Revealing pyrolysis chemistry for biofuels production: Conversion of cellulose to furans and small oxygenates†
Matthew S.
Mettler
ab,
Samir H.
Mushrif
a,
Alex D.
Paulsen
b,
Ashay D.
Javadekar
a,
Dionisios G.
Vlachos
a and
Paul J.
Dauenhauer
*b
aDepartment of Chemical Engineering and Catalysis Center for Energy Innovation, University of Delaware, 150 Academy Street, Newark, DE 19716, USA
bDepartment of Chemical Engineering and Catalysis Center for Energy Innovation, University of Massachusetts, 686 North Pleasant Street, Amherst, MA 01003, USA. E-mail: dauenhauer@ecs.umass.edu
First published on 21st November 2011
Abstract
Biomass pyrolysis utilizes high temperatures to produce an economically renewable intermediate (pyrolysis oil) that can be integrated with the existing petroleum infrastructure to produce biofuels. The initial chemical reactions in pyrolysis convert solid biopolymers, such as cellulose (up to 60% of biomass), to a short-lived (less than 0.1 s) liquid phase, which subsequently reacts to produce volatile products. In this work, we develop a novel thin-film pyrolysis technique to overcome typical experimental limitations in biopolymer pyrolysis and identify α-cyclodextrin as an appropriate small-molecule surrogate of cellulose. Ab initio molecular dynamics simulations are performed with this surrogate to reveal the long-debated pathways of cellulose pyrolysis and indicate homolytic cleavage of glycosidic linkages and furan formation directly from cellulose without any small-molecule (e.g., glucose) intermediates. Our strategy combines novel experiments and first-principles simulations to allow detailed chemical mechanisms to be constructed for biomass pyrolysis and enable the optimization of next-generation biorefineries.
Broader contextThermochemical conversion of lignocellulosic biomass provides a sustainable process for producing carbon-based liquid biofuels or biopower. Pyrolysis, gasification, and combustion utilize high temperatures to produce vapors or gases such as bio-oils or carbon monoxide which can be subsequently upgraded to green gasoline or synthetic diesel. The development of safe and efficient reactors, while critical to the overall success of thermochemical biomass conversion platforms, is hindered by a lack of molecular-level understanding of condensed-phase pyrolysis chemistry. The extreme size of polymers within biomass (such as cellulose) prevents direct simulation using first-principles simulations. Additionally, existing experimental techniques have been incapable of studying the chemistry of condensed phase cellulose without transport limitations. We describe a new experimental technique referred to as thin-film pyrolysis that can examine the reactions of cellulose under isothermal conditions without transport limitations. Thin-film pyrolysis is then used to identify α-cyclodextrin as a small carbohydrate surrogate of cellulose. This discovery enables first-principles simulation techniques to be used to identify condensed-phase pyrolysis chemistry by reducing the required simulation time from decades to weeks allowing for the condensed-phase cellulose pyrolysis pathways to be revealed for the first time. These mechanisms can serve as the first step toward developing detailed chemical models which can be used to design the next generation of biomass conversion reactors. |
1. Introduction
Thermochemical biomass conversion processes, such as pyrolysis, operate at high temperatures (400–800 °C) and break down large biopolymers (20,000 to 400,000 a.m.u.) into smaller molecules (less than 200 a.m.u.) with higher energy content and improved transportability.1,2 While existing pyrolysis technologies may be economical,1 their widespread commercialization depends on accurate process design and optimization which in turn require detailed understanding of the underlying chemistry. As shown in Fig. 1, the transformation of biomass is initiated with a network of largely unidentified solid-phase reactions which fractionate and depolymerize the solid to a short-lived intermediate liquid.2 Within the liquid-phase, a network of unknown depolymerization, rearrangement, and dehydration reactions occur until products volatilize, repolymerize to form char or are ejected in the form of aerosols.3 Vapor products are condensed to form bio-oil which can be upgraded catalytically to renewable liquid fuels or chemicals.4 Understanding the condensed-phase chemistry is critical to the widespread commercialization of pyrolysis, since the former determines the quality of the bio-oil intermediate and therefore the overall economics of the process.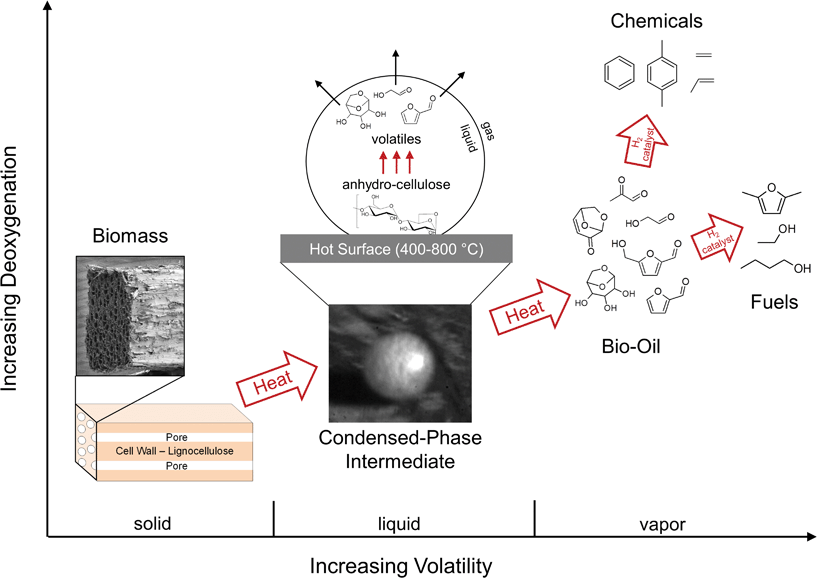 | ||
| Fig. 1 Biomass pyrolysis process. Biomass pyrolysis is comprised of thousands of solid, liquid, gas and catalytic reactions which deoxygenate and volatilize lignocellulosic material to generate liquid fuels and chemicals. The microstructure of poplar wood, an abundant form of biomass, is illustrated in the electron micrograph (far left image). The initial thermal decomposition of solid biomass generates a short-lived liquid phase, which is shown here as a molten droplet (200 microns in diameter, center image) produced during cellulose pyrolysis on an iron (Fecralloy) surface (650 °C). Further heating of the liquid intermediate produces a bio-oil intermediate which can be catalytically upgraded to fuels and chemicals. | ||
The evolution of volatiles from cellulose is a complex process. Consequently, the entire transformation of cellulose to bio-oil has so far been described with few overall reactions5,6 that, due to their lumped nature, fall short of describing varying feedstocks and pyrolysis conditions. Thermochemical cellulose conversion involves thousands of elementary reactions across three phases which generate hundreds of volatile species. While the product distribution varies substantially with reaction conditions, the most abundant pyrolysis products are levoglucosan, furans and light oxygenates (such as glycolaldehyde and formic acid). Detailed information on how these species form from cellulose, while critical to optimizing pyrolysis processes, is largely unavailable in the literature. A heterolytic depolymerization mechanism has been postulated for levoglucosan formation from cellulose7 based on low temperature, aqueous-phase acid hydrolysis chemistry; however, its relevance to high-temperature pyrolysis has not been established. While the mechanism of levoglucosan formation from cellulose involves only a few reactions (glycosidic bond cleavage and anhydro-bridge formation), furan and light oxygenate production is less straightforward. Previous researchers have speculated that glycolaldehyde and furan production proceeds through an anhydrosugar (e.g., levoglucosan) intermediate,8,9 but no fundamental chemical mechanisms have been experimentally or computationally confirmed to the best of our knowledge. It is also logical that glucose (the monomer of cellulose) is an intermediate in the conversion of cellulose to furans as observed in low temperature (100–200 °C) aqueous-phase processes.10–12
The lack of mechanistic understanding of pyrolysis chemistry is a product of the complex reaction environment. Experimental insight is hindered by a number of technical difficulties including: the substantial functionality (oxygen-rich) of biomass starting materials, intermediates and products; the temperature sensitivity of many products;7,13 the short lifetime (less than 0.1 s) of condensed-phase intermediates;2 the relatively slow heat transfer which makes isothermal pyrolysis challenging; and the dependence of product yields on the residence time of volatiles within the liquid-phase (which is often controlled by mass transfer). Overcoming all of these difficulties simultaneously requires new, advanced experimental techniques specifically tailored for biomass pyrolysis.
To gain insight into the fundamental chemistry of cellulose pyrolysis, we introduce a new thin-film pyrolysis technique that overcomes the above experimental challenges. Thin-film pyrolysis experiments identify a small molecule (α-cyclodextrin) as a surrogate for cellulose that is studied using ab initio molecular dynamics. These simulations then identify for the first time the condensed-phase pyrolysis pathways through which α-cyclodextrin (and cellulose) reacts to form major volatile products (bio-oil components) and shed light into the long standing debate of the mechanism of cellulose breakdown.
2. Methods
The pyrolysis of cellulose and carbohydrates was examined using a novel thin-film pyrolysis technique as well as conventional powder pyrolysis and thermogravimetric analysis. The experimental results were then used to direct ab initio (Car-Parrinello) molecular dynamics simulations to reveal the reaction mechanisms of cellulose pyrolysis.2.1 Design of thin-film pyrolysis experiments
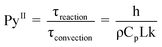
The development of the thin-film pyrolysis technique was motivated by a need for isothermal and well-mixed condensed phase conditions at high temperature (>400 °C). By utilizing a micrometer-scale thin-film, volatile products diffuse through the intermediate liquid in less than one millisecond, or 2–4 orders of magnitude faster than in powder or particle pyrolysis (see supplementary information for details†). In addition to removing unstable volatiles quickly, thin-film pyrolysis also generates extremely rapid temperature increases (greater than 1,000,000 °C min−1) due to the microscale dimensions of the sample. This mega-scale temperature ramp rate is 3–5 orders of magnitude faster than traditional pyrolysis techniques, such as thermogravimetric analysis (typically 1–150 °C min−1) and powder pyrolysis (approximately 1000 °C min−1).8 Rapid heating in thin-film pyrolysis allows for isothermal reaction conditions where overall biomass conversion is limited by chemical kinetics rather than heat transfer. The criteria for isothermal pyrolysis are defined by the reaction map shown in Fig. 2A compares conductive and convective heat transfer with reaction kinetics using the dimensionless Pyrolysis (Py) and Biot (Bi) numbers. PyI is the ratio of reaction (τreaction) and conduction (τconduction) times, PyII compares of reaction (τreaction) and convection (τconvection) time scales, and Bi relates conduction (τconduction) and convection (τconvection) times.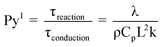 | (1) |
 | (2) |
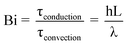 | (3) |
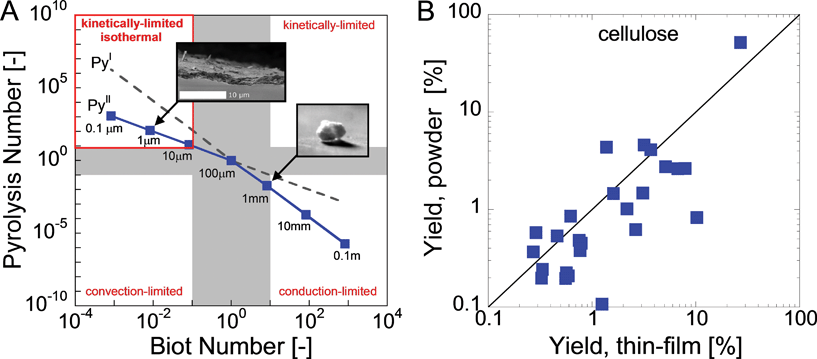 | ||
| Fig. 2 Thin-film pyrolysis design. Reaction map for cellulose fast pyrolysis (A) and comparison of powder and thin-film pyrolysis (B). Pyrolysis and Biot Numbers (eqn (1)–(3)) are used to compare heat transfer and reaction time scales; four pyrolysis regimes are identified (clockwise from top left): isothermal and kinetically-limited, kinetically-limited, conduction-limited and convection-limited. The top inset shows a SEM image of a cellulose thin-film while the bottom is a photograph of a reacting cellulose particle.2 | ||
2.2 Thin-film preparation and product identification
Thin-films were prepared by dissolving or suspending microcrystalline cellulose (Alfa Aesar), glucose (Sigma-Aldrich), α-cyclodextrin (Sigma-Aldrich) or cellohexaose (from SeikagakuBioBusiness) powders in water. Dilute (approximately 1%wt) mixtures or suspensions were transferred into a 4 mm × 8 mm (diameter × height) cylindrical pyrolysis crucible. Water was then removed via room temperature evacuation. Once the water was removed, a micrometer-scale thin-film was created on the inner wall of the crucible, as indicated by Scanning Electron Microscopy (SEM). The thin-film samples were pyrolyzed using a Frontier 2020 micropyrolyzer, and volatile products were analyzed using an Agilent 7890A Gas Chromatograph Mass Spectrometer. Multidimensional chromatography was used to characterize both volatiles and permanent gases, and the system was equipped with an oxygen injection valve to quantify char via burn-off. In general, products were confirmed by comparing analyte retention time to pure standards. For three of the products, pure samples were unavailable and identification was made using only mass spectrometry. In these cases, calibrations were performed using a structurally similar molecule with the same number of carbons. Starting materials were characterized using liquid chromatography (except cellulose) and in general were found to contain relatively little (<5%) non-volatile impurities. Volatile content (principally water) was quantified using a thermogravimetric analyzer (TGA).Thin-film experiments were repeated three times and average values are shown in the figures of the main paper. Generally, the standard deviation for a particular pyrolysis product was 5–10% of the yield value. Overall carbon balances (including permanent gases, char and volatiles) for thin-film pyrolysis of glucose, cellohexaose, cellulose and α-cyclodextrin, closed to 77%, 95%, 98% and 88%, respectively. For complete characterization details of starting materials and volatile products, see the supplementary information.†SEM imaging indicates our cellulose sample (from Alfa Aesar) is comprised of fibrous sheets with dimensions of approximately 20 × 10 × 1 μm3 (length × height × thickness). Additionally, viscosity tests (conducted by Doble Engineering Company) determined our cellulose sample has an average degree of polymerization of 133.
2.3 Car-Parrinello molecular dynamics (CPMD) simulations
Ab initio molecular dynamics simulations of α-cyclodextrin (ACD) were performed in a simulation cell of 14 × 14 × 9 Å3, with periodic boundary conditions to mimic the condensed phase environment. Experimental pyrolysis temperature ranges from 400–700 °C with millisecond reaction time scales. However, even with the state of the art computational resources, it is not possible to run ab initio molecular dynamics simulations of ACD for more than a few picoseconds. In order to explore the mechanism of fast pyrolysis reactions of ACD (where multiple products are formed from the decomposition of a single molecule in a large number of parallel reactions) without a priori knowledge of any of the pathways or use of biased reaction coordinates, the simulations were performed at higher temperatures (1700–2700 °C). Running simulations at these temperatures accelerates the reactions from the millisecond to the picosecond scale. Given the small simulation size, it is not possible to obtain the entire range of products observed experimentally. Hence, in an attempt to explore multiple pathways leading to multiple products (from a single starting reactant), simulations were conducted at different temperatures. Our methodology can provide useful insights into the chemical pathways for most major products. Prediction of the entire spectrum of products is beyond the scope of this work due to the limited number of reacting monomers and the short time scales studied (both arising from the extremely high computational cost).Simulations were performed using the CPMD package 3.13.2, which provides an implementation of the first-principles Car-Parrinello molecular dynamics scheme.17 The first-principles calculations were performed using the planewave-pseudopotential implementation of the Kohn–Sham formulation of density-functional theory. The Troullier–Martins pseudopotentials with the Perdew–Burke–Ernzerhof generalized gradient approximation, which have been validated for carbohydrates,18 were used. Only the Γ-point was used for integration over the Brillouin zone in reciprocal space. The planewave energy cut-off for the pseudopotential was determined from the variation of the energy of the system with an energy cut-off. A cut-off of 100 Ryd produced a converged energy and was used thereafter. Temperature control was achieved using the Nosé-Hoover thermostat. The frequency for the ionic thermostat was 1800 cm−1 (characteristic of a C–C bond vibration frequency) and that for the electron thermostat was 10000 cm−1. The fictitious electron mass was taken as 600 a.m.u. Short MD runs were performed without the thermostat to obtain an approximate value around which the electronic kinetic energy oscillates. The MD time step used in the simulation was 0.0964 femtoseconds. The fictitious electronic kinetic energy oscillates around the chosen mean value, confirming that the electrons do not “heat up” in the presence of the “hot” nuclei and the system remains in the Born–Oppenheimer ground state.
3. Results and discussion
Isothermal thin-film pyrolysis experiments are first discussed followed by the identification of a small-molecule surrogate (α-cyclodextrin) that closely follows the condensed-phase cellulose pyrolysis chemistry. The results of CPMD simulations are then presented.3.1 Thin-film pyrolysis of cellulose
Scanning electron microscopy indicates our thin-film is several micrometers thick (Fig. 2A, inset), which is well below the isothermal limit of 20 μm. Additionally, Fig. 2B indicates that this length scale is a critical parameter since cellulose thin-film and cellulose powder pyrolysis experiments generate very different product yields. This disagreement is especially pronounced for major products, such as levoglucosan (27% vs. 48%) and glycolaldehyde (7.9% vs. 1.9%). The disparity results from the reaction-limited nature of thin-film pyrolysis (no heat or mass transport limitations) compared to powder pyrolysis where temperature and concentration gradients develop within the biomass sample thereby altering condensed-phase chemistry and enabling product breakdown within the intermediate liquid. Of note is that even though powder pyrolysis experiments utilized sufficiently small cellulose particles (approximately 10 μm), the characteristic transport length-scale is approximately one millimeter (the size of the packed bed reaction chamber), which is well above the 20 μm isothermal limit.In both powder and thin-film pyrolysis experiments, 27 products (including char) were identified and are summarized in Table 1. Compounds were identified through gas chromatography retention time analysis, mass spectroscopy and with comparison to products reported in previous work.8,16,19–33Table 1 shows that three products were identified through mass spectrometry only since they are not commercially available. Of these three, two had been identified in previous work: 1,6-anhydroglucopyranose (AGP) and 1,4;3,6-dianhydro-α-D-glucopyranose (DHGP).29,34 These compounds were easily confirmed with our mass spectrometer. The third compound identified using Mass Spectrometry was 1,2-cyclopentanedione (see supplementary information† for details on MS-Only Identification methods).
| Compound | Cellulose powder yield [%C] | Cellulose thin film yield [%C] | α-Cyclodextrin thin film yield [%C] | Glucose thin film yield [%C] | Cellohexaose thin film yield [%C] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Confirmed by mass spectra only; pure standard unavailable. b Confirmed by retention time analysis and mass spectrometry. c Confirmed previously by Shanks and co-workers.28,29 d Confirmed previously by Shafizadeh and co-workers.34 e Confirmed previously by Silveston and co-worker.27 f Confirmed previously by Piskorz and co-workers.26 g Confirmed previously by Torri and co-workers.32 Abbreviations: ADGH - 1,5-anhydro-4-deoxy-D-glycero-hex-1-en-3-ulose, CPHM -2-cyclopenten-1-one,2-hydroxy-3-methyl, DHGP -1,4;3,6-dianhydro-α-D-glucopyranose. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Charbc | 9 ± - | 12 ± - | 14 ± - | 28 ± - | 31 ± - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Levoglucosan bcd | 48 ± 4 | 27 ± 2 | 24 ± 1 | 3.0 ± 1 | 17 ± 0.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hydroxymethylfurfural bcd | 3.9 ± 0.5 | 3.7 ± 0.1 | 3.6 ± 0.2 | 12 ± 1 | 11 ± 0.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Glycoaldehyde bc | 1.9 ± 0.6 | 7.9 ± 0.4 | 8.3 ± 0.6 | 6.0 ± 0.6 | 5.5 ± 0.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Methylglyoxal bg | 2.0 ± 0.7 | 6.7 ± 0.3 | 6.9 ± 0.5 | 5.9 ± 2 | 2.2 ± 0.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Formic acid bcf | 2 ± 1 | 10 ± 2 | 2.1 ± 0.2 | 2.8 ± 2 | 1.5 ± 0.6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ADGH ad | 3.8 ± 0.6 | 3.2 ± 0.1 | 5.2 ± 0.3 | 0.4 ± 0.06 | 2.1 ± 0.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1,6 Anhydroglucofuranoseacd | 4.0 ± 0.3 | 1.4 ± 0.4 | 1.5 ± 0.03 | 2.6 ± 0.4 | 1.6 ± 0.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Carbon dioxide bcf | 2.0 ± 0.6 | 3.4 ± 0.2 | 2.9 ± 0.4 | 2.8 ± 0.2 | 3.7 ± 0.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Furfuralbce | 1.6 ± 0.1 | 1.6 ± 0.2 | 1.2 ± 0.1 | 3.7 ± 0.4 | 2.4 ± 0.06 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Carbon monoxide bcf | 1.4 ± 0.1 | 3.1 ± 0.2 | 2.4 ± 0.2 | 1.6 ± 0.1 | 2.3 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2-Furanmethanol bc | 0.4 ± 0.4 | 0.6 ± 0.06 | 0.7 ± 0.1 | 0.8 ± 0.2 | 1.6 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Formaldehyde bfg | 4.4 ± 0.9 | 2.6 ± 0.2 | 2.1 ± 0.1 | 0.3 ± 0.02 | 3.0 ± 0.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Glyoxal bfg | 0.3 ± 0.1 | 1.2 ± 0.04 | 0.9 ± 0.1 | 0.3 ± 0.06 | 0.4 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Acetic acid bcefg | 0.27 ± 0.04 | 0.6 ± 0.06 | 0.5 ± 0.2 | 0.7 ± 0.06 | 0.4 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hydroxyacetone bcfg | 0.54 ± 0.08 | 2.6 ± 0.3 | 2.4 ± 0.1 | 0.6 ± 0.3 | 0.9 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2,5 Dimethyl furanbe | 0.34 ± 0.09 | 0.8 ± 0.1 | 0.5 ± 0.1 | 0.5 ± 0.06 | 0.5 ± 0.06 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2,3 Butanedionebf | 0.37 ± 0.02 | 0.8 ± 0.06 | 0.8 ± 0.1 | 0.4 ± 0.1 | 0.5 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5-Methyl furfuralbc | 0.48 ± 0.4 | 0.7 ± 0.2 | 0.6 ± 0.2 | 0.7 ± 0.3 | 0.9 ± 0.05 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DHGP acd | 0.98 ± 0.09 | 2.2 ± 0.06 | 2.7 ± 0.2 | 0.4 ± 0.1 | 2.0 ± 0.06 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Levoglucosenone bc | 0.3 ± 0.2 | 0.5 ± 0.06 | 0.4 ± 0.06 | 0.1 ± 0.05 | 0.2 ± 0.04 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Catechol b | 0.25 ± 0.04 | 0.3 ± 0.03 | 0.7 ± 0.4 | 0.1 ± 0.1 | 0.2 ± 0.03 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2(5H) Furanoneb | 0.20 ± 0.01 | 0.6 ± 0.05 | 0.5 ± 0.06 | 0.3 ± 0.06 | 0.5 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1,2-Cyclopentanedione a | 0.20 ± 0.01 | 0.6 ± 0.05 | 0.6 ± 0.1 | 0.2 ± 0.02 | 0.5 ± 0.02 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Furan bce | 0.65 ± 0.06 | 0.3 ± 0.03 | 0.1 ± 0.02 | 0.3 ± 0.06 | 0.3 ± 0.05 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2-Methyl furan bce | 0.20 ± 0.04 | 0.3 ± 0.02 | 0.2 ± 0.01 | 0.5 ± 0.06 | 0.5 ± 0.03 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CPHM bc | 0.2 ± 0.1 | 0.3 ± 0.03 | 0.2 ± 0.02 | 0.1 ± 0.01 | 0.2 ± 0.005 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total | 86 ± 3 | 95 ± 3 | 86 ± 2 | 75 ± 6 | 93 ± 3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
One product which was identified without a strong mass spectrometry match or retention time comparison is 1,5-anhydro-4-deoxy-D-glycero-hex-1-en-3-ulose (ADGH). While the mass spectrum for this compound is not available, the largest ion detected was 144, revealing the molecular weight of the compound. Shafizadeh and co-workers identified ADGH as a major product from cellulose pyrolysis using a combination of techniques (H-NMR, IR spectrometry, UV spectrometry, and mass spectrometry) and reported a mass spectrum for the compound34 that matches ours, confirming ADGH as a major product in our experiments.
3.2 Identification of a small-molecule cellulose surrogate
Next we search for a low molecular weight species which generates pyrolysis products in yields similar to cellulose. Identification of a small-molecule cellulose surrogate is critical to understanding condensed-phase chemistry since it allows for additional experiments (e.g., isotope labeling) and first-principles simulations which can reveal the underlying reaction pathways. Implementation of advanced ab initio molecular modeling methods, such as CPMD, is only possible for small molecules and short reaction time scales. The required computational time for first-principles molecular dynamics simulations of a typical cellulose polymer is on the order of years even when hundreds of processors are used in parallel. This extremely high computational time makes direct simulation of cellulose using ab initio methods all but impossible. Smaller molecules such as glucose or α-cyclodextrin require only days or weeks to simulate. This time scale is fully realizable and therefore identifying a cellulose surrogate enables first-principles simulations to facilitate identification of condensed-phase pyrolytic chemistry. To identify a cellulose surrogate, we use thin-film pyrolysis experiments to test several small-molecule carbohydrates.We find that most intuitive molecules poorly represent cellulose. Fig. 3A–B compares the thin-film pyrolysis products of glucose (A) and cellohexaose (B) to cellulose. Consistent with previous conventional pyrolysis experiments,29 we find that glucose and cellulose generate very different product distributions; glucose produces more furans, char and light oxygenates, whereas cellulose gives a higher levoglucosan yield (Fig. 3A). Cellohexaose, comprised of six β-linked glucose monomers, has the highest degree of polymerization of commercially-available cellodextrins but is still two orders of magnitude smaller than cellulose. Product yields for cellohexaose and cellulose (Fig. 3B) are in closer agreement than those for glucose and cellulose, but significant differences are still present in major products, such as levoglucosan, glycolaldehyde and many furanosic products (e.g., 5-hydroxymethylfurfural and furfural).
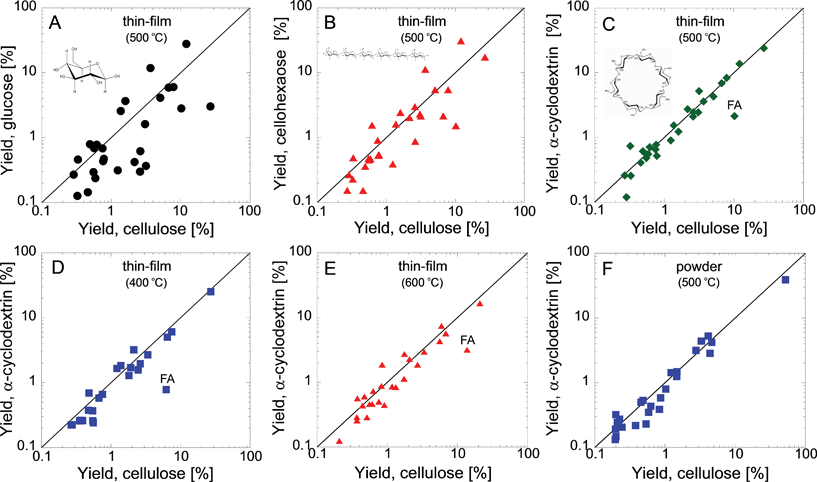 | ||
| Fig. 3 Identification of α-cyclodextrin as a cellulose surrogate. Comparison of pyrolysis product yields for (A) glucose and cellulose (thin-film, 500 °C); (B) cellohexaose and cellulose (thin-film, 500 °C); (C–E) α-cyclodextrin and cellulose thin-films at 500 (C), 400 (D) and 600 °C (E); (F) α-cyclodextrin and cellulose powders at 500 °C. Product yield is shown as percent of initial carbon for 27 identified products and char. FA indicates formic acid. | ||
We find that cyclodextrins match the pyrolysis product distribution of cellulose. These cyclic polymers are comprised of six, seven, or eight α-linked glucose monomers and are similar to cellulose in terms of end group-to-monomer ratio (cyclodextrins: 0%, cellulose: 0.01–2%). Fig. 3C shows that α-cyclodextrin yields agree well with those of cellulose for nearly all major products (with the exception of formic acid). Results for β-, and γ-cyclodextrin are presented in the supplementary information.†Fig. 3D–E shows that this agreement holds for 400 and 600 °C, i.e., at temperatures typical for pyrolysis reactors. In addition to thin-film pyrolysis, products yields are similar between cellulose and α-cyclodextrin for both powder pyrolysis (Fig. 3F) and slow pyrolysis (i.e., thermogravimetric analysis; see supplementary information for details†). This indicates that α-cyclodextrin can be a surrogate for reactor design where transport limitations exist.
One important difference between α-cyclodextrin and cellulose is the linkage type. Glucose monomers that make up cellulose are connected by equatorial ether bridges (β-linkage) while α-cyclodextrin is constructed from axial ones (α-linkage). To evaluate the effect of linkage type on pyrolysis products, we compare two sets of maltodextrins (α-linked) and cellodextrins (β-linked). Fig. 4A shows pyrolysis product yield for maltobiose and cellobiose at 500 °C, whereas Fig. 4B compares maltohexaose and cellohexaose. Both comparisons show, consistent with previous work,29 a negligible effect of glycosidic linkage type on product yield. This information indicates that end-group-to-monomer ratio, which is very different between glucose (100%), cellohexaose (33%) and cellulose (0.01–2%) but similar between α-cyclodextrin (0%) and cellulose, is a vital descriptor of cellulose pyrolysis chemistry. Based on this finding and the demonstrated similarities in product yields, α-cyclodextrin is found to the appropriate surrogate for cellulose and is employed in ab initio simulations to predict condensed phase reaction pathways.
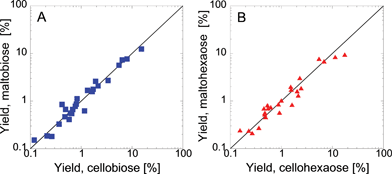 | ||
| Fig. 4 Role of glycosidic linkage. The effect of glycosidic linkage is shown by comparing α-linked (maltodextrins) and β-linked (cellodextrins) glucose polymers. Pyrolysis products are used to compare maltobiose and cellobiose (A) as well as maltohexaose and cellohexaose (B). The reaction temperature was 500 °C. | ||
3.3 Revealing cellulose pyrolysis chemistry using first-principles simulations
CPMD 17 simulations of α-cyclodextrin were performed to identify for the first time how major volatile products form from cellulose. Fig. 5 shows the mechanism of formation of the five-member furan ring and that of glycolaldehyde and 2,3-hydroxyl-succinaldehyde. The formation of the furan ring is initiated by thermal cleavage of the glycosidic linkage between two glucose monomers and is accompanied by the formation of a carbonyl group at C1 (see numbering in Fig. 5) in order for oxygen to satisfy its valency. The formation of the C1 carbonyl group simultaneously results in the opening of the pyran ring since all four valencies of C1 are satisfied without the ring oxygen, i.e., concerted chemistry prevails over the conventional stepwise pathway along a one-dimensional reaction coordinate. The need for the pyran oxygen (OP) to fulfil its valency results in re-closure of the ring at C2, which is accompanied by dehydration at the C2 carbon. The five-member ring is then separated from the cyclodextrin molecule with the cleavage of the second glycosidic linkage and hydrogen from C (formerly part of the glycosidic bond and shown in blue) of the adjacent glucose unit forms a hydroxyl group with the oxygen. Analysis of reaction time scales reveals that the first glycosidic cleavage and ring opening constitute the first step of the furan ring formation, followed by dehydration at C2 and furan ring closure. Our mechanism explains why 5-hydroxymethylfurfural (HMF) is the dominant furanosic product (rather than 2,5-furandicarboxaldehyde or 2,5-furandimethanol, which are not observed experimentally29,35) since the chemical pathway is initiated by carbonyl formation at C1 and this functionality is observed in the final product (5-hydroxymethylfurfural). Additionally, our pyrolytic furan formation mechanism is substantially different from aqueous-phase analogs,10–12 since furans form directly from cellulose without any small-molecule (e.g., glucose, levoglucosan) intermediates.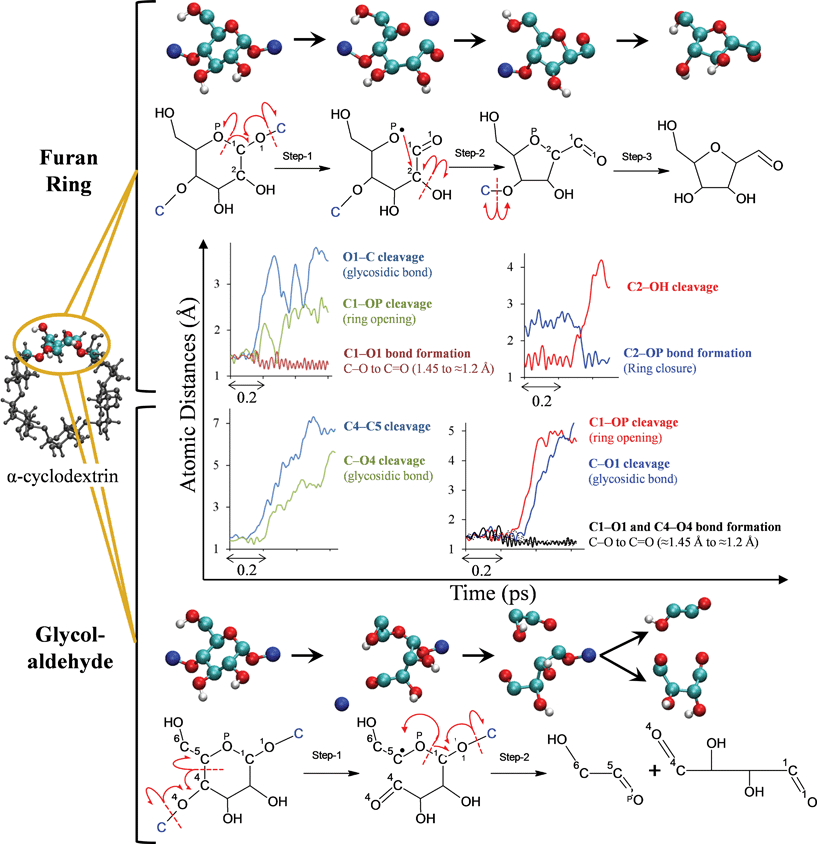 | ||
| Fig. 5 Reaction pathways of α-cyclodextrin (cellulose) pyrolysis . Chemical pathway for the formation of 5-membered furan ring (top) and glycolaldehyde (bottom), obtained using ab initio molecular dynamics (MD) simulations of α-cyclodextrin, a surrogate for cellulose. Cyan balls indicate C, red indicates O and white indicates H. Carbon atoms of the neighboring glucose unit are shown in blue. Time evolution of atomic distances along ab initio MD trajectories that result in the formation of furan ring and glycolaldehyde are depicted. In the interest of clarity, only the portion of the trajectory in which the event happens is shown. The red arrows in the molecular structures indicate the electron flow, e.g., the glycosidic linkage cleavage is homolytic. | ||
While conjectures exist about heterolytic fission of glycosidic linkage during cellulose depolymerization9 resulting in the formation of ionic intermediates, electronic structure analysis along the CPMD trajectory suggests homolytic cleavage. Topological analysis of the spin dependent (α and β) electron localization function (ELF)36,37 performed herein (additional details about the ELF analysis can be found in the supplementary information section†) shows the formation of short-lived radical species, a mechanism which is consistent with condensed-phase pyrolysis mechanisms proposed for other oxygenated polymers, such as polyethylene glycol38–41 and polyvinyl alcohol.42,43Fig. 6 shows a short-lived radical intermediate which forms during step-1 of the furan ring formation mechanism (the reaction scheme depicted in Fig. 5). The highlighted ELF-α and ELF-β isosurface regions above the (blue) carbon atom show the presence of an unpaired β electron, indicating that the intermediate is radical in nature (rather than ionic). Additionally, the topology of the ELF basin around the corresponding glycosidic oxygen atom is different between ELF-α and ELF-β isosurfaces. The presence of a toroidal ELF-β isosurface indicates the existence of an unpaired β electron on the oxygen atom. The detailed analysis of the intermediates in furan ring formation, as well as the ELF analyses of intermediates in other reaction pathways (see supplementary information for additional ELF analysis†), indicates that cellulose pyrolysis is governed by homolytic processes.
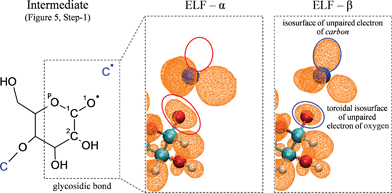 | ||
| Fig. 6 Homolytic cleavage of glycosidic linkage. Electron localization function (ELF) analysis of a short-lived radical intermediate produced immediate after glycosidic bond cleavage and in the process of converting α-cyclodextrin to furans (Fig. 5, Step-1). | ||
Decomposition of another glucose monomer within the α-cyclodextrin molecule results in the formation of glycolaldehyde (Fig. 5), a major pyrolysis product reported in our work and previous efforts.7,9,13Glycolaldehyde formation is initiated with C4–C5 homolytic bond scission, accompanied by the breaking of one of the glycosidic linkages. Since the C5 carbon is short of one valence bond, it forms another carbonyl group with OP, resulting in the homolytic cleavage of the C1–OP bond and the formation of the C1–O1 carbonyl group, which breaks the second glycosidic linkage. This process leads to glycolaldehyde (a major volatile product in experiments) and 2,3-hydroxyl-succinaldehyde (a non-volatile intermediate).
Fig. 7A shows the reaction mechanism of the formation of formic acid and carbon dioxide. Cleavage of both the glycosidic linkages initiates the separation of the glucose unit from the α-cyclodextrin molecule. Through a short-lived intermediate, the 1C carbon atom is separated from the glucose unit as formic acid. Similar to that of the furan and glycolaldehyde formation, this formic acid mechanism does not support conjectures7 about the formation of ionic intermediates after glycosidic bond cleavages. In the reaction mechanism shown in Fig. 7A, the species labeled as radical intermediates exist for a relatively longer simulation time (0.4–0.6 ps), particularly the one formed along with formic acid, possibly because the thermolytic cleavage of an O–H bond (which has very high bond dissociation energy) is needed for the radical to be converted to a stable product.
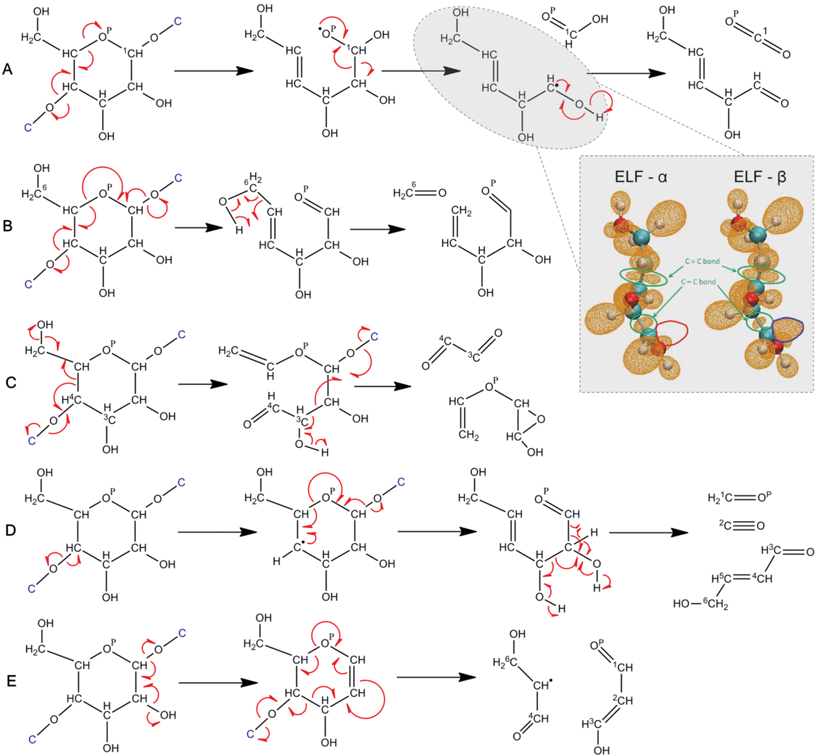 | ||
| Fig. 7 Cellulose pyrolysis pathways to volatile oxygenates. Glycosidic, homolytic cleavage produces formic acid and eventually CO2 and 2,5-dihydroxy-3-pentenal (A) through a radical intermediate identified by ELF analysis (inset). Glycosidic cleavage leads to formaldehyde and 2,3-hydroxy-4-pentenal (B). Glycosidic cleavage leads to glyoxal and 3-(vinyloxy)-2-oxiranol (C). Glycosidic cleavage leads to formaldehyde, carbon monoxide and 4-hyroxy-2-butenal (D). Glycosidic cleavage leads to the formation of malondialdehyde and a radical intermediate (E). Arrows shown assume homolytic chemistry as indicated by ELF analysis of the furan formation mechanism (see Fig. 6 and supplementary information for details†). | ||
To further confirm the nature of the intermediate (radical vs. ionic), we perform ELF analysis. Fig. 7A(inset) shows the ELF-α and ELF-β isosurfaces of one of the intermediate species. The topology of the highlighted disynaptic valence basins between the carbon atoms shows a dumbbell shaped basin and a spherical basin, revealing a double bond and a single bond, respectively. No difference between ELF-α and ELF-β isosurfaces can be seen in the bonding basins since there exists an electron sharing interaction in that region and the localization of electrons of both the spins would be the same. However, in the case of radical species, there has to be a region in the space where the unpaired electron should be found. The region highlighted by the blue contour in Fig. 7A shows the localization of the unpaired β-spin electron, which cannot be seen in the ELF-α isosurfaces in Fig. 7A(inset). Thus, the ELF topological analysis clearly shows that there exists an unpaired electron in the species shown in the inset of Fig. 7. The fact that the unpaired electron is of β-spin is an artifact of the electronic density calculation method and the probability of finding an electron of either spin should be the same. The conclusion from the topological analysis is that the ELF isosurface highlighted with the blue contour represents the region of high probability of finding just one unpaired electron, irrespective of its spin.
Fig. 7B shows the mechanism of formation of formaldehyde. After the glycosidic bond cleavage, the hydrogen from the hydroxyl group of carbon atom 6C is transferred to 5C. This is accompanied by cleavage of 5C–6C bond, thus resulting into formaldehyde and 2,3-hydroxy-4-pentenal. The mechanism of formation of glyoxal is shown in Fig. 7C. The glycosidic bond cleavage at 4C is accompanied by the cleavage of 4C–5C bond and the loss of the hydroxyl group from carbon atom 6C. The resulting species then further undergoes the second glycosidic bond cleavage and a C–C bond cleavage, resulting in the formation of glyoxal and 3-(vinyloxy)-2-oxiranol. Unlike the reaction mechanism depicted in Fig. 7B, Figure 7D shows the formation of formaldehyde from carbon atom 1C. It also shows the reaction scheme for carbon monoxide. The glycosidic bond cleavage at carbon4C is followed by the ring opening of the glucose unit and the cleavage of another glycosidic link at 1C. Additionally, hydride transfer from 2C to 1C results in the cleavage of 2C–1C bond and the production of formaldehyde. Completely satisfied valencies of 3C would not allow 2C to modify its bonding structure with it, thus resulting into breaking of 3C–2C bond and the formation of carbon monoxide with 2C carbon. Fig. 7E shows that the formation of malondialdehyde is again initiated by the cleavage of a glycosidic linkage at 1C, accompanied by the loss of hydroxyl group at 2C. The resulting species further disintegrates with the cleavage of another glycosidic linkage and ring opening to form malondialdehyde and a radical species. It has to be noted that all the reaction mechanisms shown in Fig. 7 are deduced from the bond distance analysis of CPMD trajectories.
The formation mechanisms presented here for furans and light oxygenates (e.g., glycolaldehyde, formic acid) not only describe how major pyrolysis products form, but also represent mechanisms for cellulose glycosidic bond cleavage, a critical process in the depolymerization of cellulose. We find that glycosidic bond cleavage and intra-pyran chemistry are interrelated, and thus, a comprehensive chemical model describing both of these is needed to accurately represent cellulose pyrolysis chemistry.
4. Conclusions
Contrary to previous pyrolysis work using millimeter-scale cellulose samples, thin-film pyrolysis enables the study of condensed-phase pyrolysis chemistry under isothermal conditions and minimizes the breakdown of volatile products. Using this new technique, we show that the condensed-phase chemistry of cellulose is similar to that of α-cyclodextrin over a range of reaction temperatures resulting in nearly identical product distributions. Additionally, the similarity in pyrolysis product yields holds for a variety of experimental systems (e.g. thin-film pyrolysis, powder pyrolysis, thermogravimetric analysis). Our findings provide a testimony that a vital descriptor of cellulose pyrolysis chemistry is the end-group-to-monomer ratio. Based on these findings α-cyclodextrin is an excellent cellulose surrogate for first-principles (CPMD) simulations.CPMD simulations of α-cyclodextrin show that furans form directly from cellulose without any small-molecule (e.g., glucose, levoglucosan) intermediates. Additionally, both furan and glycolaldehyde formation is initiated by homolytic cleavage of glycosidic bonds, in contrast to ionic mechanisms hypothesized previously. Finally, our analysis suggests that glycosidic bond cleavage and intra-pyran chemistry are interrelated, and thus, a comprehensive chemical model describing both of these is needed to accurately represent cellulose pyrolysis chemistry.
This work combines thin-film technology with first-principles computations to elucidate major condensed-phase pyrolysis pathways. While several other pathways remain to be identified, our approach can potentially be applied to study pyrolysis chemistry not only of cellulose, but also of other biomass components, such as lignin and hemicellulose. It also provides a platform that can account for the well-known influence of natural catalysts (such as calcium or magnesium),7,30 which are present in biomass. Our methodology is a first step in developing chemical models to describe biomass pyrolysis at the molecular level; a non-trivial improvement over today's lumped kinetic expressions. It can eventually enable design and optimization of next-generation thermochemical biofuel and chemical production processes.
Acknowledgements
This material is financially supported as part of the Catalysis Center for Energy Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, and Office of Basic Energy Sciences under Award Number DE-SC0001004. Additionally, the CPMD calculations were performed using the TeraGrid resources provided by University of Illinois' National Center for Supercomputing Applications (NCSA). S. H. M. is the recipient of the Natural Sciences and Engineering Research Council of Canada (NSERC) post-doctoral fellowship.References
- R. P. Anex, A. Aden, F. K. Kazi, J. Fortman, R. M. Swanson, M. M. Wright, J. A. Satrio, R. C. Brown, D. E. Daugaard, A. Platon, G. Kothandaraman, D. D. Hsu and A. Dutta, Fuel, 2010, 89, S29–S35 CrossRef CAS.
- P. J. Dauenhauer, J. L. Colby, C. M. Balonek, W. J. Suszynski and L. D. Schmidt, Green Chem., 2009, 11, 1555–1561 RSC.
- A. R. Teixeira, K. G. Mooney, J. S. Kruger, C. L. WIlliams, W. J. Suszynski, L. D. Schmidt, D. P. Schmidt and P. J. Dauenhauer, Energy Environ. Sci., 2011, 4, 4306–4321 CAS.
- T. P. Vispute, H. Y. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS.
- A. Broido and M. A. Nelson, Combust. Flame, 1975, 24, 263–268 CrossRef CAS.
- A. G. W. Bradbury, Y. Sakai and F. Shafizadeh, J. Appl. Polym. Sci., 1979, 23, 3271–3280 CrossRef CAS.
- M. Essig and G. N. Richards, in Cellulose and Wood: Chemistry and Technology, ed. C. Schuerch, John Wiley & Sons, Inc., 1989 Search PubMed.
- Y. C. Lin, J. Cho, G. A. Tompsett, P. R. Westmoreland and G. W. Huber, J. Phys. Chem. C, 2009, 113, 20097–20107 CAS.
- J. Piskorz, D. Radlein and D. S. Scott, J. Anal. Appl. Pyrolysis, 1986, 9, 121–137 CrossRef CAS.
- M. Moliner, Y. Roman-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–986 CrossRef CAS.
- J. Piskorz, D. S. Radlein, D. S. Scott and S. Czernik, J. Anal. Appl. Pyrolysis, 1989, 16, 127–142 CrossRef CAS.
- C. DiBlasi, Ind. Eng. Chem. Res., 1996, 35, 37–46 CrossRef CAS.
- K. Papadikis, S. Gu and A. V. Bridgwater, Fuel Process. Technol., 2010, 91, 68–79 CrossRef CAS.
- G. Varhegyi, E. Jakab and M. J. Antal, Energy Fuels, 1994, 8, 1345–1352 CrossRef CAS.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471–2474 CrossRef CAS.
- X. Biarnes, A. Ardevol, A. Planas, C. Rovira, A. Laio and M. Parrinello, J. Am. Chem. Soc., 2007, 129, 10686–10693 CrossRef CAS.
- S. L. Madorsky, V. E. Hart and S. Straus, J. Res. Natl. Bur. Stand., 1956, 56, 343–354 CAS.
- F. Shafizad. and Y. L. Fu, Carbohydr. Res., 1973, 29, 113–122 CrossRef.
- M. J. Antal and G. Varhegyi, Ind. Eng. Chem. Res., 1995, 34, 703–717 CrossRef CAS.
- H. P. Y. H. P. Yang, R. Yan, H. P. Chen, D. H. Lee and C. G. Zheng, Fuel, 2007, 86, 1781–1788 CrossRef CAS.
- M. R. Hajaligol, J. B. Howard, J. P. Longwell and W. A. Peters, Ind. Eng. Chem. Process Des. Dev., 1982, 21, 457–465 CAS.
- Y. Halpern, R. Riffer and A. Broido, J. Org. Chem., 1973, 38, 204–209 CrossRef CAS.
- P. W. Arisz, J. A. Lomax and J. J. Boon, Anal. Chem., 1990, 62, 1519–1522 CrossRef CAS.
- J. Piskorz, P. Majerski, D. Radlein, A. Vladars-Usas and D. S. Scott, J. Anal. Appl. Pyrolysis, 2000, 56, 145–166 CrossRef CAS.
- T. Funazukuri, R. R. Hudgins and P. L. Silveston, Ind. Eng. Chem. Process Des. Dev., 1986, 25, 172–181 CAS.
- P. R. Patwardhan, D. L. Dalluge, B. H. Shanks and R. C. Brown, Bioresour. Technol., 2011, 102, 5265–5269 CrossRef CAS.
- P. R. Patwardhan, J. A. Satrio, R. C. Brown and B. H. Shanks, J. Anal. Appl. Pyrolysis, 2009, 86, 323–330 CrossRef CAS.
- P. R. Patwardhan, J. A. Satrio, R. C. Brown and B. H. Shanks, Bioresour. Technol., 2010, 101, 4646–4655 CrossRef CAS.
- H. P. Yang, R. Yan, H. P. Chen, C. G. Zheng, D. H. Lee and D. T. Liang, Energy Fuels, 2006, 20, 388–393 CrossRef CAS.
- C. Torri and D. Fabbri, Microchem. J., 2009, 93, 133–139 CrossRef CAS.
- F. J. Kilzer and A. Broido, Pyrodynamic, 1965, 2, 151 CAS.
- F. Shafizadeh, T. T. Stevenson, T. G. Cochran and R. H. Furneaux, Carbohydr. Res., 1978, 67, 433–447 CrossRef CAS.
- F. Shafizadeh, T. T. Stevenson, T. G. Cochran and R. H. Furneaux, Carbohydr. Res., 1978, 67, 433–447 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS.
- H. Arisawa and T. B. Brill, Combust. Flame, 1997, 109, 87–104 CrossRef CAS.
- S. L. Madorsky and S. Straus, J. Polym. Sci., 1959, 36, 183–194 CrossRef CAS.
- K. J. Voorhees, S. F. Baugh and D. N. Stevenson, J. Anal. Appl. Pyrolysis, 1994, 30, 47–57 CrossRef CAS.
- R. P. Lattimer, J. Anal. Appl. Pyrolysis, 2000, 56, 61–78 CrossRef CAS.
- J. W. Gilman, D. L. Vanderhart and T. Kashiwagi, in Fire and Polymers II: Materials and Tests for Hazard Prevention, ed. G. L. Nelson, American Chemical Society, Washington, 1995, vol. 599, pp. 161–185 Search PubMed.
- J. L. Shie, Y. H. Chen, C. Y. Chang, J. P. Lin, D. J. Lee and C. H. Wu, Energy Fuels, 2002, 16, 109–118 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ee02743c |
| This journal is © The Royal Society of Chemistry 2012 |