Electrochemistry for biofuel generation: Electrochemical conversion of levulinic acid to octane
Peter
Nilges
,
Tatiane R.
dos Santos
,
Falk
Harnisch
and
Uwe
Schröder
*
Institute of Environmental and Sustainable Chemistry, Technische Universität Braunschweig, Hagenring 30, 38106, Braunschweig, Germany. E-mail: uwe.schroeder@tu-bs.de; Fax: +49 (0) 531918424; Tel: +49 (0) 531918425
First published on 7th November 2011
Abstract
By means of a two-step electrochemical conversion of levulinic acid to octaneviavaleric acid we propose the use of electrochemistry for the production of renewable chemicals and biofuels. The reactions can be performed in water and at room temperature and thus fulfil the major criteria of green chemistry.
Broader contextTo assure future mobility, the search for efficient and sustainable routes for the production of fuels from biomass, i.e. biofuels, has not lost significance. On the other hand, electricity based on wind power and photovoltaics reaches an increasing market share. Their fluctuating nature calls for new technologies to convert excess electricity during overproduction into a storable form. Here we propose to combine both needs by using electrochemistry for the upgrading of biomass-based components into liquid biofuels. |
Despite the strong political and scientific efforts to develop and to establish the fundamentals for electromobility (a domain of electrochemistry) combustion engines will still, and possibly for decades, be the dominant technology on our streets.1 Thus, to assure mobility under the considerations of sustainability, the search for new and efficient routes to produce fuels from biomass, i.e. biofuels, has not lost significance. On the other hand, electricity based on wind power and photovoltaics has reached a significant market share. However, the fluctuating nature of these renewable energies calls for new technologies to convert the so far mainly unused electricity during overproduction into a storable form. An example is the current strong effort to convert wind energy into gaseous fuels like hydrogen or methanevia electrolysis2 (e.g., the Wind-to-Hydrogen project, by the US national Renewable Energy Laboratory,3 the Windgas project by Greenpeace, Germany4).
The two future requirements – the need for liquid biofuels and for biomass derived chemicals, and the need to convert electricity from renewable resourses into a storable form leads to the conclusion that electrochemical routes could also be used for the generation of liquid biofuels.
Levulinic acid, produced from ligno-cellulosic biomass is considered to be an important platform intermediate in the production of renewable chemicals and of liquid fuels for the transportation sector.5–7 Based on the transformation of levulinic acid, hydrocarbons as well as derivates of valeric acids like valeric esters (“valeric biofuels”) have been proposed as future transportation biofuels.8,9
The conversion of levulinic acid into hydrocarbons or into valeric esters is usually achieved via multi-step processes, a major reaction being the catalytic hydrogenation of levulinic acid. This reaction usually requires temperatures between 250–400 °C and high hydrogen pressures between 10–35 bar.9–11
In this communication we propose the use of electrochemistry for the production of biofuels – by demonstrating the electrochemical conversion of levulinic acid into n-octane using a two-step process. The involved reactions can be performed at room temperature and in aqueous solutions; a hydrogen atmosphere or strong oxidizing or reducing chemicals are not required. Furthermore, an occurring natural phase separation allows a simple separation of the final product, n-octane.
Fig. 1 illustrates the two principle reaction pathways for the electrochemical conversion of levulinic acid into n-octane. Fundamentally, the process consists of two types of reactions: an electrochemical reduction step and an oxidative electrochemical decarboxylation/dimerization (i.e., the Kolbe reaction).
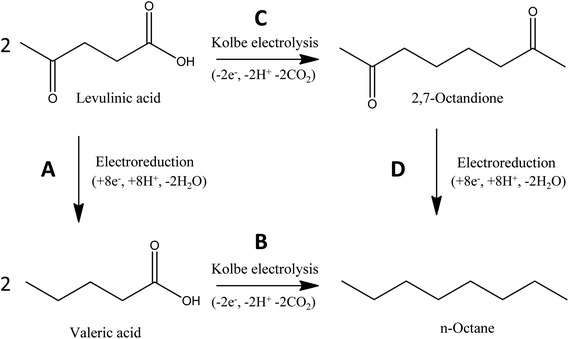 | ||
| Fig. 1 Schematic illustration of the two electrochemical routes for the conversion of levulinic acid to n-octane. | ||
As Fig. 1 illustrates, the electrochemical conversion of levulinic acid into n-octane can be achieved via two alternative directions:
(I) The electroreduction of levulinic acid to valeric acid (reaction A), followed by the Kolbe-coupling of valeric acid to n-octane (reaction B).
(II) The Kolbe reaction of levulinic acid to 2,7-octanedione (reaction C), followed by the electroreduction of 2,7-octanedione to n-octane (reaction D).
Quite remarkably, the fundamentals of this reaction scheme have been laid in the distant past: First of all, the Kolbe reaction is one of the oldest and best-known electro-organic reactions12–14 that goes back more than 160 years.15 What is more, the electroreduction of levulinic acid was described one hundred years ago,16,17 yet apparently never found a suitable application. Here we can demonstrate that these old reactions – and especially the combination of these reactions – provide a promising path for the future conversion of renewable compounds into biofuels or platform chemicals.
In this study, for the electrochemical reduction of ketones/keto groups (reactions A & D, Fig. 1) lead was chosen as electrode material,18 whereas that of the oxidative Kolbe reaction (reactions B & C) was platinum.14,19
Fig. 2 shows cyclic voltammograms of the electrochemical reduction of levulinic acid (Fig. 2A) and of the anodic decarboxylation/dimerization of valeric acid (Fig. 2B). In the voltammograms the on-set potentials are visible, at which the respective reduction and oxidation currents increase and hence the reduction and oxidation reactions commence. In electrochemical reactions the current flow is proportional (connected via the Faraday Law) to the amount of compound converted per time. Thus, for the example of the Kolbe reaction of valeric acid (reaction B) a current density of 50 mA cm−2 would correspond to a conversion rate of 190 mg cm−2 h−1 (1.9 kg m−2 h−1) valeric acid when assuming 100% Coulombic efficiency.
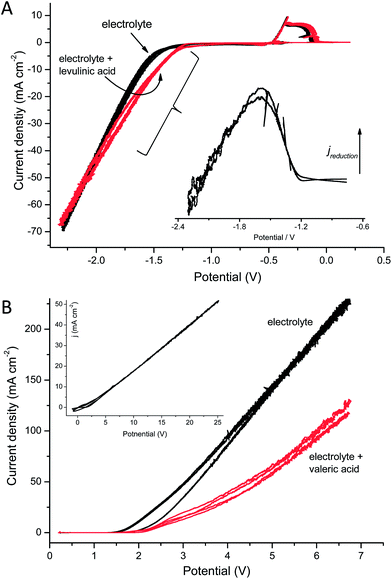 | ||
| Fig. 2 (A) Cyclic voltammograms for the electrochemical reduction of 100 mM levulinic acid in 500 mM sulphuric acid, recorded at a scan rate of 20 mV s−1. The inset Figure depicts the voltammogram corrected against the base current. (B) Main Figure: Cyclic voltammograms of the Kolbe reaction of 500 mM valeric acid in aqueous solution (the scan rate was 20 mV s−1); inset Figure: the Kolbe reaction of valeric acid in methanol (the scan rate was 250 mV s−1). The black curves denote the cyclic voltammograms recorded in the pure electrolyte solutions, the red curves were recorded in the presence of the respective educt. | ||
For the Kolbe reaction the on-set potential is about 2–2.5 V (vs.Ag/AgCl reference electrode), whereas that for the electrochemical reduction is approximately −1 V. In the experiments, the slope of the current (as a function of the potential) of the Kolbe reaction carried out in methanol, the classical solvent for this reaction, was much lower than that of reaction in aqueous solution as well as that of the electrochemical reduction (2 mA cm−2 V−1 for the Kolbe reaction in methanol and 60 mA cm−2 V−1 for the Kolbe reaction in water as well as for the electroreduction). Consequently, the overpotential that was necessary to achieve sufficiently high current densities and thus reaction rates was much higher for the Kolbe reaction in methanol than for all reduction and oxidation reactions that were performed in water.
Fig. 2B further illustrates a behaviour that may be peculiar for general electrochemistry but is typical for the Kolbe reaction: the currents obtained for the educt containing solution is considerably lower than that of the of the pure electrolyte solution. The reason for this is that presence of the organic acid suppresses the electrolyte decomposition (i.e., the oxygen evolution from water), the main reaction in the absence of the organic compounds. From the mechanistic side it is further interesting to note that for the reduction of the ketone no separate electrochemical processes become visible (see the net current curve, inset of Fig. 2A), which indicates that all four electrons are transferred at one potential.
Table 1 summarizes the major experimental results of this study. It illustrates that in principle both reaction paths, reactions A & B and reactions C & D, lead to the formation of n-octane from levulinic acid. Yet, the pathway A & B delivered the by far better results. The overall reaction selectivity as well as the Coulombic efficiency are superior to that of path C & D. This is in particular due to the excellent performance of the electrochemical reduction of levulinic acid to valeric acid (reaction A). When carried out using conventional chemical/catalytic routes, this transformation requires several steps and has to be performed at high temperature and hydrogen pressure.9 Using electrochemistry it is a simple reaction that proceeds with a selectivity of 97.2% (Table 1). Even in a single compartment cell practically no side reactions of levulinic acid take place.
| Solvent/electrolyte | E a/V | j/mA cm−2 | S b [%] | CEc [%] | |
|---|---|---|---|---|---|
| a Working electrode potential/vs.Ag/AgCl. b Selectivity of the product formation. c Coulombic efficiency, related to the educt conversion. For the reduction reaction a 4 electron reaction was assumed, for the oxidation a one electron step. | |||||
| A | Water/H2SO4 | −1.8 | 20–40 | 97.2 | 27.0 |
| B | MetOH/KOH | 10 | 15–25 | 50.2 | 69.0 |
| Water/K2CO3 | 3.5 | 40–50 | 51.6 | 66.5 | |
| C | MetOH/KOH | 5 | 3–5 | 47.0 | 86.2 |
| 10 | 3–5 | 37.5 | 92.9 | ||
| D | Water/H2SO4 | −1.8 | 20–40 | 27.5 | 11.2 |
Similar to this reduction reaction the Kolbe-coupling of the resulting valeric acid (reaction B) in methanol did not form any detectable liquid by-products. The absence of liquid by-products in combination with a reaction selectivity of 50% indicates the formation of gaseous butene by a non-Kolbe side reaction.19,20
The lower efficiency of reactions C & D is mainly due to the comparatively low selectivity and Coulombic efficiency of reaction D – the reduction of 2,7-octanedione to n-octane. In contrast to the quantitative and highly selective reduction of levulinic acid (reaction A), here GC-MS analysis revealed multiple side products. Even in a two-chamber system, side products like 1-methylcycloheptanol, 7-octen-2-ol, 1-cyclopentylethanol, 7-octen-2-one, 2-octanol, 2-octanone, 1-methylcycloheptanol, 1-methyl-2-methylenecyclohexane,1-ethyl-5-methylcyclopentene, 1-octene, 2-octene, 3-octene were detected. The major side product of the Kolbe reaction of levulinic acid (reaction D) was 4-hydroxy-2-butanone.
A special emphasis of this study was to choose experimental conditions obeying the rules of sustainable chemistry.21 For this reason we also studied the possibility to replace methanol as the usual solvent used for Kolbe reactions by an aqueous electrolyte solution.20,22
The replacement with a greener solvent had a number of positive side effects: First of all, the aqueous environment allowed us to perform the reaction at a considerably lower overpotential. Thus, an electrode potential of 3.5 V was sufficient to run the reaction at a current density of 50 mA cm−2, whereas in methanol at 10 V 25 mA cm−2 were not exceeded. In addition to the reduction of the oxidation potential, it is to be expected that due to the considerably better ionic conductivity of the aqueous environment the overall cell voltage of a respective electrolysis cell will considerably decrease.
A technologically very interesting side effect of the use of an aqueous reaction environment for the Kolbe-coupling is illustrated in Fig. 3. The decarboxylation/dimerization of the water soluble precursor valeric acid leads to the formation of the water insoluble organic product phase.
 | ||
| Fig. 3 Photographic image of the phase separation between the precursor containing aqueous electrolyte solution and the organic product phase (n-octane) produced during the Kolbe reaction of 1 M valeric acid in an aqueous potassium carbonate solution. | ||
The separation of the organic product from the aqueous reaction solution not only provides the opportunity to very easily isolate the product and to recycle the aqueous electrolyte solution, but may in future also allow the entire process to be performed in a continuously running process.
Beside octane, the organic product phase contained certain amounts of n-butyl valerate (see Fig. 4) and n-butanol. The amount of these side products was dependent on the starting concentration of valeric acid. Thus, from 500 mM valeric acid a mixture of 36% n-octane, 29% n-butyl valerate and 16% butanol was formed, whereas using 1 M valeric acid the composition was shifted towards 72% n-octane, 18% butylvalerate and 2% butanol.
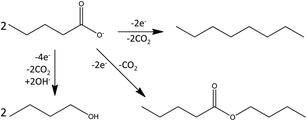 | ||
| Fig. 4 Competitive oxidative reactions of valeric acid, leading to the formation of n-octane, butylvalerate and butanol. | ||
Prospects
This study demonstrates the principle possibility of using electrochemical routes for the production of biofuels. The development of this new technology will require a multitude of further steps and tasks. Thus, to optimize the electrochemical process the overall selectivity and Coulombic efficiency has to be further improved. Three-dimensional electrodes have to be developed to increase the conversion rates at low reaction overpotentials. The impact of educt impurities that stem from the primary biomass conversion step e.g.,23 (e.g., the production of levulinic acid from cellulose) have to be studied, and electrochemical reactors have to be developed that allow an efficient conversion process at minimum capital and operational costs.A major task to be tackled in a forthcoming study will be the analysis of the energy efficiency of the electrochemical process in comparison to existing chemical/catalytic routes and in relation to the increase of energy density during the electrochemical upgrading process (thus, the energy density of levulinic acid is 20.9 MJ kg−1, whereas that of octane is 44.4 MJ kg−1). Since at the current stage all reactions were performed in half-cell experiments, an exact evaluation is not yet possible. The used electrode potentials may provide a first reference for the required theoretical energy input. From the electrode potentials and the number of transferred electrons the theoretical energy requirement of respective half-reactions can be determined. Thus, at 100% selectivity and Coulombic efficiency a reduction of one mole of levulinic acid to valeric acid at −1.8 V would require 695 kJ mol−1, whereas the Kolbe reaction of two moles of valeric acid to one mole of octane would consume 675 kJ mol−1 at 3.5 V and 1930 kJ mol−1 at 10 V. Consider that these energy values reflect only the half reaction and need to be paired with a suitable counter reaction.
A logical prospect of this work shall be to transform the so far separate batch reactions into a combined continuous electrochemical process, as illustrated in Fig. 5. Combining electrochemical reduction and oxidative Kolbe reaction in one electrochemical system will allow the electrochemical conversion process to run with optimized energy efficiency. For this, however, further work is required to unify the composition of the electrolytes for the anode and cathode reaction in order to avoid any intermediate product isolation steps and to avoid cross-over reactions.
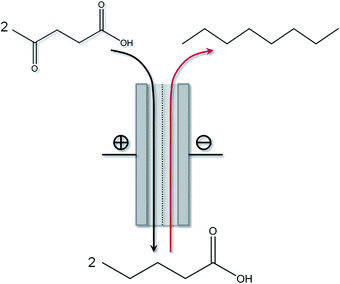 | ||
| Fig. 5 Schematic setup of a potential integrated electrochemical cell for the continuous consecutive conversion of levulinic acid into n-octane. | ||
Conclusions
This fundamental study demonstrates that electrochemistry represents a promising tool for the sustainable production of biofuels and chemicals. The proposed electrochemical approach fulfils major criteria of green chemistry:18 (i) it uses mild conditions and (ii) water as environmentally friendly solvent, (iii) electrons as immaterial agent replace the use of any oxidizing or reducing agents, and (iv) waste products are minimized.In the distant past – the 19th and the early 20th century – the fundamentals for numerous (electrochemical) processes and reactions have already been created. These reactions can now serve as the basis for the innovative development of future sustainable/green chemistry processes.
Finally, the use of electrochemistry for the upgrading of biofuels has a further advantage: Renewable energies like wind power and photovoltaics are characterized by a fluctuating electricity production. The increasing share of these energies calls for new technologies to convert the so far unused electricity during overproduction into a storable form. Here electrochemical synthesis and upgrading pathways, like the presented reactions, may play an important role.
Experimental
Materials
Levulinic acid (4-oxopentanoic acid, 98%, Sigma-Aldrich, Germany), valeric acid (pentanoic acid, 99%, Alfa Aesar GmbH & Co. KG, Germany), potassium carbonate (K2CO3, 99%, Grüssing, Germany), potassium hydroxide (KOH, ≥99.5%, Roth), methanol (≥99.8%, Sigma Aldrich) were used as purchased, without further purification. 2,7-Octandione was produced by Kolbe-electrolysis and purified by re-crystallization in heptane and confirmed by in-house NMR (1H and 13C) measurements. N-Octane (99%, Sigma–Aldrich, Germany), 2-octanone (98%, Alfa Aesar GmbH & Co. KG, Germany), (±)-2-octanol (≥99.5%, Sigma-Aldrich, Germany), levulinic acid, valeric acid and 2,7-octanedione were used for the preparation of reference sample for GC and HPLC calibration curves. Deionized water was used for the preparation of aqueous solutions.Electrochemical procedures
All electrochemical reactions were conducted under potentiostatic control using an AMEL 7050 potentiostat (Amel srl, Milano, Italy) or a SP50 potentiostat (Bio-Logic SAS, Claix, France). Ag/AgCl sat. KCl reference electrodes (Sensortechnik Meinsberg, Germany, 0.195 V vs.SHE) were used throughout the study. All potentials provided in this article refer to this reference electrode. As electrochemical cells either one chamber cells (50 mL–100 mL two-necked flasks) or two-chamber H-type electrochemical glass cell were used. The latter consisted of a 30 mL anodic chamber and 30 mL for the cathodic chamber separated from each other via a cation exchange membrane (Fumasep®FKE, Fumatech, St. Ingbert, Germany). The distance between the working and counter electrode was 61 mm. All experiments were performed at least as duplicates, usually as triplicates.For the reduction reactions lead rods (8 mm diameter, 99.9%, PHYWE Systeme GmbH & Co. KG, Göttingen, Germany) with a projected surface area of 11.60 cm2 served as working electrodes. Here, the counter electrode was a platinum sheet (99.9%, chemPUR, Karlsruhe, Germany) with a projected surface area of 5.65 cm2. To assure a constant reaction temperature the reduction experiments were run in an ice-bath at about 8 °C. Evolving gas was passed through an ether solution, also held at about 8 °C. The anodic Kolbe reactions were conducted in a one chamber glass cell (100 mL volume) equipped with a reflux condenser. The anode and cathode were platinum sheets (99.9%, chemPUR, Karlsruhe, Germany) with projected surface areas of 5.65 cm2. The distance between both electrodes was 15 mm. For constant temperature the experiments were run in a water-bath at about 18 °C.
For the reduction reactions 100 mM of the precursor (either levulinic or valeric acid) was applied in 500 mM aqueous sulfuric acid. The duration of the electrolysis varied between 4 h (reduction of levulinic acid, reaction A, Fig. 1) and 23 h (reduction of octanedione, reaction D, Fig. 1). The reaction time in all cases is mainly governed by the ratio of electrode size to the volume of the educt solution. Thus, appropriate shortening of the reaction time can be achieved by using larger electrodes.
For the Kolbe reaction in methanol the respective organic acids (levulinic acid, valeric acid) were used in concentrations of 500 mM, dissolved in 100 mL methanol. KOH was added until the pH of about 5.5 was reached. For the conduction of the Kolbe reaction in water, 500 mM or 1 M of the valeric acid was dissolved in water and neutralized with potassium carbonate to reach a pH of 5.5.
All experiments were run under stirring using magnetic stirrers.
Analysis
Qualitative and quantitative analysis was carried out using GC/MS (Trace GC Ultra, DSQ II, Thermo Scientific, Germany) equipped with a TR-WaxMS (30 m × 0.25 mm ID × 0.25 μm film GC column, Thermo Scientific, Germany) or TR-5MS (30 m × 0.25 mm ID × 0.25 μm film GC column Thermo Scientific, Germany). External calibration curves with different concentration levels in a range from 0 to 600 ng μL−1 were used for the quantification.Quantitative Analysis was also carried out by GC/FID (Hewlett Packard Series II 5890, Hewlett Packard, United States of America) equipped with a DB-5 (30 m × 0.25 mm ID × 0.25 μm film GC column from Agilent JW Scientific, United States of America). External calibration curves with different concentration levels in a range from 0 to 600 ng μL−1 were used for the quantification.
A HPLC system (Spectrasystem P4000, Finnigan Surveyor RI Plus Detector, Fisher Scientific, Germany) equipped with a HyperREZ XP Carbohydrate H+ 8 μm (S/N: 026/H/012-227) column was used for routine substance quantification (in the aqueous phase). Sulfuric acid (0.005 N, flow rate 0.5 mL min−1) served as the eluent. The column was kept at room temperature; the refractory index detector was set at 40 °C during measurements. Educt and product concentrations were determined using calibration curves in a range from 0 to 200 mmol L−1 that were obtained from reference samples.
References
- M. Contestabile, G. J. Offer, R. Slade, F. Jaeger and M. Thoennes, Energy Environ. Sci., 2011, 4, 3754, 10.1039/C1EE01804C.
- E. Troncoso and M. Newborough, Int. J. Hydrogen Energy, 2011, 36, 120–134 CrossRef CAS.
- http://www.nrel.gov/docs/fy09osti/44082.pdf .
- http://www.greenpeace-energy.de/windgas.html .
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 CAS.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef.
- R. Leonard, Ind. Eng. Chem., 1956, 48, 1330–1341 CrossRef.
- D. J. Braden, C. A. Henao, J. Heltzel, C. C. Maravelias and J. A. Dumesic, Green Chem., 2011, 13, 1755–1765 RSC.
- J.-P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479–4483 CrossRef CAS.
- J. C. Serrano-Ruiz, D. Wang and J. A. Dumesic, Green Chem., 2010, 12, 574–577 RSC.
- Y. Gong, L. Lin and Z. Yan, Bio-Research, 2011, 6, 686–699 CAS.
- H. J. Schäfer, Top. Curr. Chem., 1990, 152, 91–151 CrossRef.
- B. E. Conway and A. K. Vijh, J. Anal. Chem., 1966, 224, 149–159 Search PubMed.
- H. J. Schäfer, Chem. Phys. Lipids, 1979, 24, 321–333 CrossRef.
- H. Kolbe, Justus Liebigs Ann. Chem., 1849, 69, 257–294 CrossRef.
- J. Tafel and B. Emmert, Z. Elektrochem., 1911, 17, 569–572 Search PubMed.
- S. Swann, Trans. Electrochem. Soc., 1932, 62, 177–182 CrossRef.
- J. Tafel, Z. Elektrochem., 1912, 17, 569 CAS.
- E. Klocke, A. Matzeit, M. Gockeln and H. J. Schäfer, Chem. Ber., 1993, 126, 1623–1630 CrossRef CAS.
- P. F. Levy, J. E. Sanderson and L. K. Cheng, J. Electrochem. Soc., 1984, 131, 773–777 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Praxis, Oxford University Press, New York, 1998 Search PubMed.
- G. S. Pande and S. N. Shukla, Electrochim. Acta, 1961, 4, 215–231 CrossRef CAS.
- M. Möller
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) , P. Nilges, F. Harnisch and U. Schröder, ChemSusChem, 2011, 4, 566–579 CrossRef.
, P. Nilges, F. Harnisch and U. Schröder, ChemSusChem, 2011, 4, 566–579 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |