Self-standing positive electrodes of oxidized few-walled carbon nanotubes for light-weight and high-power lithium batteries†
Seung Woo
Lee‡
a,
Betar M.
Gallant‡
a,
Youngmin
Lee
a,
Noboru
Yoshida
b,
Dong Young
Kim
b,
Yuki
Yamada
b,
Suguru
Noda
b,
Atsuo
Yamada
b and
Yang
Shao-Horn
*ac
aDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
bDepartment of Chemical System Engineering, The University of Tokyo, Tokyo, Bunkyo-ku, Japan
cDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA. E-mail: shaohorn@mit.edu; Tel: +1 617 253-2259
First published on 6th December 2011
Abstract
Binder-free and self-standing carbon nanotube (CNT) electrodes of tens of microns in thickness have been assembled via a vacuum-filtration process of oxidized few-walled CNTs (FWNTs), with different amounts of oxygen functional groups on FWNTs. Sub-millimetre long FWNTs can provide high electrical conductivity and mechanical strength in self-standing porous networks by reducing the number of junctions among FWNTs. We show that the gravimetric capacity of FWNT electrodes in lithium cells can be enhanced by increasing oxygen functional groups on FWNTs, which results from Faradaic reactions between lithium ions and surface oxygen functional groups. These self-standing FWNT electrodes (free of binder/additive and current collector) can provide a high gravimetric energy of ∼200 W h kg−1 at a high power of ∼10 kW kg−1, showing promise as the positive electrode for light-weight, high-power lithium batteries.
Broader contextNext-generation energy storage applications such as load-leveling and electrified propulsion have power and energy demands that exceed the power capability of conventional lithium-ion batteries and the energy output of electrochemical capacitors. While lithium-ion battery positive electrodes have high gravimetric energy of ∼500 W h per kgelectrode at ∼1 kW per kgelectrode, high-power performance (>10 kW per kgelectrode) can only be achieved at the expense of energy (<100 W h per kgelectrode). One promising strategy to meet increasing demands for high-energy and high-power positive electrodes is to store energy in oxygen-containing sites (functional groups) on carbon nanotubes, capable of storing substantially higher energy through Faradaic reactions on the surface of capacitive materials. Functionalized carbon nanotubes, which can be modified using acid treatments, can then be assembled using a simple vacuum-filtration method to yield free-standing, binder-free (carbon-only) electrodes with gravimetric energy on the order of that of Li-ion batteries, yet obtainable at substantially higher power (10 kW per kgelectrode). In this work, we show that the quantity of functional groups can be controlled, leading to “tunable” energy and power. More broadly, we highlight design principles for functionalized carbon nanotube-based electrodes, where factors such as carbon structure, electrode thickness, oxygen content, and electrical conductivity play critical roles in modulating the energy–power characteristics. |
Introduction
Carbon nanotubes (CNTs) have become promising materials to design electrodes for electrochemical energy conversion and storage devices, including electrochemical capacitors (ECs)1–4 and lithium-ion batteries,5,6 due to their physical characteristics such as high electrical conductivity, high surface area, high aspect ratio, chemical and mechanical stability.7 Significant research efforts have focused on developing nanostructured CNT electrodes to facilitate electrical and ionic transport for high-power EC applications.1,3 State-of-the-art nanostructured CNT electrodes have been prepared by water-assisted chemical vapor deposition,8 spray,9 Meyer rod,10 and layer-by-layer (LbL) assembly,2,6,11 all of which avoid the use of insulating polymer binders to maximize the electrochemically active surface area. However, using electrical double layer capacitance (5–20 μF cm−2)1,12 on the surface of CNT-based electrodes8 alone yields gravimetric energy values significantly lower than lithium-ion battery electrodes.A promising strategy to further increase the gravimetric energy of CNT electrodes is the incorporation of fast Faradaic reactions (pseudocapacitance) into a CNT conducting matrix.1 Previous studies have introduced pseudocapacitive materials, including nano-size RuO2,2,13MnO2,3,14,15MoO3,16 conducting polymers,15,17,18 and surface-oxygen and -nitrogen functional groups3,6 into nanostructured CNT electrodes, successfully increasing the gravimetric energy density of electrodes. Recently we have shown the redox of oxygen functional groups on functionalized multi-walled CNTs (MWNTs) for LbL-assembled thin-film electrodes, which are derived from electrostatically self-assembled, negatively charged (carboxylic acid-functionalized) and positively charged (amine-functionalized) MWNTs.2,6,11 These thin electrodes can deliver both high gravimetric energy and power (200 W h per kgelectrode at 100 kW per kgelectrode) in lithium cells, competitive with solid-state thin-film lithium battery technologies.19 The high gravimetric energy can be attributed to Faradaic reactions between lithium ions and carboxylic acid functional groups detected by XPS, via the mechanism C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + Li+ + e− ↔ C–OLi at ∼3 V vs. Li.6 Although LbL-MWNT thin-film electrodes (with thicknesses from several nanometres to ∼3 μm) can be promising energy sources for micro-scale electronic devices and micro-electro-mechanical systems (MEMS), scale-up to tens or hundreds of microns in thickness is required for transportation and load-leveling applications.2
O + Li+ + e− ↔ C–OLi at ∼3 V vs. Li.6 Although LbL-MWNT thin-film electrodes (with thicknesses from several nanometres to ∼3 μm) can be promising energy sources for micro-scale electronic devices and micro-electro-mechanical systems (MEMS), scale-up to tens or hundreds of microns in thickness is required for transportation and load-leveling applications.2
In this work, we demonstrate the synthesis of free-standing, binder-free CNT electrodes of tens of microns in thickness, where the amount of oxygen functional groups on CNTs and stored energy per unit weight of CNTs can be controlled by the oxidation time. Here we use few-walled CNTs (FWNTs), which can be synthesized efficiently by fluidized bed chemical vapor deposition (CVD) at high yield and productivity.20 FWNTs have higher specific surface area (SSA) than MWNTs as SSA is inversely proportional to the number of walls in CNTs. Upon the introduction of surface functional groups to the outer walls, FWNTs can retain the electrical conductivity by their inner walls unlike single-walled CNTs (SWNTs).8 In addition, sub-millimetre long FWNTs can minimize the number of junctions in the porous electrode network, and thus reduce junction and electrode sheet resistances, which can enable the development of self-standing electrodes free of current collector to enhance the gravimetric capacity of practical electrodes. Surface morphology and oxygen-to-carbon atomic ratio of oxidized FWNTs are investigated using high-resolution transmission electron microscopy (HRTEM) and X-ray photoelectron spectroscopy (XPS), respectively. Oxidized FWNTs are assembled successfully into free-standing electrodesvia a simple VF method, creating much thicker electrodes (VF-FWNT electrodes, 15–55 μm, Fig. 3) compared to ∼3 μm of LbL-MWNT electrodes reported previously6 (Fig. S1†). Scanning electron microscopy (SEM) shows the interpenetrating network structure of the assembled FWNT electrodes. We correlate the amount of oxygen functional groups on FWNTs with gravimetric capacitances of electrodes using potential-dependent cyclic voltammetry. Furthermore, the gravimetric energy and power density of VF-FWNT electrodes are measured using rate-dependent galvanostatic tests. Finally, we show that Faradaic reactions on oxygen functional groups in lithium cells are very stable over 1000 galvanostatic cycles.
Experimental procedure
Electrode preparation
Sub-millimetre long FWNTs (6–10 nm diameter, 0.4 mm length, 99 wt% purity, 400 m2 g−1 SSA, triple-walled on average) were synthesized by CVD in a single fluidized bed reactor.20 FWNTs were refluxed in concentrated H2SO4/HNO3 (3/1 v/v, 96% and 70%, respectively) at 70 °C to introduce oxygen functional groups on the surface of FWNTs.11Oxidation time was controlled from 2 to 6 hours to control the amount of oxygen functional groups on FWNTs. FWNTs in H2SO4/HNO3 were diluted in 5 vol% HCl solution (1 L) after the oxidation process, and filtered via a polycarbonate membrane filter (Whatman membrane, 0.05 μm pore size). Remaining FWNT powders on the filter were washed with deionized Milli-Q water (18 MΩ cm) water (3 L). Pristine FWNTs were dispersed in ethanol, and oxidized (2 hours and 6 hours). FWNTs were dispersed in Milli-Q deionized water at a concentration of 0.5 mg mL−1 with unadjusted pH, and were sonicated for 15–30 minutes to improve the quality of dispersion. Binder- and additive-free electrodes were synthesized by the VF method on Whatman polycarbonate track-etched membranes (0.2 μm pore diameter), which resulted in interwoven and mechanically robust FWNT films attached to the membrane. Free-standing electrodes were obtained by removing the air-dried FWNT films using a razor blade, and electrodes were further dried overnight (70 °C) in air, then cut or punched to the desired size. The typical thickness of VF-FWNT electrodes was 15–55 μm, depending on the initial volume of FWNT dispersion used for filtration.Characterization
A Kratos Axis Ultra XPS instrument (Kratos Analytical) with a monochromatized Al Kα X-ray source was used to investigate the surface chemistry of pristine and oxidized FWNTs. Curve fitting of the photoemission spectra was performed following a Shirley-type background subtraction. Using C 1s peak from sp2 hybridized carbons centered at 284.5 eV as a reference, all other peaks were fitted by the Gaussian–Lorentzian function. The relative sensitivity factors used to scale the peaks of C 1s and O 1s were 0.278, and 0.780, respectively. A JEOL 2010 transmission electron microscope (TEM) with a LaB6 thermal emission electron gun was used for high resolution imaging of pristine and oxidized FWNT samples. The microscopes were operated at 200 kV and were able to ultimately achieve 0.19 nm point-to-point resolution. Microstructure of the VF-FWNT electrode was investigated using a scanning electron microscope (JEOL 6320 SEM) operating at 5 kV. Electrical conductivities of VF-FWNT electrodes were measured by using a standard four-point probe configuration (LUCAS LABS model Pro 4). A series of 3 measurements were taken at different positions on each electrode, and then were averaged to give the final reported value with the standard deviation as an error range.Electrochemical measurement
Two-electrode electrochemical cells (Tomcell, Japan) were assembled inside an Argon-filled glovebox (Vacuum Atmosphere Co., O2 level below 1 ppm and H2O level below 0.1 ppm) and consisted of a free-standing VF-FWNT positive electrode, a lithium metal negative electrode, two porous Celgard 2500 separators, and 1 M LiPF6 in an EC:DMC (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 volume ratio, 3.5 ppm H2O) electrolyte (Kishida Chemical Corporation, Japan). VF-FWNT electrodes were tested electrochemically using a Solartron 1470 test unit in the voltage range 1.5–4.5 V vs. Li. For galvanostatic rate capability tests, the gravimetric current ranged from 0.1 to 100 A g−1, and the voltage was held constant at either 1.5 or 4.5 V vs. Li for 30 minutes prior to charge or discharge, respectively. Cycling tests consisted of accelerated galvanostatic cycling at 10 A g−1 for 99 cycles, followed by a slower charge and discharge (0.1 A g−1) every 100 cycles up to 1000 cycles, with a 30 min voltage hold at 1.5 V or 4.5 V vs. Li prior to low-rate charge or discharge, respectively. Only low-rate (0.1 A g−1) discharge capacity is reported. In all cases, as-assembled cells allowed to rest for at least 8 hours prior to testing in order to allow full wetting of porous VF-FWNT films by the electrolyte.
:7 volume ratio, 3.5 ppm H2O) electrolyte (Kishida Chemical Corporation, Japan). VF-FWNT electrodes were tested electrochemically using a Solartron 1470 test unit in the voltage range 1.5–4.5 V vs. Li. For galvanostatic rate capability tests, the gravimetric current ranged from 0.1 to 100 A g−1, and the voltage was held constant at either 1.5 or 4.5 V vs. Li for 30 minutes prior to charge or discharge, respectively. Cycling tests consisted of accelerated galvanostatic cycling at 10 A g−1 for 99 cycles, followed by a slower charge and discharge (0.1 A g−1) every 100 cycles up to 1000 cycles, with a 30 min voltage hold at 1.5 V or 4.5 V vs. Li prior to low-rate charge or discharge, respectively. Only low-rate (0.1 A g−1) discharge capacity is reported. In all cases, as-assembled cells allowed to rest for at least 8 hours prior to testing in order to allow full wetting of porous VF-FWNT films by the electrolyte.
Results and discussion
The amount of oxygen functional groups on oxidized FWNTs was quantified using XPS. An XPS wide scan survey showed that O 1s peaks increased relative to C 1s peaks with longer oxidation time until 4 hours, and maintained similar ratios from 4 hours to 6 hours (Fig. 1a), indicating that oxygen incorporation was saturated after 4 hours of oxidation time. Atomic ratios of oxygen to carbon (O/C) of FWNTs (obtained from high-resolution C 1s and O 1s peaks) were found to increase from 0.11 (2 h oxidation) to 0.20 (4 h oxidation), indicating the oxidation time can be used to control the amount of oxygen functional groups on FWNTs. High-resolution C 1s XPS spectra tracked detailed changes in surface chemistry of FWNTs during the oxidation process (Fig. 1b). Pristine FWNT C 1s peaks consisted of mostly sp2 hybridized graphitic carbons centered at 284.5 eV (in green) and included small contents of sp3 hybridized diamond-like carbon centered at 285.2 eV (in orange) (Fig. 1b).11,21 As the oxidation time increased, the intensities of carbon atom peaks connected to oxygen atoms (C–O centered at 286.0 ± 0.1 eV (in magenta), CO centered at 286.9 ± 0.2 eV (in dark cyan), and COOH(COOR) centered at 288.9 ± 0.2 eV (in dark yellow))21,22 as well as sp3 hybridized carbon peaks were found to increase, indicating that the oxidation process gradually breaks the exterior graphitic wall of FWNTs and incorporates oxygen functional groups (Fig. 1b and S2†). The introduced carboxylic acid groups (COOH) on the surface of FWNTs exist as carboxylate anions (COO−) in aqueous solution, which can prevent aggregation and precipitation of CNTs.11
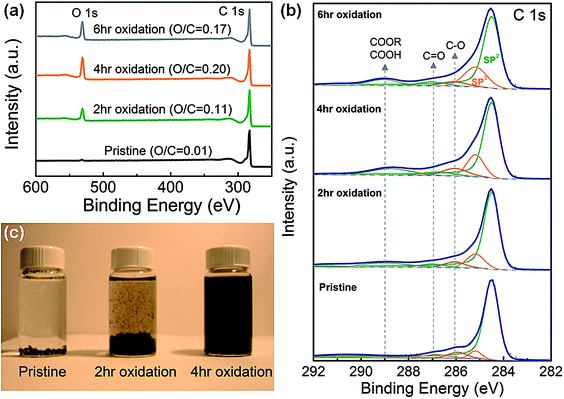 | ||
| Fig. 1 (a) X-Ray photoelectron spectroscopy (XPS) wide scan survey of pristine and oxidized few-walled carbon nanotubes (FWNTs). C/O indicates atomic ratio of carbon to oxygen of FWNTs. (b) C 1s XPS spectra of pristine and oxidized FWNTs. (c) Optical images of FWNT solutions after 30 min from sonication. | ||
Fig. 1c compares the dispersion of FWNTs in deionized water (0.5 mg mL−1) after 30 min following sonication for 15 minutes. Pristine FWNTs immediately precipitated after sonication, but 4 h oxidized FWNTs maintained a stable dispersion, showing that electrostatic repulsions between anions (COO−) on FWNTs can overcome van der Waals interactions among FWNTs. 2 h oxidized FWNTs showed moderate dispersion quality and precipitated gradually after the sonication process. It is interesting to note that previously reported 2 h oxidized MWNTs (10–20 nm in diameter, 1–5 μm in length)6,11 have a similar oxygen content (O/C = 0.11) but show stable dispersion in water. This difference of dispersion could be attributed to different aspect ratios (∼50000 (ref. 20) for FWNTs used in this work vs. ∼167 for previously reported MWNTs6,11). HRTEM measurements showed that pristine FWNTs had smooth outer surfaces (Fig. 2a), but the oxidation process gradually roughened the exterior walls of FWNTs with longer oxidation time (Fig. 2b and c), which can be related to the enhanced solubility of FWNTs in water (Fig. 1c). This is in good agreement with the XPS results showing that more sp3 carbons and oxygenated species are detected on FWNTs as the oxidation time increases. Thin FWNTs (Fig. 2a) became less visible in HRTEM and only large FWNTs were observed with increasing oxidation time, as shown in Fig. 2b and c.
 | ||
| Fig. 2 Transmission electron microscopy (TEM) images of (a) pristine, (b) 2 h oxidized and (c) 4 h oxidized FWNTs. | ||
Pristine and oxidized FWNTs were dispersed in ethanol and deionized water, respectively, then assembled via the VF method, yielding films of interwoven FWNTs that could be removed from the membrane to create free-standing VF-FWNT electrodes (Fig. 3). The origin of mechanical stability of free standing electrodes can be attributed to the highly interpenetrating network structure of high aspect ratio FWNTs, which was revealed by SEM images of VF-FWNT electrodes (Fig. 4). A cross-sectional SEM image of a free-standing electrode from 4 h oxidized FWNTs clearly shows that the individual FWNTs assume random orientations within the film. It should be mentioned that it is difficult to detach thin VF-FWNT electrodes (<∼5 μm) from the membranes due to mechanical brittleness, which leads to crumbling of the film. Densities of free-standing electrodes were ∼0.2 g cm−3 for pristine FWNTs, ∼0.4 g cm−3 for 2 h or 4 h oxidized FWNTs, and ∼0.8 g cm−3 for 6 h oxidized FWNTs, corresponding to electrode thicknesses of 55, 22, 20, and 22 μm, respectively, which were found to be typical of electrodes synthesized using the VF process. The increasing density with oxidation time indicates that FWNTs can create a more densely packed structure owing to good dispersion of FWNTs in water. It is interesting to note that the densities (0.4–0.8 g cm−3) of oxidized VF-FWNT electrodes are lower than 0.83 g cm−3 of electrostatically self-assembled LbL-MWNT electrodes.6
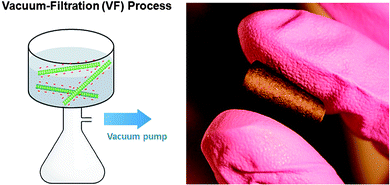 | ||
| Fig. 3 A schematic illustration of the vacuum-filtration process of oxidized FWNTs (left) and a digital picture image (right) of a folded FWNT electrode assembled via the VF method (2 h oxidized VF-FWNT electrode). | ||
 | ||
| Fig. 4 Scanning electron microscopy (SEM) top view images of electrodesviaVF of (a) pristine, (b) 2 h oxidized, and (c) 4 h oxidized FWNT solutions. (d) SEM cross-section view image of electrodesviaVF of 4 h oxidized FWNT solutions. | ||
The electrical conductivities of VF-FWNT electrodes were measured by a 4-point probe as a function of the oxidation time (Fig. 5). The electrical conductivity of VF-FWNT electrodes was found to increase from ∼40 S cm−1 of pristine FWNTs to ∼100 S cm−1 of 2 h oxidized FWNTs and finally decrease to ∼50 S cm−1 of 4–6 h oxidized FWNTs. The initial increase of the electrical conductivity for 2 h oxidized VF-FWNT electrodes can be attributed to higher packing density giving more continuous pathways for fast electron transport, and the later decrease of 4–6 h oxidized VF-FWNT electrodes can be related to the disruption of the conjugated carbon sp2 orbitals on the FWNT walls with the incorporation of surface functional groups. It is interesting to note that all of the VF-FWNT electrodes have much higher electronic conductivities compared to 2–10 S cm−1LbL-MWNT electrodes.11 This difference can be explained by sub-millimetre long length and high aspect ratio of FWNTs that can provide more continuous pathways for electron transport in VF-FWNT electrodes compared to those in LbL-MWNT electrodes (Fig. S3†).
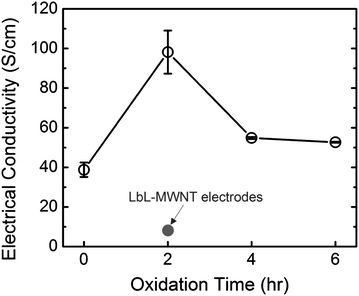 | ||
| Fig. 5 Electrical conductivities of VF-FWNT electrodes as a function of the oxidation time. The grey circle indicates electrical conductivity of LbL-MWNT electrodes (ref. 11). | ||
Electrochemical testing of VF-FWNT electrodes in lithium cells showed that FWNT electrode performance is strongly influenced by oxidation time and the corresponding oxygen content of FWNTs. Cyclic voltammetry at 1 mV s−1 of pristine VF-FWNTs (Fig. 6a) exhibited characteristic double-layer charging, with an average capacitance corresponding to 33 F g−1 (1.5 to 4.5 V vs. Li) for both the forward and backward scans, which was measured by dividing the gravimetric current by the scan rate. The pristine VF-FWNT capacitance was found to be largely independent of the voltage scan range, yielding nearly overlapping curves over several different scan windows. In contrast, oxidized VF-FWNTs exhibited substantially higher gravimetric currents and capacitances compared to pristine VF-FWNTs, as shown in Fig. 6b and c. The 2 h oxidized VF-FWNTs exhibited an average capacitance of 94 and 90 F g−1 for the forward and backward scan (1.5–4.5 V vs. Li), respectively. Increasing oxidation time to 4 hours, the forward and backward capacitances were increased to 120 and 116 F g−1, respectively. Additionally, the volumetric capacitance, as determined from the gravimetric capacitance and density of electrodes, was found to increase with oxidation time in accordance with the trend for gravimetric capacitance, yielding 8, 41, and 50 F cm−3 for pristine, 2 h and 4 h oxidized FWNTs for the forward scan. The potential-dependent gravimetric currents obtained from a 6 h oxidized electrode (Fig. S4a†) were comparable to that of the 4 h oxidized sample, consistent with the fact that 4 hour and 6 hour oxidation times yielded similar oxygen to carbon ratios (Fig. 1a).
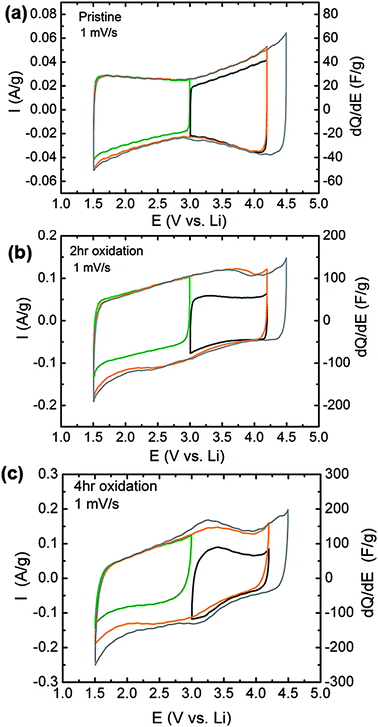 | ||
| Fig. 6 Potential-dependent cyclic voltammetry and differential capacitance of VF-FWNT electrodes in lithium cells for electrodes consisting of (a) pristine, (b) 2 h oxidized, and (c) 4 h oxidized VF-FWNT electrodes. The examined potential windows were 3.0–4.2 V vs. Li (black), 1.5–3.0 V vs. Li (green), 1.5–4.2 V vs. Li (orange), and 1.5–4.5 V vs. Li (gray) at a scan rate of 1 mV s−1. The thicknesses of the FWNT electrodes were 55 μm for pristine FWNT, 22 μm for 2 h oxidized FWNT, and 15 μm for 4 h oxidized FWNT electrodes. The corresponding loading density of VF-FWNT electrodes ranges from 0.8 to 1.7 mg cm−2. | ||
The capacitances measured from oxidized VF-FWNTs were found to be highly potential-dependent. Lowering the upper voltage limit from 4.5 to 4.2 V vs. Li (1.5–4.2 V), no substantial reduction in the average capacitance was noted. However, narrowing the voltage range to 3.0–4.2 V vs.Li, the average forward-scan capacitances of oxidized VF-FWNTs were reduced considerably to 50 and 67 F g−1 for 2 h and 4 h oxidized FWNT electrodes, respectively. A similar reduction was observed when the voltage window was limited to 1.5–3.0 V vs. Li. The potential-dependent capacitances obtained from functionalized FWNTs indicate that additional charge storage mechanisms are accessible during the first discharge below 3.0 V vs. Li and subsequent charging above 3.0 V vs.Li, which can be attributed to Faradaic reactions between lithium ions and oxygen functional groups on FWNTs such as carboxylic acid and carbonyl groups as reported previously.6 The evidence of these Faradaic reactions is supported by the appearance of redox peaks centered at ∼3.2 V vs. Li emerges with increasing oxidation time.
The relative contributions of Faradaic reactions and double-layer charging as a function of oxidation time can yield deeper insight into the underlying mechanisms that influence electrode performance. The role of these two charge storage mechanisms was quantified by analyzing the pure double-layer contribution (taken as the average capacitance of the green curves in Fig. 6a–c over a restricted voltage range of 2.5–2.0 V vs.Li, corresponding to Li+ adsorption on FWNTs) compared to the total Faradaic + double-layer capacitance contributions (taken as the average of the orange curves over the same voltage range) for the negative CV scans (Fig. 7). For pristine FWNTs without oxygen functional groups, the total capacitance (∼25 F g−1) is attributed almost entirely to double layer capacitance. With 2 h oxidation, the double-layer capacitance increased from ∼25 to 75 F g−1, while the total capacitance increased further to ∼110 F g−1, showing that introduced oxygen groups (O/C = 0.11) increase the capacitance through Faradaic reactions. The increase in the double-layer capacitance is thought to result from an increase in the surface area of FWNTs resulting from opening of the sidewalls during oxidation, which was clearly observed from TEM (Fig. 2). Increasing the oxidation time to four hours led to a slight additional increase in the Faradaic capacitance compared to the 2 h oxidation, while the double-layer component remained constant. Finally, with an additional increase from 4 h to 6 h oxidation time, the double-layer and total capacitance increased by roughly the same amount. This finding is consistent with the fact that 4 h and 6 h oxidation times yielded similar O/C ratios (0.20 and 0.17, respectively), and therefore the additional 2 hours of oxidation for 6 h oxidized electrodes was expected to result predominantly in damaging the outer walls of FWNTs, leading to newly exposed surfaces for double layer charging.
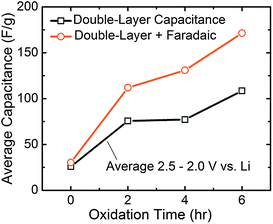 | ||
| Fig. 7 Contributions to the gravimetric capacitance as a function of oxidation time. The double-layer capacitance was determined as the average capacitance of the green curves in Fig. 6a–c over a restricted voltage range of 2.5–2.0 V vs.Li, corresponding to Li+ adsorption on FWNTs. The total Faradaic + double-layer capacitance was determined as the average of the orange curves over the same voltage range. | ||
Galvanostatic testing of VF-FWNT electrodes in the range 1.5–4.5 V vs. Li (Fig. 8a–c) showed that the gravimetric capacities at low rates (<1 A g−1) increased with increasing oxidation time. As shown in Fig. 8d, a pristine electrode delivered a capacity of 26 mA h g−1 at 0.1 A g−1 whereas 2 h and 4 h functionalized electrodes had capacities of 87 and 118 mA h g−1, respectively. This slight advantage in capacity observed for the 4 h oxidized VF-FWNT electrodes could be related to a larger amount of oxygen functional groups on the carbon surface resulting from longer oxidation time (Fig. 1a). 6 hour functionalized FWNTs (Fig. S4b†) were found to have comparable gravimetric capacitances to those of 4 h oxidized FWNTs at 0.1 A g−1 (Fig. 8d), indicating that no further advantage in discharge capacity was obtained with oxidation time beyond 4 hours. With increasing rate, 4 h oxidized VF-FWNT electrodes exhibited more rate-sensitive capacities than pristine and 2 h oxidized VF-FWNT electrodes. At 50 A g−1, 4 h oxidized VF-FWNT electrodes delivered only 9 mA h g−1 compared to 34 mA h g−1 for 2 h oxidized VF-FWNTs. The reduced power performance of 4 h oxidized VF-FWNTs could be related to poor electric conductivity resulting from the introduction of a large number of defect sites on FWNTs during the oxidation process. Gravimetric capacities were sustained for FWNT electrodes over 1000 cycles (within each 100 cycles, 99 cycles at 10 A g−1, followed by 1 cycle at 0.1 A g−1; discharge capacities are shown) without any capacity fade (Fig. 9). The cycle life is comparable to that of LbL-MWNT electrodes reported recently6 and conventional carbon-based double-layer capacitors.1
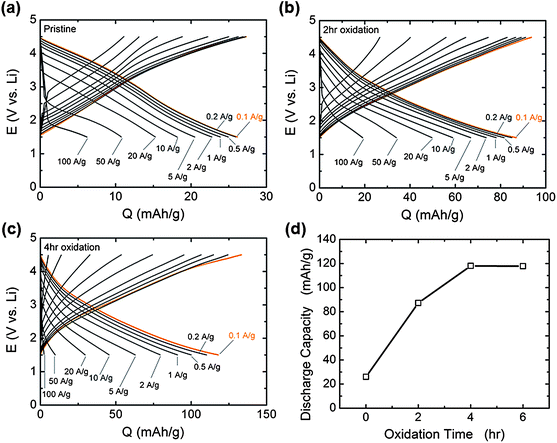 | ||
| Fig. 8 Galvanostatic rate capability of VF-FWNT electrodes in lithium cells for (a) pristine, (b) 2 h oxidized, and (c) 4 h oxidized VF-FWNT electrodes. The voltage window was 1.5–4.5 V vs. Li and the gravimetric currents ranged from 0.1 A g−1 to 100 A g−1. Prior to charge or discharge, the cells were held at a constant voltage of 4.5 or 1.5 V vs.Li, respectively, for 30 minutes. (d) Gravimetric discharge capacity at 0.1 A g−1 upon galvanostatic discharge as a function of oxidation time, as obtained from (a–c). | ||
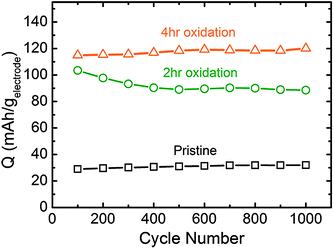 | ||
| Fig. 9 Cycling performance of VF-FWNT electrodes tested galvanostatically in the voltage range of 1.5–4.5 V vs. Li. The data points correspond to discharge capacity obtained at a gravimetric current of 0.1 A g−1 following voltage hold at 4.5 V vs. Li for 30 minutes. Between data points, the cells were cycled at an accelerated gravimetric current of 10 A g−1 for 99 cycles. | ||
The gravimetric energy and power characteristics of FWNT electrodes are shown in Fig. 10, which were computed based on data in Fig. 8a–c. At a low power of ∼300 W kg−1, 2 hour, 4 h and 6 h oxidized VF-FWNT electrodes delivered gravimetric energies of ∼200 W h kg−1 up to a power of ∼10 kW kg−1. On further increasing the power, a substantial decrease in attainable energy occurred. The lower gravimetric energy (78 W h kg−1) of pristine VF-FWNT electrodes compared to oxidized VF-FWNT electrodes indicates that Faradaic reactions between lithium ions and functional groups contribute substantially to the gravimetric energy of FWNT electrodes. These oxidized VF-FWNT electrodes show higher gravimetric energy density than nanostructured CNT electrodes for electrochemical capacitors (50–100 W h kg−1).8,10,23 In addition, at high power (greater than 10 kW kg−1), VF-FWNT electrodes deliver higher gravimetric energy than high-power lithium battery materials.24–26 Although these VF-FWNT electrodes with more practical thicknesses of ∼20 μm have lower gravimetric energy than thin LbL-MWNT electrodes of a few microns reported previously6 (gravimetric energies up to 400 W h kg−1 at 1 kW kg−1), they are free of current collector, and thus higher gravimetric energy is expected in practical devices.
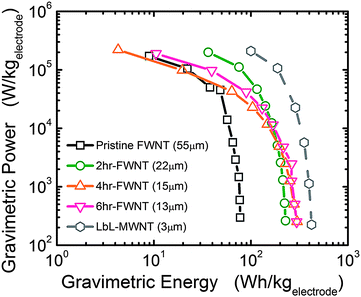 | ||
| Fig. 10 Ragone plot comparing energy and power performance of VF-FWNT electrodes, calculated from galvanostatic data in Fig. 7(a)–(c) and ref. 6. Only positive electrode weight was considered in the gravimetric energy and power density calculations. | ||
Conclusion
In summary, we systematically introduced oxygen functional groups onto sub-millimetre long FWNTs by a surface oxidation process. Pristine and oxidized FWNTs are assembled by the VF method to yield mechanically robust free-standing electrodes of tens of microns in thickness under ambient conditions. The interwoven microstructure of FWNTs not only supports the free-standing structure of the electrodes, but also provides a fast electric-conduction network for high-power applications. We correlated the amount of oxygen functional groups with gravimetric capacitances of oxidized VF-FWNT electrodes in lithium cells, confirming the role of Faradaic reactions between oxygen groups on FWNTs and lithium ions. Utilization of Faradaic reaction with oxygen groups yields a high gravimetric energy of ∼200 W h kg−1 at a high power of ∼10 kW kg−1 in lithium cells, indicating that self-standing films of oxidized FWNTs (free of current collector) are promising positive electrodes for high power applications. Future research will focus on the investigation of specific oxygen functional groups having higher redox potentials and incorporation of surface functional groups in the nanostructured FWNT matrix to further increase the gravimetric energy and power densities of electrodes. Finally, we believe incorporating surface oxygen functional groups on FWNTs having high electrical conductivity can open up various opportunities for the development of novel engineered electrodes for energy storage applications.Acknowledgements
The authors acknowledge the support from Contour Energy Systems and Mitsubishi Motor Company for this project. The research made use of the Shared Experimental Facilities supported by the MRSEC Program of the National Science Foundation under award number DMR 08-019762. The assistance of S. Chen in the SEM image collection, and of E. Crumlin in XPS measurement is greatly appreciated. S.W.L. acknowledges a Samsung Scholarship from the Samsung Foundation of Culture and B.M.G. acknowledges a graduate research fellowship from the National Science Foundation. We would like to thank P.T. Hammond and M. N. Hyder at MIT for helpful input and discussion on this paper.Notes and references
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS
.
- S. W. Lee, B. M. Gallant, H. R. Byon, P. T. Hammond and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 1972–1985 CAS
- G. Lota, K. Fic and E. Frackowiak, Energy Environ. Sci., 2011, 4, 1592–1605 CAS
- E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937–950 CrossRef CAS
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Energy Environ. Sci., 2009, 2, 638–654 CAS
- S. W. Lee, N. Yabuuchi, B. M. Gallant, S. Chen, B. S. Kim, P. T. Hammond and Y. Shao-Horn, Nat. Nanotechnol., 2010, 5, 531–537 CrossRef CAS
-
P. M. Ajayan and O. Z. Zhou, in Carbon Nanotubes, Springer-Verlag Berlin, Berlin, 2001, vol. 80, pp. 391–425 Search PubMed
- D. N. Futaba, K. Hata, T. Yamada, T. Hiraoka, Y. Hayamizu, Y. Kakudate, O. Tanaike, H. Hatori, M. Yumura and S. Iijima, Nat. Mater., 2006, 5, 987–994 CrossRef CAS
- M. Kaempgen, C. K. Chan, J. Ma, Y. Cui and G. Gruner, Nano Lett., 2009, 9, 1872–1876 CrossRef CAS
- L. B. Hu, J. W. Choi, Y. Yang, S. Jeong, F. La Mantia, L. F. Cui and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21490–21494 CrossRef CAS
- S. W. Lee, B.-S. Kim, S. Chen, Y. Shao-Horn and P. T. Hammond, J. Am. Chem. Soc., 2009, 131, 671–679 CrossRef CAS
- E. Pollak, N. Levy, L. Eliad, G. Salitra, A. Soffer and D. Aurbach, Isr. J. Chem., 2008, 48, 287–303 CrossRef CAS
- X. Qin, S. Durbach and G. T. Wu, Carbon, 2004, 42, 451–453 CrossRef CAS
- S. W. Lee, J. Kim, S. Chen, P. T. Hammond and Y. Shao-Horn, ACS Nano, 2010, 4, 3889–3896 CrossRef CAS
- H. Zhang, G. P. Cao and Y. S. Yang, Energy Environ. Sci., 2009, 2, 932–943 CAS
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS
- Y. Hou, Y. W. Cheng, T. Hobson and J. Liu, Nano Lett., 2010, 10, 2727–2733 CrossRef CAS
- H. Zhang, G. P. Cao, Z. Y. Wang, Y. S. Yang, Z. J. Shi and Z. N. Gu, Electrochem. Solid-State Lett., 2008, 11, A223–A225 CrossRef CAS
- J. N. Dudney, Electrochem. Soc. Interface, 2008, 17, 44–48 Search PubMed
- D. Y. Kim, H. Sugime, K. Hasegawa, T. Osawa and S. Noda, Carbon, 2011, 49, 1972–1979 CrossRef CAS
- H. Ago, T. Kugler, F. Cacialli, W. R. Salaneck, M. S. P. Shaffer, A. H. Windle and R. H. Friend, J. Phys. Chem. B, 1999, 103, 8116–8121 CrossRef CAS
- C. Kozlowski and P. M. A. Sherwood, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 2745–2756 RSC
- A. Izadi-Najafabadi, S. Yasuda, K. Kobashi, T. Yamada, D. N. Futaba, H. Hatori, M. Yumura, S. Iijima and K. Hata, Adv. Mater., 2010, 22, E235–E241 CrossRef CAS
- Y. J. Lee, H. Yi, W. J. Kim, K. Kang, D. S. Yun, M. S. Strano, G. Ceder and A. M. Belcher, Science, 2009, 324, 1051–1055 CAS
- X. L. Wu, L. Y. Jiang, F. F. Cao, Y. G. Guo and L. J. Wan, Adv. Mater., 2009, 21, 2710–2714 CrossRef CAS
- K. S. Kang, Y. S. Meng, J. Breger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ee02409d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |