68.0 | ||||||||
| Fe/K–SiO2 | 46.1 | 18.5 | 81.5 | ||||||||
| Fe/K–Al2O3 | 55.3 | 24.5 | 75.5 | ||||||||
| 15.9Fe/1.6Cu/3.2K/79.3Al2O3 | Impregnation | IN, CuN, KC | 300 | 10 | 2000 ml gcat−1 h−1/2.0 cm s−1 | 2.0 | 96.1/95.8 | 51.5/68.3 | 48.5/31.7 | ||
| 15.9Fe/1.6Cu/3.2K/79.3SiO2 | Impregnation | IN, CuN, KC | 2000 ml/gcath/1.0 cm s−1 | 37.2/60.7 | 41.3/63.3 | 58.7/36.7 | |||||
| 3.2K/79.2Fe/5.2Cu/12.4Al | Co-Precipitation | IN, CuN, AlN, KCA | 2000 ml gcat−1 h−1/1.0 cm s−1 | 95.4/97.2 | 40.8/61.3 | 59.2/38.7 | |||||
| 6K/94FeOx | Impregnation | KC | Fixed bed reactor/bubbling fluidized-bed reactor | 2000 ml gcat−1 h−1/1.0 cm s−1 | 96.5/97.5 | 33.4/68.4 | 66.6/31.6 | S.-H. Kang, 2011 (ref. 139f) | |||
| 100Fe/5.64Cu/2La | Fe/Cu: Co-precipitation | — | Fixed Bed microreactor | 290 | 17 | 4900 Nml gFe−1 h−1 | 1.0 | 64.1 | 52.4 | 47.6 | A. Nakhaei Pour, 2010 (ref. 60) |
| 100Fe/5.64Cu/2Ca | Ca, Mg, La: impregnation | 73.5 | 48.5 | 51.5 | |||||||
| 100Fe/5.64Cu/2Mg | 77.3 | 44.5 | 55.5 | ||||||||
| 15.7Fe/94K/AC (0Cu) | Incipient-wetness impregnation | IN, CuN | Fixed Bed reactor | 280 | 20 | 3000 ml gcat−1 h−1 | 0.9 | 85.7 | 43.5 | 56.5 | W. Ma, 2011 (ref. 74) |
| 15.7Fe/2Cu/0.94K/AC (2Cu) | 52.5 | 43.1 | 56.9 |
On the other hand, due to the higher cost of Co as compared to Fe, the primary goal is to produce a significant and stable metal surface exposure (Co). Hence, the support should have a relatively high specific surface area in order to achieve both good dispersion of the active phase, good mechanical and hydrothermal resistances and additionally, a high chemical inertness in order to reduce the fraction of hardly reducible phase during the thermal treatment steps.91 The use of support-precursor pairs with intermediate interaction strengths and the slow and controlled reduction of impregnated precursors were pointed to be the most appropriate approaches to synthesize high loaded supported Co catalysts with relatively low dispersions.24
The support material strongly influenced CO conversion and chain growth probability, which was found to mainly depend on the particle size (Table 6). It is known that both support pore sizes and overall cobalt content could affect cobalt dispersion in the supported catalysts. Many of the studies reported to date suggest that a smaller pore size led to lower reducibility of Co species, whereas a larger pore size caused the formation of larger Co particles. Therefore, the balance between the reducibility and the particle size of Co results in the optimum activity and selectivity for catalysts with a medium pore size.92 The preparation of relatively large cobalt particles (8–10 nm) requires support materials with an average pore diameter between 10 and 15 nm. However a few studies suggest that an optimum pore size should exist. Some of them performed with SiO2 and γ-Al2O3 showed that catalysts with mean pore sizes of close to 10 nm displayed higher FT activity and higher C5+ selectivity.93 Accordingly, MCM-41 and SBA-15 type mesoporous silica showed a maximum catalytic performance for a value of pore size equal to 9 nm.94 On the other hand, narrow pore size distribution in periodic mesoporous silica seemed to prevent cobalt particles from sintering whereas in amorphous silica with broader pore size distribution, cobalt dispersion decreased to some extent with an increase in cobalt surface density. For this reason, the active phase is typically supported on Al2O3, TiO2 or SiO295 as well as on zeolites,96 other mesoporous materials (>500 m2 g−1, 2–30 nm pore size),97 molecular sieves98 and carbonaceous (CNF/CNT) materials.89,99 The catalyst activity decreased in the following order: TiO2 > Al2O3 > SiO2 > C > MnO > MgO.100 However, in spite of the high intrinsic activity observed for Co/TiO2, a low activity per unit mass of cobalt was displayed. Therefore, among the different supports, alumina is one of the most appropriate to fulfil catalytic requirements. Also SiO2 has been often considered the optimum support for Co FT catalysts due to its high surface area (which favors high Co dispersion at high Co loadings) and interesting surface chemistry (which facilitates the reduction of Co3+ or Co2+ to Co0).101 Nevertheless, the low thermal conductivity of these supports renders them more sensitive to hot spot formation, which may compromise the plant security and decrease the selectivity towards liquid hydrocarbons.91 Regarding those requirements, new support materials such as SiC are now being developed. It has been demonstrated that SiC can replace those cited above, since it provides an improvement of not only physical properties but also both catalytic performance and long hydrocarbon products selectivity.91,102
In general cobalt catalysts are less influenced by the presence of promoters with respect to iron materials. Nevertheless, these catalysts often contain small amounts (0.05–0.1 wt%90) of a second metal promoter (usually noble metals such as Pd, Pt or Ru) that improves the aforementioned Co reduction and leads to an increase of their activity and selectivity to C5+ products via enhancement of the hydrogenolysis of the carbonaceous deposits (thus cleaning the catalytic surface, avoiding deactivation).30a,32a,103 Storsæter et al.104 observed that the addition of Re to Co/Al2O3, Co/SiO2 and Co/TiO2 increased the hydrocarbon formation rate per gram of catalyst and slightly enhanced the selectivity to C5+ hydrocarbons. Early studies proposed that Re increased the Co dispersion on TiO2 by preventing agglomeration of CoOx particles during calcination treatments and oxidative regenerations.24 However, some research groups reported that the intrinsic activity of the metallic cobalt crystallites was not influenced by Re.105
The addition of Group 11 metals (Cu, Ag and Au) as promoters in the preparation and FT activity of Co/Al2O3 catalysts106 has been reported to improve the Co reducibility as well as the surface Co metal active site densities (especially in the cases of Ag and Au).107 At lower Au levels (1.51 wt%) and relatively high Ag levels (0.83–2.76 wt%), significant gains in Co active site densities were achieved resulting in improved CO conversion levels relative to the unpromoted catalyst.107 An overloading of metal promoter (5 wt%) was again found to be detrimental as it appeared to block the Co surface sites and decreased the CO conversion rates. The addition of small amounts of Au to Co/TiO2 was also found to enhance the catalyst activity.108 As compared to Ag and Au, the Co-promoted reduction by Cu did not provide improved active site densities, apparently related to a partial Cu covering of the rim of Co clusters, resulting in decreases in CO conversion rates and a decrease in light product selectivity.109
However, due to their high prices, the industrial application of these promoters is restricted. Thus, promotion with less expensive metal oxides (1–10 wt%90) has been also one of the methods to improve activity and hydrocarbon selectivity of FT catalysts. Among the oxide promoters, ZrO2, La2O3, MnO and CeO2 have been most often employed. ZrO2 was evidenced as a good promoter to improve the CO conversion activity and C5+ selectivity over Co/SiO2.110ZrO2 may form an active interface with Co, which may facilitate CO dissociation. Goodwin et al.111 demonstrated that the addition of ZrO2 to γ-Al2O3 could improve cobalt reducibility by preventing the formation of Co-aluminates. The presence of certain amounts of MnOx was reported to lead to an improved C5+ selectivity and a decrease in CH4 selectivity.112 The addition of CeO2 (4.5–38 wt%) into Co/SiO2 was found to modify the product selectivity with slight variations of the catalytic activity.113 Finally, addition of La2O3 was found to decrease the reducibility of cobalt species but dispersion was enhanced. It has been proposed that the presence of these species does not change the intrinsic activity of the Co species but increases the concentration of active sites or active intermediates.114 Other transition metal oxides considered have been MgO, MoO3, Nb2O5 or V2O5.115 Addition of oxide promoters26 could modify the catalyst texture and porosity, reduce formation of hardly reducible cobalt mixed oxides, increase cobalt dispersion, reducibility and fraction of different cobalt metal crystalline phases, enhance mechanical and chemical attrition resistance of cobalt FT catalysts and improve the chemical stability of the support. More information about the effect of promotion on cobalt based catalysts is available in recent reviews.26,90
On the other hand, as aforementioned, there are numerous papers studying the influence of addition of alkali promoters on FTS over iron-based catalysts.116 However, the literature regarding the addition of alkali promoter on cobalt catalysts is scarce.117 Studies of K and other alkalis to cobalt on various metal surfaces are limited because they have been shown to severely reduce catalyst activity.13 Alkaline earth elements are also found in biomass ash118 but again research is limited. Therefore, there are only some separate studies119 where chain growth probability was found to increase significantly, while the activity can be negatively influenced depending on the promoter concentration level. Physical blocking of active cobalt sites by an alkali/alkali-earth promoter is one possibility to explain this behavior. However, it is unlikely that physical blocking is responsible for the activity decline as the amount of promoter is too small. Particle size effects are a second possible reason for the decrease in activity. It can be considered that alkali/alkali-earth promoter introduction decreases the cobalt metal particle size to a region where the size affects catalytic activity.120 Finally, electronic effects, which can be attributed to changes in the adsorption of the reactants on the catalyst surface, seem more likely to be responsible for the decrease in activity when Ca is added to the catalytic formulation. Electropositive alkalis added to the Co catalyst transfer charge to the catalyst surface, inducing “long-range electronic effects” and influencing H2 and CO adsorption and dissociation.121 As a result, a decrease of the surface hydrogen concentrations and an increase of CO adsorption and dissociation have been observed.
On the other hand, regarding hydrocarbon selectivity, recent studies122 found a higher chain-growth probability for alkali/alkali-earth metals, in contrast to that by Gaube and Klein.117b A reduced hydrogen concentration, as suggested by Balonek et al.,121 can explain this effect. It is likely that the addition of those promoters increases the CO2 selectivity at a constant conversion level, enhancing the water–gas-shift reaction, although the effect is small. Nevertheless, it is clear this behavior is irrespective of support nature.
Finally, similarly to Fe-based catalysts, Co materials have also a high sensitivity towards sulfur compounds that requires a thorough cleaning of the syngas prior to its submission for the Fischer–Tropsch process.26 S contents below 100 ppm123 in syngas are therefore a prerequisite for FT synthesis using the Co-catalyst. However, despite Fe materials being in theory more resistant to sulfur28 and ammonia poisoning124 with respect to Co catalysts, Co catalysts are generally the choice for GTL and CTL processes, which employ natural gas and coal as feedstocks, due to their low activity in the WGS reaction under required LTFT conditions for synthetic fuels production.
Biomass-To-Liquid Fischer–Tropsch (BTL-FT) process: choice of catalysts
The chemical composition of biomass is not uniform. Therefore, the composition of biomass-gasification derived syngas (mainly H2 and CO, with lesser amounts of CO2, H2O, CH4, higher hydrocarbons (C2+) and N2) will vary according to biomass nature and other many factors, including reactor type, feedstock and processing conditions (temperature, pressure, etc.).Few studies have investigated in detail the suitability of different catalysts in the BTL-FT process, starting from syngas generated from biomass gasification.28,125,126 Configurations currently investigated for the BTL process require high overall and per pass CO-conversion and high C5+-selectivity.4,12
Cobalt-based materials have been so far the chosen catalysts to evaluate the feasibility (both economically and efficiency) of the BTL process, due to the superior activity of cobalt as compared to iron. However, there are several recent studies that point to the selection of Fe or Co-materials depending on the conditions.2,28 There are several key aspects that have to be considered in BTL-FT processes from biomass derived gaseous feeds.
Firstly, the syngas produced from biomass gasification contains low hydrogen content. In this context, Fe-catalysts can operate with lower hydrogen content syngas (such as that coming from biomass gasification) so this can be advantageous in the use of these catalysts as compared to Co-based catalysts. It can be also observed from Table 2 that depending on the origin of biomass different percentages of S and ashes could be obtained, generally lower than that from coal, coke or lignite. For the particular case of thoroughly cleaned and conditioned biomass syngas, cobalt-based materials could be the catalyst of choice for the BTL-FT synthesis of linear heavier hydrocarbons due to their higher productivity at high conversion levels. However, as some contamination of the syngas entering the FT reactor cannot be avoided most of the time, the use of iron catalysts should be promoted due to their inherent economical and stability advantages compared to Co materials, in spite of their lower productivity under the same operational conditions.
Furthermore, a water gas shift reactor after the biomass gasification unit may be required in order to obtain a good productivity regardless of the catalyst utilised in the reaction (Fe or Co).127
Secondly, the production of water vapour from biomass gasification has to be taken into account, especially when wet biomass feedstocks are gasified. In general, water has been reported to inhibit the reaction rate of FTS processes, although its effect is highly dependent on concentration, type of catalyst (e.g. supported or unsupported), type of metal (e.g.Co, Fe, Ru), metal particle size and the support (e.g. a positive effect of water was reported for Co-silica supported catalysts while it has a negative effect for alumina supported catalysts and little effect for titania). Fe and Co-based catalysts seem to be similarly affected by water vapour so other factors have to be considered in this particular case. An example of such water effects on Co catalysts has been recently compiled by Dalai and Davis.101 In terms of stability, both iron and cobalt materials have been shown to be similarly stable and capable of operating for 6 months or more under optimised conditions.2
Thirdly, and possibly most importantly, the operational mode of the FTS process may recommend the utilisation of Fe or Co in the reaction. Harsher and more severe conditions (in terms of temperatures, presence of impurities, etc.) require the utilisation of iron-based materials as these actually show a superior activity and selectivity in BTL-FT processes (Co materials generate high quantities of methane). Cobalt-based catalysts are comparatively more active at low-severity conditions and thus should be the catalysts of choice under these conditions. Nevertheless, and in spite of many available reports, there are still considerable differences in defining catalyst activity, selectivity to methane and ultimately the selection of Fe or Co-based catalysts in BTL processes.
In view of these premises, iron and cobalt catalysts should both in principle be considered for BTL-FT processes. A number of scenarios for the BTL process should be developed with both types of catalysts, where the overall process design should be integrated with catalyst developments to clearly prove the superiority of the optimum catalytic system in view of its commercial and/or industrial application.12 Further in-depth investigations are also needed to ascertain the effect of several parameters on the performance and stabilities of these FT catalysts, namely, the variability of feedstocks (source and time of the year), the effect of different biomass feedstocks and biomass composition in the gasification process.128
Details on the choice of reactors and chemical engineering challenges that the FTS industry faces in future years is out of the scope of this perspective, but there are some recent reports in which some of these issues have been highlighted. Readers are kindly referred to the reviews by Lappas and Heracleous12 and Dalai and Davis101 as well as references included therein.
Prospects and challenges
One of the main challenges in the field is the development of more efficient, cheaper and tailored Fe-based catalysts that can compete with related Co-based materials in terms of performance, selectivity and costs as well as with the possibility to be fine-tuned for the specific and selective production of different hydrocarbons. Some comments in this regard have been mentioned in the previous section.Another relevant challenge to be addressed in FTS processes is the utilisation of alternative feedstocks to syngas for the production of liquid hydrocarbons. These will include other gaseous feeds including CO2 (significant quantities generated by the production of syngas from coal or in a lesser extent by biomass), which could be an interesting alternative to other reported carbon dioxide sequestration methods by geological and/or biological means.
In this regard, a promising approach in this field was recently reported by Dorner et al. that successfully utilized CO2 as feedstock for FTS transformations into jet fuel-like hydrocarbons using different metal catalysts, firstly via FTS to selected alkenes and then subsequent upgrading to hydrocarbons using other catalysts (e.g.zeolites). Their methodology could effectively recycle CO2 into energy-rich molecules viahydrogenation using a two-stage process.129–131 Initially, they employed a traditional Co FTS catalyst (Co–Pt/Al2O3) and product distribution was investigated at different H2 and CO2 feed gas ratios (3:1, 2:1 and 1:1) as well as pressures (450–150 psi).129 Reducing H2/CO2 ratios (from 3:1 to 1:1), the product distribution was shifted to the production of higher chain C2–C4hydrocarbons at the expense of methane production (mainly observed at H2/CO2 3:1 ratios). A similar effect was also observed at decreasing pressures. Nevertheless, and despite these interesting findings, the Co material behaved as a methanation catalyst in the hydrogenation of CO2. These authors then moved on to investigate related promoted and/or doped Fe catalysts in the process.130,131 Over 40% conversion levels could be achieved with Mn and K-doped Fe/γ-Al2O3 catalysts, along with an alkene/alkane ratio higher than 4. The quantity of Mn and K promoters had to be carefully controlled as over-doping with these metals remarkably reduced the CO2 conversion as well as suppressed the formation of the targeted unsaturated hydrocarbons.130 Additional promotion of Mn–Fe/γ-Al2O3 with ceria showed the almost negligible effect on CO2 conversion and selectivity to alkenes at low ceria loadings (typically 2 wt% and below) turned into a slight increase in the formation of CO at high loadings (10 wt%), a phenomenon that has been proposed to be due to a reduction in the available chain-growth active sites on the iron.131
There is a lot of potential in this approach to CO2 valorisation and the development of more active, selective and stable Fe catalysts for these processes (e.g. through metal doping, choice of suitable supports for Fe, different metal loadings, synthesis procedure, etc.) should be attempted in the near future.
On the other hand, as commented before, biomass has lower energy content per kg if compared to other raw materials (e.g. coal). The use of biomass is consequently most economically effective if transportation costs are low. Hence, gasification of coal and biomass mixtures could provide an opportunity to build more economically and larger plants in case insufficient biomass is available in the surrounding areas132 as coal has been used as an oil substitute since the 1970/80s.133 However, as previously mentioned, not only an oil substitute is now being required but also a way to minimize carbon dioxide emissions, which many countries agreed to in the Kyoto protocol. These two fuels, when co-gasified, exhibit synergy with respect to overall emissions, including greenhouse gas emissions, without sacrificing the energy content of the product gas. In this sense, additional environmental benefits such as reduced sulfur and nitrogen emissions, when adding biomass, are recently increasing the interest in the process of co-gasification of biomass and coal.
Several reports concerning co-gasification are available.134 However, little literature is available regarding tests using different ratios of biomass and coal.135 Several authors have presented studies about the co-gasification of coals and different types of biomass resources and demonstrated the presence of synergic effects.136 For example, Kumabe et al.135a observed an increase in syngas yield, a decrease in char and tar conversion and in the extent of the water–gas shift reaction after co-gasification of wood and coal with air and steam in a downdraft gasifier. Fermoso et al.137j also studied the effect of operation variables and gasifying agent composition on several process parameters by considering a bituminous coal and a binary and ternary mixture of coal, biomass and petcoke. The main result was that the addition of biomass (up to 10%) to the gasification process led to an increase of H2 and CO production.
Conclusions
FTS processes for hydrocarbon production from syngas obtained from biomass gasification are becoming increasingly trendy in the past years as a suitable alternative to produce various high quality fuels for different applications. Many investigations are ongoing to test the suitability of biomass syngas for FTS using the traditionally employed catalysts. The choice of catalysts for these processes is normally restricted to Fe and Co-based materials as they provide the best compromise between performance and price. However, in spite of the promotion of Co-based materials for BTL, Fe materials have been shown to have comparable advantages to Co catalysts in such processes. All these facts should foster more in-depth investigation with the aim of improving the performance of Fischer–Tropsch processes. In addition, the integration of biomass gasification processes and catalyst design is also a crucial step to be optimized for the successful implementation of BTL processes for biomass valorization and biofuels production.Acknowledgements
Rafael Luque gratefully acknowledges Ministerio de Ciencia e Innovacion, Gobierno de España for the concession of a Ramon y Cajal contract (ref: RYC-2009-04199) and funding under project and CTQ2011 as well as Consejeria de Ciencia e Innovacion, Junta de Andalucia for funding project P10-FQM-6711. The authors also acknowledge funding from projects CTQ-2010-18126 (MICINN) and P09-FQM-4781 (Consejeria de Ciencia e Innovacion, Junta de Andalucia). Financial support from the Ministerio de Industria, Turismo y Comercio of Spain (CENIT-PiIBE project) and ELCOGAS S.A. are gratefully acknowledged by the UCLM research group.References
- http://www.fischer-tropsch.org/ .
- B. H. Davis, Ind. Eng. Chem. Res., 2007, 36, 8938–8945 CrossRef.
- A. R. de la Osa, A. de Lucas, J. L. Valverde, A. Romero and P. Sánchez, Rev. Ing. Quim., 2010, 489, 42–48 Search PubMed.
- A. Dutta and B. Acharya, Handbook of Biofuels Production: Processes and Technologies, ed. R. Luque, J. M. Campelo and J. H. Clark, Woodhead Publishing Ltd., Cambridge, UK, 2010, ch. 16 Search PubMed.
- D. Leckel, Energy Fuels, 2009, 23, 2342–2358 CrossRef CAS.
- S. Milmo, Chem. Ind., 2011, 9, 17–19 Search PubMed.
- http://www.shell.com.my/ .
- http://www.oryxgtl.com/ .
- http://www.shell.com.qa/ .
- http://theenergycollective.com/ .
- http://www.biofuelstp.eu/ .
- A. Lappas and E. Heracleous, Handbook of Biofuels Production: Processes and Technologies, ed. R. Luque, J. M. Campelo and J. H. Clark, Woodhead Publishing Ltd., Cambridge, UK, 2010, ch. 18 Search PubMed.
- M. M. Yung, W. S. Jablonski and K. A. Magrini-Bair, Energy Fuels, 2009, 23, 1874–1887 CrossRef CAS.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Fuel, 2010, 89, 1763–1783 CrossRef CAS.
- (a) J. Fermoso, B. Arias, M. V. Gil, M. G. Plaza, C. Pevida, J. J. Pis and F. Rubiera, Bioresour. Technol., 2010, 101, 3230–3235 CrossRef CAS; (b) A. Romero, P. Sánchez, N. Mota, F. García y and P. Casero, Rev. Ing. Quim., 2009, 467, 100–103 Search PubMed.
- J. Singh and S. Gu, Renewable Sustainable Energy Rev., 2010, 14, 1367–1378 CrossRef CAS.
- A. Kumar, D. D. Jones and M. A. Hanna, Energies, 2009, 2, 556–581 CrossRef CAS.
- M. Balat, M. Balat, E. Kirtay and H. Balat, Energy Convers. Manage., 2009, 50, 3158–3168 CrossRef CAS.
- V. Kirubakaran, V. Sivaramakrishnan, R. Nalini, T. Sekar, M. Premalatha and P. Subramanian, Renewable Sust. Energy Rev., 2009, 13, 168–175 CAS.
- K. Ptasinski, Biofuels, Bioprod. Biorefin., 2008, 2, 239–253 CrossRef CAS.
- C. H. Bartholomew, Catal. Lett., 1990, 7, 303–316 CrossRef CAS.
- (a) A. Tavasoli, A. N. Pour and M. G. Ahangari, J. Nat. Gas Chem., 2010, 19, 653–659 CrossRef CAS; (b) J. Huyser, M. Janse van Vuuren and G. Kupi, Advances in Fischer-Tropsch Synthesis, Catalysts, and Catalysis (Chemical Industries), ed. James G. Speight, CD & W, Inc., Laramie, Wyoming, 2009, ch. 10, pp 185–198 Search PubMed.
- J. Patzlaff, Y. Liu, C. Graffmann and J. Gaube, Appl. Catal., A, 1999, 186, 109–119 CrossRef CAS.
- E. Iglesia, Appl. Catal., A, 1997, 161, 59–78 CrossRef CAS.
- E. de Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758–2781 RSC.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS.
- R. Guettel, U. Kunz and T. Turek, Chem. Eng. Technol., 2008, 31, 746–754 CrossRef CAS.
- E. van Steen and M. Claeys, Chem. Eng. Technol., 2008, 31, 655–666 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884–890 CAS.
- (a) E. Iglesia, S. L. Soled, R. A. Fiato and G. H. Via, J. Catal., 1993, 143, 345–368 CrossRef CAS; (b) L. Fan, K. Yokota and K. Fujimoto, AIChE J., 1992, 38, 1639–1648 CrossRef CAS; (c) M. A. Vannice and R. L. Garten, J. Catal., 1980, 63, 255–260 CrossRef CAS.
- (a) H. Schulz, Appl. Catal., A, 1999, 186, 3–12 CrossRef CAS; (b) D. A. Simonetti, J. Rass-Hansen, E. L. Kunkes, R. R. Soares and J. A. Dumesic, Green Chem., 2007, 9, 1073–1083 RSC; (c) M. Claeys and E. van Steen, Catal. Today, 2002, 71, 419–427 CrossRef CAS.
- (a) M. E. Dry, Stud. Surf. Sci. Catal., 2004, 152, 533–600 CrossRef CAS; (b) J. G. Goodwin, Jr, Prep. ACS Div. Pet. Chem., 1991, 36, 156–159 Search PubMed.
- (a) J.-S. Girardon, A. Constant-Griboval, L. Gengembre, P. A. Chernavskii and A. Y. Khodakov, Catal. Today, 2005, 106, 161–165 CrossRef CAS; (b) L. Guczi, D. Bazin, I. Kovacs, L. Borko, Z. Schay, J. Lynch, P. Parent, C. Lafon, G. Stefler, Z. Koppany and I. Sajo, Top. Catal., 2002, 20, 129 CrossRef CAS; (c) G. Jacobs, T. K. Das, P. M. Patterson, J. Li, L. Sanchez and B. H. Davis, Appl. Catal., A, 2003, 247, 335–343 CrossRef CAS; (d) A. Kogelbauer, J. G. Goodwin, Jr and R. Oukaci, J. Catal., 1996, 160, 125–133 CrossRef CAS.
- D. Schanke, S. Vada, E. A. Blekkan, A. M. Hilmen, A. Hoff and A. Holmen, J. Catal., 1995, 156, 85–95 CrossRef CAS.
- B. Jongsomjit, J. Panpranot and J. G. Goodwin, Jr, J. Catal., 2001, 204, 98–109 CrossRef CAS.
- H. Boerrigter and R. Rauch, in Handbook Biomass Gasification, Biomass Technology Group (BTG), ed. H. Knoef, The Netherlands, 2005 Search PubMed.
- M. E. Dry, Catal. Today, 2002, 71, 227–241 CrossRef CAS.
- B. H. Davis, Catal. Today, 2009, 141, 25–33 CrossRef CAS.
- (a) M. E. Dry, Catalysis: Science and Technology, ed. J. R. Anderson and M. Boudart, Springer-Verlag, Berlin, 1981, vol. 1, pp. 159–255 Search PubMed; (b) S. H. Kang, J. W. Bae, P. S. Sai Prasad, S. J. Park, K. J. Woo and K. W. Jun, Catal. Lett., 2009, 130, 630–636 CrossRef CAS; (c) S. Z. Li, S. Krishnamoorthy, A. W. Li, G. D. Meitzner and E. Iglesia, J. Catal., 2002, 206, 202–217 CrossRef CAS.
- M. Esmaieli, A. Khodadadi and Y. Mortazavi, Int. J. Chem. React. Eng., 2009, 7, A36 Search PubMed.
- K. Pasanga, N. Lohitharn, A. C. Y. Chien, E. Lotero, J. Panpranot, P. Praserthdam and J. G. Goodwin, Appl. Catal., A, 2007, 332, 130–137 CrossRef.
- W. P. Ma, E. L. Kugler and D. B. Dadyburjor, Energy Fuels, 2007, 21, 1832–1842 CrossRef CAS.
- (a) E. V. Steen and F. F. Prinsloo, Catal. Today, 2002, 71, 327 CrossRef; (b) L. Guczi, G. Stefler, O. Geszti, Z. Koppány, Z. Kónya, E. Molnár, M. Urbán and I. Kiricsi, J. Catal., 2006, 244, 24–32 CrossRef CAS.
- W. Chen, Z. Fan, X. Pan and X. Bao, J. Am. Chem. Soc., 2008, 130, 9414–9419 CrossRef CAS.
- (a) F. J. Pérez-Alonso, T. Herranz, S. Rojas, M. Ojeda, M. López Granados, P. Terreros, J. L. G. Fierro, M. Graciab and J. R. Gancedo, Green Chem., 2007, 9, 663–670 RSC; (b) M. Luo and B. H. Davis, Fuel Process. Technol., 2003, 83, 49–65 CrossRef CAS; (c) T. Herranz, S. Rojas, F. J. Pérez-Alonso, M. Ojeda, P. Terreros and J. L. G. Fierro, J. Catal., 2006, 243, 199–211 CrossRef CAS; (d) R. J. O'Brien, L. Xu, R. L. Spicer and B. H. Davis, Energy Fuels, 1996, 10, 921–926 CrossRef CAS; (e) F. J. Pérez-Alonso, M. López Granados, M. Ojeda, T. Herranz, S. Rojas, P. Terreros, J. L. G. Fierro, M. Gracia and J. R. Gancedo, J. Phys. Chem. C, 2006, 110, 23870–23880 CrossRef.
- (a) D. B. Bukur, M. Koranne, X. Lang, K. R. P. M. Rao and G. P. Huffman, Appl. Catal., A, 1995, 126, 85–113 CrossRef CAS; (b) D. B. Bukur, X. Lang and Y. Ding, Appl. Catal., A, 1999, 186, 255–275 CrossRef CAS.
- (a) P. Khemthong, W. Klysubun, S. Prayoonpokarach, F. Roessner and J. Wittayakun, J. Ind. Eng. Chem., 2010, 16, 531–538 CrossRef CAS; (b) S. A. Eliason and C. H. Bartholomew, Appl. Catal., A, 1999, 186, 229–243 CrossRef CAS; (c) A. Nakhaei Pour, S. M. K. Shahri, Y. Zamani, M. Irani and S. Tehrani, J. Nat. Gas Chem., 2008, 17, 242–248 CrossRef.
- M. D. Shroff, D. S. Kalakkad, K. E. Coulter, S. D. Köhler, M. S. Harrington, N. B. Jackson, A. G. Sault and A. K. Datye, J. Catal., 1995, 156, 185–207 CrossRef CAS.
- (a) N. Sirimanothan, H. H. Hamdeh, Y. Zhang and B. H. Davis, Catal. Lett., 2002, 82, 181–191 CrossRef CAS; (b) T. R. Motjope, H. T. Dlamini, G. R. Hearne and N. J. Coville, Catal. Today, 2002, 71, 335–341 CrossRef CAS.
- R. A. Dictor and A. T. Bell, J. Catal., 1986, 97, 121–136 CrossRef CAS.
- Y. Q. Zhang, N. Sirimanothan, R. J. O'Brien, H. H. Hamdeh and B. H. Davis, Stud. Surf. Sci. Catal., 2001, 139, 125–132 CrossRef CAS.
- (a) M. E. Dry, CHEMTECH, 1982, 744–750 CAS; (b) M. D. Shroff, D. S. Kalakkad, K. E. Coulter, S. D. Köhler, M. S. Harrington, M. B. Jackson, A. G. Sault and A. K. Datye, J. Catal., 1993, 156, 185–207 CrossRef.
- (a) M. Ojeda, R. Nabar, A. U. Nilekar, A. Ishikawa, M. Mavrikakis and E. Iglesia, J. Catal., 2010, 272, 287–297 CrossRef CAS; (b) C. G. Visconti, E. Tronconi, L. Lietti, P. Forzatti, S. Rossini and R. Zennaro, Top. Catal., 2011, 54, 786–800 CrossRef CAS.
- J. Gracia, F. Prinsloo and J. Niemantsverdriet, Catal. Lett., 2009, 133, 257–261 CrossRef CAS.
- Z. Tao, Y. Yang, M. Ding, T. Li, H. Xiang and Y. Li, Catal. Lett., 2007, 117, 130–136 CrossRef CAS.
- E. I. Mabaso, E. van Steen and M. Claeys, DGMK Tagungsber., 2006, 4, 93 Search PubMed.
- M. V. Cagnoli, S. G. Marchett, N. G. Gallegos, A. M. Alvarez, A. A. Yeramián and R. C. Mercader, J. Catal., 1990, 123, 21–30 CrossRef CAS.
- L. D. Mansker, Y. Jin, D. B. Bukur and A. K. Datye, Appl. Catal., A, 1999, 186, 277–296 CrossRef CAS.
- S. A. Eliason and C. H. Bartholomew, in Catalyst Deactivation, ed. C. H. Bartholomew and J. B. Butt, Elsevier, Amsterdam, 1991, p. 211 Search PubMed.
- A. Nakhaei Pour, M. Reza Housaindokht, J. Zarkesh and S. Faramarz Tayyari, J. Ind. Eng. Chem., 2010, 16, 1025–1032 CrossRef CAS.
- V. R. R. Pendyala, G. Jacobs, J. C. Mohandas, M. Luo, H. H. Hamdeh, Y. Ji, M. C. Ribeiro and B. H. Davis, Catal. Lett., 2010, 140, 98–105 CrossRef CAS.
- P. S. Sai Prasad, J. Wook Bae, K. W. Jun and K. W. Lee, Catal. Surv. Asia, 2008, 12, 170–183 CrossRef.
- (a) M. Luo and B. H. Davis, Appl. Catal., A, 2003, 246, 171–181 CrossRef CAS; (b) M. E. Dry and G. J. Oosthuizen, J. Catal., 1968, 11, 18–24 CrossRef CAS.
- J. Abbot, N. J. Clark and B. G. Baker, Appl. Catal., 1986, 26, 141–153 CrossRef CAS.
- (a) D. B. Bukur, D. Mukesh and S. A. Patel, Ind. Eng. Chem. Res., 1990, 29, 194–204 CrossRef CAS; (b) H. J. Wan, B. S. Wu, C. H. Zhang, H. W. Xiang and Y. W. Li, J. Mol. Catal. A: Chem., 2008, 283, 33–42 CrossRef CAS; (c) S. Z. Li, A. W. Li, S. Krishnamoorthy and E. Iglesia, Catal. Lett., 2001, 77, 197–205 CrossRef CAS.
- (a) Z. Tao, Y. Yang, C. Zhang, T. Li, J. Wang, H. Wan, H. Xiang and Y. W. Li, Catal. Commun., 2006, 7, 1061–1066 CrossRef CAS; (b) W. N. Hoc, Y. Zhang, R. J. O. Brien, M. Luo and B. H. Davis, Appl. Catal., A, 2002, 236, 77–89 CrossRef; (c) C. H. Zhang, Y. Yang, B. T. Teng, T. Z. Li, H. Y. Zheng, H. W. Xiang and Y. W. Li, J. Catal., 2006, 237, 405–415 CrossRef CAS.
- (a) M. Luo, R. J. O'Brien, S. Bao and B. H. Davis, Appl. Catal., A, 2003, 239, 111–120 CrossRef CAS; (b) G. Zhao, C. Zhang, S. Qin, H. Xiang and Y. W. Li, J. Mol. Catal. A: Chem., 2008, 286, 137–142 CrossRef CAS.
- D. Ronald and A. T. Bell, J. Catal., 1986, 97, 121–136 CrossRef.
- S. Li, W. Ding, G. D. Meitzner and E. Iglesia, J. Phys. Chem. B, 2002, 106, 85–91 CrossRef CAS.
- G. B. McVicker and M. A. Vannice, J. Catal., 1980, 63, 25–34 CrossRef CAS.
- A. Zhang, I. Nakamura and K. Fujimoto, J. Catal., 1997, 168, 328–333 CrossRef CAS.
- A. A. Adesina, Appl. Catal., A, 1996, 138, 345–367 CrossRef CAS.
- (a) H. J. Wan, B. S. Wu, C. H. Zhang, H. W. Xiang and Y. W. Li, J. Mol. Catal. A: Chem., 2008, 283, 33–42 CrossRef CAS; (b) R. J. O'Brien and B. H. Davis, Catal. Lett., 2004, 94, 1–6 CrossRef CAS.
- W. Ma, E. L. Kugler and D. B. Dadyburjor, Energy Fuels, 2011, 25, 1931–1938 CrossRef CAS.
- W. Yu, B. Wu, J. Xu, Z. Tao, H. Xiang and Y. Li, Catal. Lett., 2008, 125, 116–122 CrossRef CAS.
- M. S. Luo, R. O'Brien and B. H. Davis, Catal. Lett., 2004, 98, 17–22 CrossRef CAS.
- R. M. Malek Abbaslou, J. Soltan and A. K. Dalai, Fuel, 2011, 90, 1139–1144 CrossRef CAS.
- Y. Yang, H. W. Xiang, L. Tian, H. Wang, C. H. Zhang, Z. C. Tao, Y. Y. Xu, B. Zhong and Y. W. Li, Appl. Catal., A, 2005, 284, 105–122 CrossRef CAS.
- J. P. Hong, P. A. Chernavskii, A. Y. Khodakov and W. Chu, Catal. Today, 2009, 140, 135–141 CrossRef CAS.
- A. Tavasoli, R. M. M. Abbaslou and A. K. Dalai, Appl. Catal., A, 2008, 346, 58–64 CrossRef CAS.
- X. Ma, Q. Sun, W. Ying and D. Fang, J. Nat. Gas Chem., 2009, 18, 354–358 CrossRef CAS.
- O. Kitakami, H. Sato and Y. Shimada, Phys. Rev. B: Condens. Matter, 1997, 56, 13849–13854 CrossRef CAS.
- J. van de Loosdrecht, B. Balzhinimaev, J. A. Dalmon, J. W. Niemantsverdriet, S. V. Tsybulya, A. M. Saib, P. J. van Berge and J. L. Visagie, Catal. Today, 2007, 123, 293–302 CrossRef CAS.
- A. M. Saib, D. J. Moodley, I. M. Ciobîcă, M. M. Hauman, B. H. Sigwebela, C. J. Weststrate, J. W. Niemantsverdriet and J. van de Loosdrecht, Catal. Today, 2010, 154, 271–282 CrossRef CAS.
- (a) S. L. Soled, E. Iglesia, R. A. Fiato, J. E. Baumgartner, H. Vroman and S. Miseo, Top. Catal., 2003, 26, 101–109 CrossRef CAS; (b) E. Iglesia, S. C. Reyes, R. J. Madon and S. L. Soled, Adv. Catal., 1993, 39, 221–302 CrossRef CAS.
- Ø. Borg, S. Eri, E. A. Blekkan, S. Storsæter, H. Wigum, E. Rytter and A. Holmen, J. Catal., 2007, 248, 89–100 CrossRef.
- (a) G. L. Bezemer, J. H. Bitter, H. P. C. E. Kuipers, H. Oosterbeek, J. E. Holewijn, X. Xu, F. Kapteijn, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2006, 128, 3956–3964 CrossRef CAS; (b) G. L. Bezemer, U. Falke, A. J. Van Dillen and K. P. de Jong, Chem. Commun., 2005, 731–733 RSC.
- (a) A. Y. Khodakov, J.-S. Girardon, A. Griboval-Constant, A. S. Lermontov and P. A. Chernavskii, Stud. Surf. Sci. Catal., 2004, 147, 295–300 CrossRef CAS; (b) J. W. Bae, Y. J. Lee, J. Y. Park and K. W. Jun, Energy Fuels, 2008, 22, 2885–2891 CrossRef CAS.
- G. L. Bezemer, P. B. Radstake, V. Koot, A. J. van Dillen, J. W. Geus and K. P. de Jong, J. Catal., 2006, 237, 291–302 CrossRef CAS.
- A. Y. Khodakov, Catal. Today, 2009, 144, 251–257 CrossRef CAS.
- M. Lacroix, L. Dreibine, B. de Tymowski, F. Vigneron, D. Edouard, D. Bégin, P. Nguyen, C. Pham, S. Savin-Poncet, F. Luck, M. J. Ledoux and C. Pham-Huu, Appl. Catal., A, 2011, 397, 62–72 CrossRef CAS.
- Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030–1058 CrossRef CAS.
- (a) D. Song and J. Li, J. Mol. Catal. A: Chem., 2006, 247, 206–212 CrossRef CAS; (b) A. M. Saib, M. Claeys and E. van Steen, Catal. Today, 2002, 71, 395–401 CrossRef CAS.
- (a) A. Y. Khodakov, A. Griboval-Constant, R. Bechara and F. Villain, J. Phys. Chem. B, 2001, 105, 9805–9811 CrossRef CAS; (b) H. Xiong, Y. Zhang, K. Liew and J. Li, J. Mol. Catal. A: Chem., 2008, 295, 68–76 CrossRef CAS.
- R. Oukaci, A. H. Singleton and J. G. Goodwin, Appl. Catal., A, 1999, 186, 129–144 CrossRef CAS.
- S. Bessell, Appl. Catal., A, 1995, 126, 235–244 CrossRef CAS.
- (a) G. Prieto, A. Martinez, R. Murciano and M. A. Arribas, Appl. Catal., A, 2009, 367, 146–156 CrossRef CAS; (b) A. Martinez, C. Lopez, F. Marquez and I. Diaz, J. Catal., 2003, 220, 486–499 CrossRef CAS.
- M. K. Gnanamani, G. Jacobs, U. M. Graham, W. P. Ma, V. R. R. Pendyala, M. Ribeiro and B. H. Davis, Catal. Lett., 2010, 134, 37–44 CrossRef CAS.
- (a) G. L. Bezemer, P. B. Radstake, U. Falke, H. Oosterbeek, H. P. C. E. Kuipers, A. J. van Dillen and K. P. de Jong, J. Catal., 2006, 237, 152–161 CrossRef CAS; (b) G. A. Tavasoli, K. Sadagiani, K. Khorashe, A. A. Seifkordi, A. A. Rohani and A. Nakhaeipour, Fuel Process. Technol., 2008, 89, 491–498 CrossRef; (c) A. Tavasoli, R. M. M. Abbaslou, M. Trepanier and A. K. Dalai, Appl. Catal., A, 2008, 345, 134–142 CrossRef CAS.
- R. C. Reuel and C. H. Bartolomew, J. Catal., 1984, 85, 78–88 CrossRef CAS.
- A. K. Dalai and B. H. Davis, Appl. Catal., A, 2008, 348, 1–15 CrossRef CAS.
- A. R. de la Osa, A. De Lucas, A. Romero, J. L. Valverde and P. Sánchez, Catal. Today, 2011, 176, 298–302 CrossRef CAS.
- D. Xu, W. Li, H. Duan, Q. Ge and H. Xu, Catal. Lett., 2005, 102, 229–235 CrossRef CAS.
- S. Storsæter, Ø. Borg, E. A. Blekkan and A. Holmen, J. Catal., 2005, 231, 405–419 CrossRef.
- C. J. Bertole, C. A. Mims and G. Kiss, J. Catal., 2004, 221, 191–203 CrossRef CAS.
- J. E. Baker, R. Burch and N. Yuqin, Appl. Catal., 1991, 73, 135–152 CrossRef CAS.
- G. Jacobs, M. C. Ribeiro, W. P. Ma, Y. Y. Ji, S. Khalid, P. T. A. Sumodjo and B. H. Davis, Appl. Catal., A, 2009, 361, 137–151 CrossRef CAS.
- K. Jalama, D. Hildebrandt, D. Glasser, L. L. Jewell, J. A. Anderson, S. Taylor, N. J. Coville and G. J. Hutchings, Top. Catal., 2007, 44, 129–136 CrossRef CAS.
- (a) L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollar, Surf. Sci., 1998, 411, 186–202 CrossRef CAS; (b) J. C. W. Swart, P. van Helden and E. van Steen, J. Phys. Chem. C, 2007, 111, 4998–5005 CrossRef CAS.
- H. P. Withers, Jr, K. F. Eliezer and J. W. Mitchell, Ind. Eng. Chem. Res., 1990, 29, 1807–1814 CrossRef.
- B. Jongsomjit, J. Panpranot and J. G. Goodwin, Jr, J. Catal., 2003, 215, 66–77 CrossRef CAS.
- F. Morales, F. M. F. de Groot, O. L. J. Gijzeman, A. Mens, O. Stephan and B. M. Weckhuysen, J. Catal., 2005, 230, 301–308 CrossRef CAS.
- B. Ernst, L. Hilaire and A. Kiennemann, Catal. Today, 1999, 50, 413–427 CrossRef CAS.
- G. J. Haddad, B. Chen and J. G. Goodwin, Jr, J. Catal., 1996, 160, 43–51 CrossRef CAS.
- (a) Y. Zhang, H. Xiong, K. Liew and J. Li, J. Mol. Catal. A: Chem., 2005, 237, 172–181 CrossRef CAS; (b) T. Wang, Y. J. Ding, J. M. Xiong, L. Yan, H. J. Zhe, Y. Lu and L. W. Lin, Catal. Lett., 2006, 107, 47–52 CrossRef CAS; (c) F. M. T. Mendes, J. Phys. Chem. B, 2006, 110, 9155 CrossRef CAS; (d) E. van Steen, E. L. Viljoen, J. van de Loosdrecht and M. Claeys, Appl. Catal., A, 2008, 335, 56–63 CrossRef CAS.
- (a) Y. Yang, H. W. Xiang, Y. Y. Xu, L. Bai and Y. W. Li, Appl. Catal., A, 2004, 266, 181–194 CrossRef CAS; (b) J. Yang, Y. Sun, Y. Tang, Y. Liu, H. Wang, L. Tian, H. Wang, Z. Zhang, H. Xiang and Y. W. Li, J. Mol. Catal. A: Chem., 2006, 245, 26–36 CrossRef CAS.
- (a) D. A. Wesner, G. Linden and H. P. Bonzel, Appl. Surf. Sci., 1986, 26, 335–356 CrossRef CAS; (b) J. Gaube and H. F. Klein, Appl. Catal., A, 2008, 350, 126–132 CrossRef CAS.
- N. E. Tsakoumis, M. Rønning, Ø. Borg, E. Rytter and A. Holmen, Catal. Today, 2010, 154, 162–182 CrossRef CAS.
- (a) A. Tavasoli, A. Khodadadi, Y. Mortazavi, K. Sadaghiani and M. G. Ahangari, Fuel Process. Technol., 2006, 87, 641–647 CrossRef CAS; (b) M. Trépanier, A. Tavasoli, A. K. Dalai and N. Abatzoglou, Appl. Catal., A, 2009, 353, 193–202 CrossRef; (c) A. R. de la Osa, A. De Lucas, J. L. Valverde, A. Romero, I. Monteagudo, P. Coca and P. Sánchez, Catal. Today, 2011, 167, 96–106 CrossRef CAS; (d) A. R. de la Osa, A. De Lucas, A. Romero, J. L. Valverde and P. Sánchez, Fuel, 2011, 90, 1935–1945 CrossRef CAS.
- A. Barbier, A. Tuel, I. Arcon, A. Kodre and G. A. Martin, J. Catal., 2001, 200, 106–116 CrossRef CAS.
- C. M. Balonek, A. H. Lillebø, S. Rane, E. Rytter, L. D. Schmidt and A. Holmen, Catal. Lett., 2010, 138, 8–13 CrossRef CAS.
- S. Eri, J. G. Goodwin, Jr, G. Marcelin, T. Riis, US pat., 4880, 763 (to Den Norske Stats Oljeselskap AS), 1989.
- C. G. Visconti, L. Lietti, P. Forzatti and R. Zennaro, Appl. Catal., A, 2007, 330, 49–56 CrossRef CAS.
- N. Koizumi, K. Murai, T. Ozaki and M. Yamada, Catal. Today, 2004, 89, 465–478 CrossRef CAS.
- N. Escalona, C. Medina, R. Garcia and P. Reyes, Catal. Today, 2009, 143, 76–79 CrossRef CAS.
- K. W. Jun, H. S. Roh, K. S. Kim, J. S. Ryu and K. W. Lee, Appl. Catal., A, 2004, 259, 221–226 CrossRef CAS.
- M. J. A. Tijmensen, A. P. C. Faaij, C. N. Hamelinck and M. R. M. van Hardeveld, Biomass Bioenergy, 2002, 23, 129–152 CrossRef CAS.
- S. V. Vassilev, D. Baxter, L. K. Andersen and C. G. Vassileva, Fuel, 2010, 89, 913–933 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams, B. H. Davis and H. D. Willauer, Energy Fuels, 2009, 23, 4190–4195 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Appl. Catal., A, 2010, 373, 112–121 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Catal. Commun., 2010, 11, 816–819 CrossRef CAS.
- I. Aigner, C. Pfeifer and H. Hofbauer, Fuel, 2011, 90, 2404–2412 CrossRef CAS.
- (a) M. J. Prins, K. J. Ptasinski and F. J. J. G. Janssen, Energy, 2007, 32, 1248–1259 CrossRef CAS; (b) A. G. Collot, Int. J. Coal Geol., 2005, 65, 191–212 CrossRef.
- (a) T. Chmielniak and M. Sciazko, Appl. Energy, 2003, 74, 393–403 CrossRef CAS; (b) J. J. Hernández, G. Aranda-Almansa and C. Serrano, Energy Fuels, 2010, 24, 2479–2488 CrossRef; (c) T. R. McLendon, A. P. Lui, R. L. Pinault, S. K. Beer and S. W. Richardson, Biomass Bioenergy, 2004, 26, 377–388 CrossRef CAS; (d) R. N. André, F. Pinto, C. Franco, M. Dias, I. Gulyurtlu and M. A. A. Matos, et al. , Fuel, 2005, 84, 1635–1644 Search PubMed.
- (a) K. Kumabe, T. Hanaoka, S. Fujimoto, T. Minowa and K. Sakanishi, Fuel, 2007, 86, 684–689 CrossRef CAS; (b) K. Li, R. Zhang and J. Bi, Int. J. Hydrogen Energy, 2009, 35, 2722–2726 CrossRef.
- (a) C. Reinoso, A. Cuevas, K. Janssen, M. Morris, K. Lassing and T. Nilsson, et al., APAS Clean Coal Technology Programme, 1994, vol. 3, pp. 1992–1994 Search PubMed; (b) E. Kurkela, J. Laatikainen and P. Stahlburg, APAS Clean Coal Technology Programme, 2000, vol. 3, pp. 1992–1994 Search PubMed; (c) Y. G. Pan, E. Velo, X. Roca, J. J. Manya and L. Puigjaner, Fuel, 2000, 79, 1317–1326 CrossRef CAS.
- (a) G. Chen, Y. Zhang, J. Zhu, Y. Cao and W. Pan, Energy Fuels, 2011, 25, 1964–1969 CrossRef CAS; (b) A. Smolinski, N. Howaniec and K. Stanczyk, Renewable Energy, 2011, 36, 1836–1842 CrossRef CAS; (c) A. Lickrastina, I. Barmina, V. Suzdalenko and M. Zake, Fuel, 2011, 90, 3352–3358 CrossRef CAS; (d) S. I. Ngo, T. D. B. Nguyen, Y.-I. Lim, B.-H. Song, U.-D. Lee, Y.-T. Choi and J.-H. Song, Appl. Energy, 2011, 88, 5208–5220 CrossRef CAS; (e) W. T. Tsai, M. K. Lee and Y. M. Chang, Bioresour. Technol., 2007, 98, 22–28 CrossRef CAS; (f) J. M. Encinar, J. F. González and J. González, Fuel Process. Technol., 2000, 68, 209–222 CrossRef CAS; (g) F. A. Agblevor, S. Besler and A. E. Wiselogel, Energy Fuels, 1995, 9, 635–640 CrossRef CAS; (h) E. Schröder, J. Anal. Appl. Pyrolysis, 2004, 71, 669–694 CrossRef; (i) A. Demirbas, J. Anal. Appl. Pyrolysis, 2006, 76, 285–289 CrossRef CAS; (j) J. Fermoso, B. Arias, M. G. Plaza, C. Pevida, F. Rubiera, J. J. Pis, F. García-Peña and P. Casero, Fuel Process. Technol., 2009, 90, 926–932 CrossRef CAS; (k) M. Lapuerta, J. J. Hernández, A. Pazo and J. López, Fuel Process. Technol., 2008, 89, 828–837 CrossRef CAS; (l) Y. S. Kim, J. J. Lee, T. S. Kim and J. L. Sohn, Energy Convers. Manage., 2011, 52, 2262–2271 CrossRef; (m) C.-C. Cormos, Int. J. Hydrogen Energy, 2011, 36, 5960–5971 CrossRef CAS.
- (a) D. J. Duvenhage and N. J. Coville, Appl. Catal., A, 2006, 298, 211–216 CrossRef CAS; (b) E. van Steen and M. Claeys, Chem. Eng. Technol., 2008, 31, 655–666 CrossRef CAS.
- (a) M. Kraum and M. Baerns, Appl. Catal., A, 1999, 186, 189–200 CrossRef CAS; (b) P. Concepción, C. López, A. Martínez and V. F. Puntes, J. Catal., 2004, 228, 321–332 CrossRef; (c) M. Trépanier, A. Tavasoli, A. K. Dalai and N. Abatzoglou, Fuel Process. Technol., 2009, 90, 367–374 CrossRef; (d) W. J. Wang and Y. W. Chen, Appl. Catal., 1991, 77, 223–233 CrossRef CAS; (e) S. Bessel, Appl. Catal., A, 1993, 96, 253–268 CrossRef; (f) S.-H. Kang, J. W. Bae, J.-Y. Cheon, Y.-J. Lee, K.-S. Ha, K.-W. Jun, D.-H. Lee and B.-W. Kim, Appl. Catal., B, 2011, 103, 169–180 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |