Quantitative assessment of Ras over-expression via shotgun deployment of vectors utilizing synthetic promoters†
Joshua P.
Ferreira
,
Ingrid E. B.
Lawhorn
,
Ryan W. S.
Peacock
and
Clifford L.
Wang
Department of Chemical Engineering, Stanford University, 381 N-S Axis, Rm 113, Stanford, CA 94305, USA. E-mail: cliff.wang@stanford.edu; Fax: +1-650-725-7294; Tel: +1-650-721-1351
First published on 23rd November 2011
Abstract
We sought to characterize and compare wild-type and oncogenic Ras over-expression. Because different levels of Ras over-expression can have different effects on cell phenotype, it was important to evaluate a wide range of expression. Different expression levels were achieved by using retroviral vectors equipped with different strength promoters. Cells were “shotgun” transduced with a mixture of these vectors to generate heterogeneous populations exhibiting a range of expression levels. We used flow cytometry to analyze the populations and generate high-resolution, nearly continuous Ras dose-response curves. These efforts revealed that a single-copy level of oncogenic Ras generated maximal imatinib resistance and activated MAPK pathway signaling as effectively as six-fold amplification of wild-type Ras. Although further increased expression lead to even greater signal transduction, this increased expression had minimal or decreasing effects on the proliferation rate. In addition, this study introduces a general method to quantify genetic dose-response relationships and identify gene expression ranges that produce an optimized phenotypic response.
Insight, innovation, integrationThis work has the potential to impact the quantitative study of molecular genetics. It is a technical innovation that enables the quantitative assessment of gene expression over a continuous range of expression levels. To vary gene expression levels, we employed promoters engineered using synthetic biology principles. Using retroviral transduction, we generate heterogeneous cell populations where all expression levels of interest are represented. We then employ flow cytometry to collect single-cell expression data for the entire population and computational regression analysis to generate expression dose-response curves. Using our approach, we assessed the dose-dependent relationships between Ras, MAPK signaling activity, and proliferation and demonstrate that resistance to the drug imatinib is optimized at an intermediate level of oncogenic Ras expression and downstream signaling. |
Introduction
Generating dose-response curves is a fundamental experiment for those evaluating drugs or other biologically active molecules. Yet experiments that generate and evaluate a continuous range of gene expression levels are less common. Partly due to the limitations of available genetic tools, researchers often address discrete expression levels, e.g., high over-expression, endogenous expression, or very low expression (i.e., “knock-down” or “knock-out”). Furthermore, because commonly used expression vectors generally utilize strong promoters, expression levels may be supra-physiological and less relevant.We aimed to measure the dose-dependent relationships between Ras, downstream signaling, and cell proliferation. Ras is a GTPase that is a key intermediate in several signal transduction pathways, including the mitogen-activated protein kinase (MAPK) pathway. When activated, Ras initiates the MAPK phosphorylation cascade. This cascade leads to the phosphorylation of Erk, which then activates transcription of genes involved in proliferation, survival, and other phenotypes. In a variety of tumors, Ras is often over-expressed or mutated;1–3 in the latter case, Ras often acquires a missense mutation (commonly at amino acids 12, 13, or 61) to form oncogenic Ras, which has minimal GTPase activity and transmits a constitutively active signal. While researchers have demonstrated that oncogenic Ras activation initiates hyperproliferation,4 others have observed that ectopic expression can induce cellular arrest and senescence in cell types such as fibroblasts.5,6 Thus the effects of Ras are not only context dependent (e.g., cell type, co-mutations, environment) but also depend on its level of expression or activity6,7
Ras and other signaling factors can be activated by constitutively active mutants of the Abelson kinase. As a result, expression of Abelson kinases such as the BCR-Abl fusion protein and v-Abl can immortalize pre-B cells and block their differentiation. In patients, BCR-Abl can cause chronic myeloid leukemia, which, if not properly treated with the Abl kinase inhibitor imatinib, can progress into a blast crisis of pre-B cells or B-cell acute lymphocytic leukemia. Since inactivation of the kinase returns such cells to a non-immortalized state8 cultivating BCR-Abl or v-Abl-expressing cell lines with imatinib could return Ras-mediated signaling to more normal levels and thus provide a cell model suitable for evaluating the ectopic expression of Ras.
In this study, our goal was to quantitatively assess the Ras dose response over a continuous range of over-expressed levels. To accomplish this we have developed an approach that utilizes different synthetic promoters. Here “synthetic promoter” refers to a constitutive promoter that has been modified so that it produces a different transcription level.9,10 Although other gene elements can be modified to achieve varying expression (e.g., mutation of polyadenylation signal11), the application of synthetic promoters is perhaps the most common method to generate various levels of constitutive expression. Synthetic promoters previously have been used to vary gene expression levels12 and optimize lycopene production13 in bacteria. In yeast, synthetic promoter libraries have been used to vary and optimize gene expression14 and glycerol yields.15 The method by Lu and Jeffries utilizes shuffled promoters to vary expression and was used to optimize the expression of multiple xylose fermentation genes for increased ethanol production.16 In mammalian cells, Tornoe et al. created a library with a 10-fold range in transcriptional activity by generating chimeric promoters.17 In this study, we demonstrate that a set of synthetic promoters can be utilized to generate heterogeneous populations of mammalian cells. These populations were then analyzed by flow cytometry to quantitatively assess a range of ectopic Ras expression levels.
Materials and methods
Cell culture and transduction
PD-31 cells were cultured in RPMI-1640 media with 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, and 55 μM β-mercaptoethanol. All media was supplemented with 1 mM glutamine, 100 U/ml penicillin, and 100 μg ml−1streptomycin (FBS: Gemini Bio-Products, Sacramento, CA, USA; all other media and supplements: Life Technologies, Carlsbad, CA, USA).Construction and characterization of the synthetic promoters and retroviral vectors has been described previously.18 Retroviral vector particles were produced by co-transfecting plasmids encoding the Moloney Murine Leukemia Virus (MLV) vectors and pCL-Eco19 into HEK-293T cells using calcium phosphate precipitation. Virus-containing supernatant was harvested and used to transduce cells. Virus was titered so that transduced cells received a single copy of the vectors. Polybrene (hexadimethrine bromide, Sigma-Aldrich, St. Louis, MO, USA) was added to cultures at a concentration of 3 μg ml−1. 24–48 h post-infection, cells were selected with 2 μg ml−1puromycin (MP Biomedicals, Solon, OH, USA). For shotgun transduction, viruses, individually produced and titered, were pooled and used to transduce a single culture of PD-31 cells. When appropriate, cells expressing mCherry, were added to the shotgun-transduced cultures so BFP-negative cells could be distinguished.
Immunoblotting
Immunoblotting was performed using standard protocols. Endogenous Ras (H, K, and N isoforms) and our BFP-Ras fusions were detected using the same anti-Ras primary antibody (E-15, #sc-68743, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and a goat anti-rabbit IgG antibody conjugated to horseradish peroxidase (HRP) secondary. β-actin was detected using an HRP-conjugated antibody (#A00730, GenScript, Piscataway, NJ, USA). The ECL Plus Western Blotting Detection Kit (#RPN2132, GE Healthcare, Piscataway, NJ, USA) enabled detection with a Typhoon PhosphorImager (GE Healthcare). Band intensities were analyzed using ImageJ software (National Institutes of Health, USA).Flow cytometry
For measuring phosphorylated Erk, 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 PD-31 cells per 1 ml volume were treated with combinations of imatinib mesylate (also known as STI-571 and Gleevec, Novartis, Basel, Switzerland) and phorbol-12-myristate-13-acetate (PMA, Sigma). Cells were treated with 0 or 3 μM imatinib for 1 h at 37 °C before antibody staining. 0.5 μg ml−1ethidium monoazide (EMA, Sigma) was added to all samples and incubated for an additional 15 min in the dark at 37 °C. Fifteen minutes prior to antibody staining, cells were stimulated with 0 or 50 nM PMA at room temperature and simultaneously exposed to light from a standard fluorescent lamp. The exposure to light activated EMA so that it could bind the DNA of dead cells, allowing us to distinguish dead cells by EMA fluorescence and exclude them from analysis. After stimulation, each sample was fixed and stained as previously described.20 Briefly, all the samples were fixed with 1.6% formaldehyde, incubated for 15 min at 37 °C, and permeabilized in ice-cold 100% methanol. The samples were stored overnight at −20 °C before washing three times with phosphate-buffered saline (PBS) and resuspending in 100 μL PBS containing 0.02% sodium azide and 0.5% BSA. The cells were then intracellularly labeled with a saturating amount of Alexa Fluor 647-conjugated phosphospecific rabbit monoclonal antibody to p44/42 ERK1/2 (#4370, Cell Signaling, Danvers, MA, USA) at room temperature for 45 min, washed twice with PBS, resuspended in PBS with 2 mM EDTA, and analyzed on a BD Biosciences LSRII flow cytometer. For experiments using the shotgun method, at least 100000 cells were analyzed for each sample (with the exception of one BFP-RasG12V no treatment sample, where loss of cells during transfer and spin steps resulted in only 20000 being available to analyze).
000 PD-31 cells per 1 ml volume were treated with combinations of imatinib mesylate (also known as STI-571 and Gleevec, Novartis, Basel, Switzerland) and phorbol-12-myristate-13-acetate (PMA, Sigma). Cells were treated with 0 or 3 μM imatinib for 1 h at 37 °C before antibody staining. 0.5 μg ml−1ethidium monoazide (EMA, Sigma) was added to all samples and incubated for an additional 15 min in the dark at 37 °C. Fifteen minutes prior to antibody staining, cells were stimulated with 0 or 50 nM PMA at room temperature and simultaneously exposed to light from a standard fluorescent lamp. The exposure to light activated EMA so that it could bind the DNA of dead cells, allowing us to distinguish dead cells by EMA fluorescence and exclude them from analysis. After stimulation, each sample was fixed and stained as previously described.20 Briefly, all the samples were fixed with 1.6% formaldehyde, incubated for 15 min at 37 °C, and permeabilized in ice-cold 100% methanol. The samples were stored overnight at −20 °C before washing three times with phosphate-buffered saline (PBS) and resuspending in 100 μL PBS containing 0.02% sodium azide and 0.5% BSA. The cells were then intracellularly labeled with a saturating amount of Alexa Fluor 647-conjugated phosphospecific rabbit monoclonal antibody to p44/42 ERK1/2 (#4370, Cell Signaling, Danvers, MA, USA) at room temperature for 45 min, washed twice with PBS, resuspended in PBS with 2 mM EDTA, and analyzed on a BD Biosciences LSRII flow cytometer. For experiments using the shotgun method, at least 100000 cells were analyzed for each sample (with the exception of one BFP-RasG12V no treatment sample, where loss of cells during transfer and spin steps resulted in only 20000 being available to analyze).
To measure proliferation, cells were stained with a CarboxyFluorescein Succinimidyl Ester (CFSE) analog, CellTrace Oregon Green (#C34555, Life Technologies) according to the manufacturer's protocol. Immediately after staining with this vital dye, cells were either maintained in media at 10% FBS or the serum was reduced to 1% FBS. Puromycin was reduced from 2 to 1 μg ml−1, and 0 or 3 μM imatinib was added. Cellular fluorescence intensity was analyzed by flow cytometry after 24 (t1) and 92–96 (t2) hours in culture. Approximately 150000 cells were analyzed for each flow cytometry sample. The proliferation rate was then determined from the following equation: rate = log2(Ft1/Ft2)/(t2 − t1), where Ft1 and Ft2 are the geometric means of fluorescence intensity for cell populations analyzed at times t1 and t2, respectively. For all flow cytometry data, cell auto-fluorescence was subtracted before further analysis.
Generation of dose-response curves from flow cytometry data
Flow cytometry data was first analyzed with FlowJo software (Tree Star, Ashland, OR, USA). Relevant cell populations were gated, and data for these populations was exported. Exported data was then analyzed using Matlab software (Mathworks, Natick, MA, USA). A regression curve was fit to each data set using the robust locally weighted scatterplot smoothing (LOWESS) method.21,22 The LOWESS fits were performed with a span of 0.1 (i.e., 10% of the data near each fit point was used to calculate the weighted linear regression). Only fit points within the middle 99.9% of the flow cytometry data on the expression axis were used. Fit curves for triplicate samples were then averaged to make the final curves plotted in this paper.Results
We previously generated a library of synthetic promoters by introducing mutations into the TATA, CAAT, and other regions of the human cytomegalovirus immediate-early promoter (CMV) and the human elongation factor 1α (EF1α) promoter. Based on prior experiments18 that evaluated the promoters' expression of green fluorescent protein (GFP) in PD-31 cells, a v-Abl immortalized pre-B cell line, we chose seven promoters from the library that would allow us to adequately sample a broad expression range (Fig. S1, ESI†). Using retroviral expression vectors that employ these promoters (plus a no promoter control) to express a fusion of blue fluorescent protein and Ras, BFP-Ras (Fig. 1a), we transduced v-Abl immortalized pre-B cells (PD-31) and generated separate cell lines, each expressing a different level of wild-type Ras (RasWT) or oncogenic Ras (RasG12V) (Fig. 1b). We also generated cell lines that expressed a membrane-localized control (BFP with farnesylation signal, as Ras is membrane-localized). Next, we confirmed ectopic expression of Ras by immunoblotting (Fig. 1c). By comparing band intensities of ectopic Ras, endogenous Ras, and the blue fluorescence of each cell line (Fig. 1d), we were able to correlate fluorescence intensity with ectopic expression levels relative to endogenous Ras levels.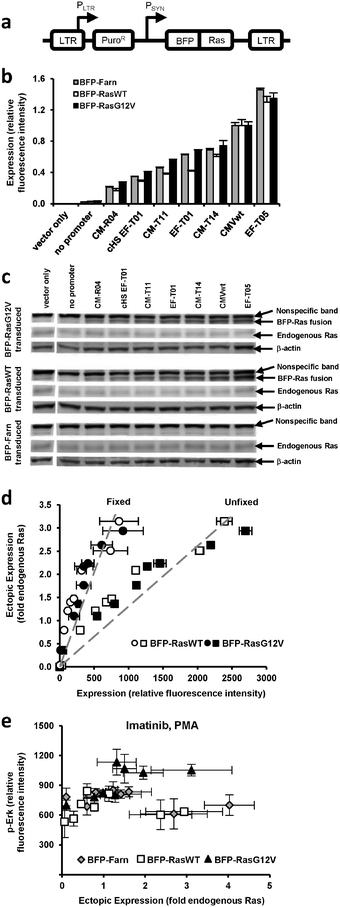 | ||
| Fig. 1 Ectopic and endogenous Ras expression levels in cells transduced with vectors equipped with synthetic promoters. (a) Retroviral vectors equipped with synthetic promoters (PSYN) were used to express blue fluorescent protein (BFP) fused to wild-type Ras (BFP-RasWT), oncogenic Ras (BFP-RasG12V), or a farnesylation signal (BFP-Farn control). LTR, retroviral long terminal repeat; PLTR, LTR promoter; PuroR, puromycin resistance gene. (b) PD-31 cells transduced with different vectors capable of different levels of expression. In the bar graph, expression by vectors with promoters are identified by a designated promoter number. One vector contained a Ras transgene but no synthetic promoter (no promoter). One vector contained neither a synthetic promoter nor transgene (vector only). Values are arithmetic means ± s.d. (n = 3) calculated from geometric means of each sample population. (c) Ectopic and endogenous Ras levels determined by immunoblot analysis of the cell lines in panel b. (d) BFP-Ras fluorescence intensities (when fixed with paraformaldehyde or unfixed) were correlated with ectopic expression levels determined by measuring the immunoblot band intensities in panel c. This correlation allowed ectopic expression levels to be reported with units of fold-endogenous expression. Fluorescence intensity data represent arithmetic means ± s.d. (n = 3) calculated from arithmetic means of each sample population. The dashed line represents a linear regression. (e) p-Erk levels of individually transduced cultures, i.e., each were transduced with a vector employing a different promoter. Cells were treated with imatinib and PMA. Values are arithmetic means ± s.d. (n = 3) calculated from geometric means of each sample population. | ||
Having established different cell lines expressing different amounts of Ras, we could assay each line and assess the dose-response relationship between ectopic expression level and MAPK signaling activity. In these experiments, we treated the cells with imatinib and phorbol myristate acetate (PMA), an activator of protein kinase C often used to amplify the signaling response before assaying for signaling activity. Because (1) PD-31 cells are immortalized by v-Abl and (2) v-Abl activates signaling factors upstream of Ras, ectopic expression of Ras in the presence of imatinib allowed us to determine the degree to which Ras over-expression contributes to cell proliferation and imatinib resistance.
To determine the level of MAPK signaling activity in each cell line, we stained cells with a fluorescently-labeled antibody that recognizes phosphorylated Erk (p-Erk) and then analyzed the cells by flow cytometry. We observed that increasing levels of RasG12V generally led to increasing levels of p-Erk. In contrast, increasing levels of wild-type Ras and the BFP control did not produce discernable changes. Furthermore, the standard deviation between replicates was on the order of the variation between the different cell lines (Fig. 1e). We concluded that with this poor reproducibility, it would be difficult to detect differences in signaling due to small changes in ectopic Ras expression (e.g., changes less than 2-fold endogenous Ras).
We aimed to improve the resolution of the dose-response experiment. Rather than analyze separate cultures (each transduced with Ras expressed from a different promoter) we developed a method whereby all expression levels could be determined from a single culture analyzed by flow cytometry (Fig. 2). With this method our results would be less influenced by culture-to-culture variations that have little to do with differences in expression levels. We produced retroviral particles from eight different vectors—seven equipped with synthetic promoters and one containing no promoter. We pooled the particles and transduced a single PD-31 culture (Fig. 2a), with cells receiving a single copy of the expression constructs. This approach, which we call “shotgun” transduction, produced a heterogeneous culture where the population consisted of cells expressing Ras from different promoters. This approach allowed us to observe cells producing fluorescence intensity levels spanning two orders of magnitude (Fig. 2b) and study ectopic expression ranging from 0 to a 6–10 fold level of endogenous Ras.
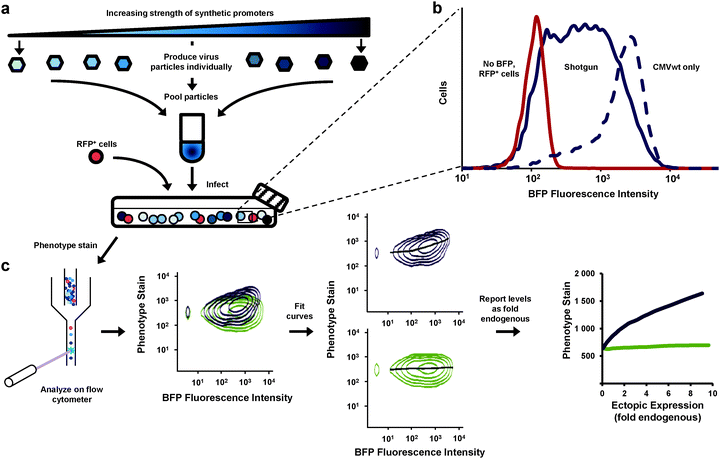 | ||
| Fig. 2 Assessment of the dose-response relationship between gene expression and cell phenotype via shotgun transduction of synthetic promoters. (a) Retroviral vectors employed different synthetic promoters to achieve different expression levels of a gene of interest and a BFP reporter. Viral particles were produced separately and pooled. Cells were transduced with this mixture. Red fluorescent protein-expressing cells (RFP+) were spiked so that zero expression (no BFP, no gene of interest) could be set on the expression scale. Cells were then stained for phenotype and analyzed by flow cytometry. (b) This shotgun-transduced culture contained cells that express a broad range of ectopic expression levels (measured by BFP fluorescence)—broader and more evenly represented than cells transduced with only a single wild-type CMV promoter (CMVwt). (c) Flow cytometry enabled single-cell analysis of the heterogeneous cultures (shown first are two samples overlaid for visual comparison). Curves were fit to the data and using immunoblotting data (Fig. 1c, d), expression levels were reported in relation to endogenous levels (fold endogenous). | ||
The heterogeneous cultures were then stained and analyzed by flow cytometry to measure p-Erk levels. Since flow cytometry analyzes the individual fluorescence of each cell in a given population, the relationship between the levels of p-Erk level and Ras ectopic expression for the entire population can be visualized as two-dimensional density contour plots (Fig. 2c). We then fit a curve to this data (Fig. 2c and 3a–d) where each point on these curves represented a geometric mean computed using values from approximately 10000 cells. Compared to the same dose-response experiment determined from individually transduced cultures (Fig. 1e), we could more clearly distinguish the dose-response curves (Fig. 3a) for each ectopically expressed gene. For these cells treated with imatinib and PMA, increasing RasG12V generated increasing p-Erk levels. At ectopic expression 6-fold that of endogenous Ras, RasG12V's contribution to signaling (i.e., change in p-Erk) was 14.5 times greater than that of wild-type (Fig. 3a). While only very small contributions to signaling due to ectopic wild-type Ras were observed with imatinib inhibition of v-Abl and PMA stimulation (Fig. 3a), without imatinib, signaling initiated by the wild-type was clearly evident (Fig. 3b). In this case, increasing wild-type ectopic expression led to increasing p-Erk until stabilizing at a level equivalent to approximately 6-fold that of endogenous Ras; although, at this point signaling could have been limited by growth factors provided by the culture media. Nonetheless, RasG12V still produced greater signaling (3.2 fold that of wild-type at 6-fold endogenous levels). Comparison of dose-response curves revealed that a single copy of RasG12V can generate MAPK signaling activity approximately equivalent to that from 6 wild-type copies (Fig. 3b). Without PMA stimulation, lesser increases in p-Erk were observed (Fig. 3c, d); with v-Abl inhibition, p-Erk levels were slightly, but discernibly, higher in cells expressing ectopic RasG12V than in those expressing the wild-type or BFP control. The two observations that (1) wild-type signaling was suppressed by the v-Abl inhibitor imatinib and (2) the G12V mutant mediated considerable activity in the presence of imatinib are consistent with the notion that v-Abl significantly contributes to activation of wild-type Ras but not the constitutively active mutant.
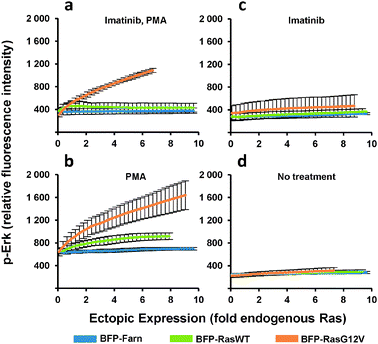 | ||
| Fig. 3 p-Erk response to increasing ectopic Ras expression. PD-31 cells were transduced with retroviral vectors that utilize synthetic promoters to express BFP fused to wild-type Ras (BFP-RasWT), oncogenic Ras (BFP-RasG12V), or a farnesylation signal (BFP-Farn control). Cells were treated with combinations of imatinib and phorbol-12-myristate-13-acetate. (a–d) Dose-response determined from analysis of cells transduced with a vector mixture employing different synthetic promoters (shotgun transduction). Values are arithmetic means ± s.d. (n = 3) calculated from geometric means of each sample population. | ||
In addition, having now presented data generated by the single-promoter and shotgun transduction approaches, we can now compare the approaches. When Ras expression was generated by the single-promoter approach, the upper limit for the evaluated level of ectopic expression was approximately three times that of endogenous (Fig. 1d and e). In contrast, when expression was generated by the shotgun approach, the upper limit was six to ten times that of endogenous (Fig. 3). The difference in the range of expression levels can be explained by the fact that in any transduced culture there will be inherent cell-to-cell variability or heterogeneity (e.g., differences arising from varying vector integrations sites in the genome). In the more traditional, single-promoter experiments, the bulk average expression levels were plotted (Fig. 1d and e). In contrast, with the shotgun transduction method (Fig. 3), the expression levels of each individual cell were evaluated and the inherent cell-to-cell variability actually expands the dynamic range of evaluated expression levels.
Although the library of promoters does provide better expression coverage compared to a single promoter (Fig. 2b), one might argue that given the inherent expression heterogeneity of transduced cell populations, the range of expression from a single promoter is sufficient as long as expression is analyzed at the single-cell level (not bulk averages). To investigate the differences in data quality between the single-promoter and the shotgun approach, we examined how p-Erk levels varied over a range of BFP-control levels. If we assume that BFP expression should not significantly affect Erk phosphorylation, then in this regard, the shotgun-transduced population yielded better results (i.e., a flatter curve, Fig. S2†). In some instances, p-Erk levels varied by as much as 50% over a BFP expression range generated by a single promoter (CMVwt top curve, Fig. S2†). Using only the wild-type CMV promoter, data variation occurred most frequently at lower expression levels that are underrepresented in the population. We believe that analysis of these outliers may enrich for phenotypes that introduce experimental noise. Thus, shotgun transduction of multiple promoters generates superior data resolution.
We next determined how the expression of ectopic Ras affected the proliferation rate of PD-31 cells. As with the previous experiment, we transduced cells with a mixture of retroviral vectors that employ the synthetic promoter to express Ras over a range of levels. Next, in order to measure cell proliferation activity, we stained the cells with a fluorescent vital dye and put the cells back into culture. Cellular fluorescence intensity was then measured by flow cytometry and a curve was fit to the population data associating single-cell levels of fluorescence staining with ectopic Ras expression. By tracking the population over time, average proliferation rates are calculated based on the decrease (i.e., halving) of fluorescence intensity that accompanies each cell division. Under standard culture conditions (10% serum), ectopic expression of both the Ras wild-type and G12V mutant had little effect on proliferation (Fig. 4a). In contrast, when we added imatinib, increasing ectopic RasG12V expression led to increased proliferation before reaching a maximum proliferation rate (46% greater than BFP control) at a level approximately 1.4-fold of endogenous Ras (Fig. 4b). Any greater expression of RasG12V led to decreasing proliferation rates. A similar optimum is seen when we take into account the corresponding p-Erk experiments (Fig. 3c, d) and plot the correlation between p-Erk and proliferation rate (Fig. S3†); note though that proliferation should not be considered dependent solely on p-Erk levels, since Ras mediates signaling through other pathways as well.
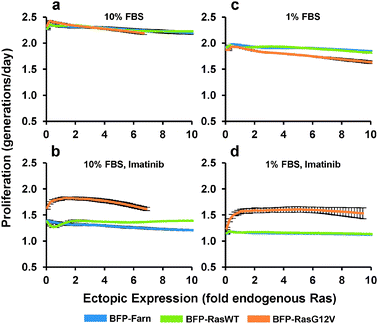 | ||
| Fig. 4 Proliferation response to increasing ectopic Ras expression. PD-31 cells were transduced with a mixture of retroviral vectors (shotgun transduction) utilizing different synthetic promoters to express BFP fused to wild-type Ras (BFP-RasWT), oncogenic Ras (BFP-RasG12V), or a farnesylation signal (BFP-Farn control). (a–d) Dose-response relationships between ectopic Ras expression and proliferation rate for cultures treated with different amounts of serum (FBS) and with or without imatinib. Values are arithmetic means ± s.d. (n = 3) calculated from geometric means of each sample population. | ||
In contrast to the cells expressing RasG12V, cells over-expressing wild-type Ras demonstrated increased proliferation at the higher expression levels (11% greater than BFP control at 10-fold endogenous). This result suggested that even with v-Abl inhibition, a non-trivial amount of wild-type Ras was activated and contributed to proliferation, which was surprising since its contribution to signaling was difficult to detect (Fig. 3c). This wild-type activation without v-Abl activity presumably occurs viagrowth factor stimulation, and indeed with low stimulation (1% serum), over-expression of wild-type Ras had minimal effect on proliferation (Fig. 4c, d). Furthermore, we observed that with 1% serum but no imatinib, expression of RasG12V actually decreased proliferation (Fig. 4c). We also evaluated expression of RasG12V in the presence of imatinib and 1% serum. Under these conditions, RasG12V clearly increased proliferation (at maximum, 43% greater than BFP control), though levels greater than that of endogenous Ras (i.e., 1-fold) made little difference (Fig. 4d).
Discussion
In summary, we have generated high-resolution, nearly continuous dose-response curves for ectopic gene expression. We developed a shotgun approach, where cells were transduced with a mixture of retroviral vectors containing different synthetic promoters. The resulting population contained cells that ectopically express a gene over a range of levels. To quantify the phenotypic response, cells were then stained with fluorescently-labeled antibodies or other dyes and analyzed by flow cytometry. In general, even when all precautions are taken, there can be significant differences in staining between identically prepared samples. Our method, like the barcoding method developed by Krutzik and Nolan23 avoided such variation by staining a mixed population in one batch. Flow cytometry, with its multi-parameter and high-throughput single-cell analysis, has long been used to characterize different cell populations. Here we analyzed the flow cytometry profile from a single, engineered population. Essentially, the shape of the two-dimensional population density contours described the relationship between genotype and phenotype.Using our method, we have assessed the dose-response relationship between ectopic Ras and two phenotypes, MAPK signaling and cell proliferation. Not only did we see qualitatively that RasG12V is more active than wild-type Ras, but also we quantified that, for this particular cell line, a single-copy level of the oncogenic mutant can activate MAPK signaling comparable to 6-fold amplification of the wild-type. However, increased signaling did not always produce increased proliferation and when v-Abl was inactivated by imatinib, the proliferation rates began to decrease when optimal levels of RasG12V were exceeded. This general suppressive effect has been documented in other cells and it is generally held that high levels of chronic Ras-mediated signaling (i.e., constitutively active mutants) can lead to stalled DNA replication forks24 and subsequent activation of DNA damage response pathways,25 which can eventually lead to slower proliferation, senescence or apoptosis.5,6,26
Yet in the presence of active v-Abl, there was no level of ectopic expression of either Ras gene that increased proliferation and this suggests that this cell line has already optimized Ras-mediated signaling for maximal proliferation. When the cells are shifted away from this optimized state through inactivation of v-Abl, only then does additional Ras expression enhance proliferation. Our results also indicate that relatively small but chronic increases in Ras activity can markedly enhance proliferation of Abelson-transformed pre-B cells treated with imatinib; this could be important in understanding the etiology of BCR-Abl+ acute leukemias that develop imatinib resistance without acquiring additional mutations to BCR-ABL.
Beyond these particular implications, this work stands to impact how one assesses the relationship between genotype and phenotype. Shotgun transduction using synthetic promoters stands to be broadly applicable as long as (1) the chosen promoters demonstrate an adequate range of expression in the cells of interest and (2) the total activity of the protein of interest can be regulated at the transcriptional level. Our approach will be helpful for applications that require evaluation of expression ranges, reproduction of physiologically-relevant expression levels, and optimization of genetically-engineered devices and systems.
Acknowledgements
We thank Scott Lowe (Cold Spring Harbor Laboratory, USA) for reagents, Tobias Meyer (Stanford University, USA) for discussion, and Wes Overton and Goutam Nistala (both from Stanford University, USA) for proofreading the manuscript. This work was supported by the NSF (#0846392) and Ellison Medical Foundation (AG-NS-0550-09).References
- K. Fujita, N. Ohuchi, T. Yao, M. Okumura, Y. Fukushima, Y. Kanakura, Y. Kitamura and J. Fujita, Frequent overexpression, but not activation by point mutation, of ras genes in primary human gastric cancers, Gastroenterology, 1987, 93, 1339–45 Search PubMed.
- J. L. Bos, The ras gene family and human carcinogenesis, Mutat. Res., Rev.Genet. Toxicol., 1988, 195, 255–71 Search PubMed.
- S. Schubbert, K. Shannon and G. Bollag, Hyperactive Ras in developmental disorders and cancer, Nat. Rev. Cancer, 2007, 7, 295–308 CrossRef CAS.
- B. S. Braun, D. A. Tuveson, N. Kong, D. T. Le, S. C. Kogan, J. Rozmus, M. M. Le Beau, T. E. Jacks and K. M. Shannon, Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 597–602 Search PubMed.
- M. Serrano, A. W. Lin, M. E. McCurrach, D. Beach and S. W. Lowe, Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a, Cell, 1997, 88, 593–602 Search PubMed.
- Q. Deng, R. Liao, B. L. Wu and P. Sun, High intensity ras signaling induces premature senescence by activating p38 pathway in primary human fibroblasts, J. Biol. Chem., 2003, 279, 1050–9 Search PubMed.
- C. J. Sarkisian, B. A. Keister, D. B. Stairs, R. B. Boxer, S. E. Moody and L. A. Chodosh, Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis, Nat. Cell Biol., 2007, 9, 493–505 Search PubMed.
- S. A. Muljo and M. S. Schlissel, A small molecule Abl kinase inhibitor induces differentiation of Abelson virus-transformed pre-B cell lines, Nat. Immunol., 2002, 4, 31–7 Search PubMed.
- C. Ruth and A. Glieder, Perspectives on synthetic promoters for biocatalysis and biotransformation, ChemBioChem, 2010, 11, 761–5 Search PubMed.
- P. R. Jensen and K. Hammer, Artificial promoters for metabolic optimization, Biotechnol. Bioeng., 1998, 58, 191–5 Search PubMed.
- Y. Yang, S. C. Ho and M. G. Yap, Mutated polyadenylation signals for controlling expression levels of multiple genes in mammalian cells, Biotechnol. Bioeng., 2009, 102, 1152–60 Search PubMed.
- I. Rud, P. R. Jensen, K. Naterstad and L. Axelsson, A synthetic promoter library for constitutive gene expression in Lactobacillus plantarum, Microbiology, 2006, 152, 1011–9 Search PubMed.
- H. Alper, C. Fischer, E. Nevoigt and G. Stephanopoulos, Tuning genetic control through promoter engineering, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12678–83 CrossRef CAS.
- F. S. Hartner, C. Ruth, D. Langenegger, S. N. Johnson, P. Hyka, G. P. Lin-Cereghino, J. Lin-Cereghino, K. Kovar, J. M. Cregg and A. Glieder, Promoter library designed for fine-tuned gene expression in Pichia pastoris, Nucleic Acids Res., 2008, 36, e76 Search PubMed.
- E. Nevoigt, J. Kohnke, C. R. Fischer, H. Alper, U. Stahl and G. Stephanopoulos, Engineering of promoter replacement cassettes for fine-tuning of gene expression in Saccharomyces cerevisiae, Appl. Environ. Microbiol., 2006, 72, 5266–73 Search PubMed.
- C. Lu and T. Jeffries, Shuffling of promoters for multiple genes to optimize xylose fermentation in an engineered Saccharomyces cerevisiae strain, Appl. Environ. Microbiol., 2007, 73, 6072–7 Search PubMed.
- J. Tornoe, P. Kusk, T. E. Johansen and P. R. Jensen, Generation of a synthetic mammalian promoter library by modification of sequences spacing transcription factor binding sites, Gene, 2002, 297, 21–32 Search PubMed.
- J. P. Ferreira, R. W. S. Peacock, I. E. B. Lawhorn and C. L. Wang, Modulating ectopic gene expression levels by using retroviral vectors equipped with synthetic promoters, Syst. Synth. Biol., 2011 DOI:10.1007/s11693-011-9089-0.
- R. K. Naviaux, E. Costanzi, M. Haas and I. M. Verma, The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses, J. Virol., 1996, 70, 5701–5 Search PubMed.
- P. O. Krutzik, J. M. Irish, G. P. Nolan and O. D. Perez, Analysis of protein phosphorylation and cellular signaling events by flow cytometry: techniques and clinical applications, Clin. Immunol., 2004, 110, 206–21 CrossRef CAS.
- W. S. Cleveland, Robust locally weighted regression and smoothing scatterplots, J. Am. Stat. Assoc., 1979, 74, 829–836 CrossRef.
- W. S. Cleveland and S. J. Devlin, Locally weighted regression: an approach to regression analysis by local fitting, J. Am. Stat. Assoc., 1988, 83, 596–610 Search PubMed.
- P. O. Krutzik and G. P. Nolan, Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling, Nat. Methods, 2006, 3, 361–8 CrossRef CAS.
- T. D. Halazonetis, V. G. Gorgoulis and J. Bartek, An oncogene-induced DNA damage model for cancer development, Science, 2008, 319, 1352–5 CrossRef CAS.
- F. A. Mallette, M. F. Gaumont-Leclerc and G. Ferbeyre, The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence, Genes Dev., 2007, 21, 43–8 Search PubMed.
- A. J. Fikaris, A. E. Lewis, A. Abulaiti, O. M. Tsygankova and J. L. Meinkoth, Ras triggers ataxia-telangiectasia-mutated and Rad-3-related activation and apoptosis through sustained mitogenic signaling, J. Biol. Chem., 2006, 281, 34759–67 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ib00082a |
| This journal is © The Royal Society of Chemistry 2012 |