In situ collection of volatile silver species in a new modular quartz tube atomizer for atomic absorption spectrometry†
Stanislav
Musil
*,
Jan
Kratzer
,
Miloslav
Vobecký
and
Tomáš
Matoušek
Institute of Analytical Chemistry of the ASCR, v.v.i., Veveří 97, 602 00 Brno, Czech Republic. E-mail: stanomusil@biomed.cas.cz
First published on 15th May 2012
Abstract
Collection of volatile Ag species on a quartz surface has been successfully achieved for the first time, improving substantially the analytical performance of a previously reported on-line atomization procedure. Ag volatile species were formed during the reaction with tetrahydroborate in the presence of chemical modifiers (Triton X-100 and Antifoam B) in a special generator, collected and atomized in a quartz tube atomizer and detected by atomic absorption spectrometry. A novel modular design of the quartz tube atomizer was employed to allow trapping of volatile Ag species inside the optical tube heated to 1000 °C in excess of O2 over H2. Fast revolatilization and atomization of Ag species were subsequently realized by the change of the gas composition to H2 excess over O2. Collection efficiency under optimized conditions reached 94.1 ± 3.2%. The limit of detection was 0.11 ng ml−1 for a 250 μl sample, nine times better than in the on-line atomization mode. Precision expressed as the relative standard deviation of measured trapping peak areas was 3% at the 4 ng ml−1 level. The method was tested using water reference materials. The present shortcomings of this approach such as quartz devitrification at the collection area are discussed and feasible solutions are suggested.
1. Introduction
A chemical reaction of an analyte with sodium or potassium tetrahydroborate (THB) in an acidic medium is employed almost universally for generation of volatile hydrides of classical hydride forming elements.1,2 Since the first report on Cu generation in 1996,3 successful volatile species generation (VSG) of about 15 transition metals by the same reaction has been also reported up till now.4,5 VSG provides several advantages in comparison to nebulization techniques: analyte separation from a matrix, higher introduction efficiency, milder atomization conditions necessary and easy preconcentration from the gaseous phase.2 However, very little is as yet known about the actual reaction mechanism and analyte release from the reacting solution.6 At least the crucial question about the real identity of those volatile species was answered when nanoparticles were detected in the gaseous phase in the case of Ag and Au VSG.7,8A remarkable improvement in the overall VSG efficiency (εVSG) of Ag was achieved in our laboratory.7,9 The generator was coupled to an atomic absorption spectrometer (AAS) with quartz tube atomizers (QTAs). An εVSG of 33% and a transport efficiency of 95% in the gaseous phase were reached when the inlet arm of the multiatomizer was directly connected to the generator and heated to 300 °C. Nevertheless, slow kinetics of analyte release from the reaction mixture was reflected in too broad flow injection (FI) signals that were found to be the main limiting factor of further limit of detection (LOD) improvement.
Analyte collection prior to atomization is a suitable solution to achieve narrow analytical signals and thus to enhance the VSG performance and reduce LOD to the values required by ultratrace analysis. Volatile hydrides have been reported to be efficiently preconcentrated either in a special collection device (usually by cryotrapping) or in-atomizer employing any of its parts.10,11 A special case of in-atomizer preconcentration is in situ collection, in which the analyte is collected directly in the atomizer part aligned in the optical path of the spectrometer. The performance of the collection method is controlled by the efficiency of collection (εcol) which can be expressed as follows:12
| εcol = εtrap × εrev × εctr | (1) |
There is another, very cheap and straightforward, approach to in-atomizer preconcentration that utilizes a quartz tube, typically the inlet arm of the QTA, as the collection device for hydrides. Ataman and coworkers were the first who analytically utilized a quartz surface for collection of plumbane,14 stibine15,16 and volatile Cd species.17 They employed different temperatures of the separated quartz tube or of the inlet arm of the QTA for trapping and revolatilization. Analyte revolatilization was supported by additional H2 flow. Kratzer and Dědina came up with a very practical and sensitive approach for collection of stibine,18,19 bismuthine,19,20 arsine,21 selenium hydride21 and plumbane.22 They found that these hydrides could be effectively captured on the quartz surface in stoichiometric excess of O2 over H2. A quartz capillary inserted into the inlet arm was usually used for O2 supply to remove (burn) all H2 originating from the chemical VSG reaction in the generator and entering as a byproduct into the atomizer. Subsequent revolatilization of trapped species was realized by the change in the gas composition (decrease of O2 flow and/or increase of H2 coming to the atomizer). The feasibility of this approach was demonstrated by 100% εcol for stibine, bismuthine and plumbane under the optimal conditions of trapping and revolatilization (temperature and gas composition).19,22,23 For arsine and selenium hydride εcol values of only 50% and 70% have been reached so far.21 Stibine and bismuthine were also effectively trapped truly in situ, i.e. within the optical tube of the QTA (not in the inlet arm).18,20
As for volatile transition metal species, in situ collection in the graphite furnaces has been the only practical way of collection up to now.8,24–29 To the best of our knowledge, the only work dealing with a different experimental arrangement for in situ collection of generated transition metal species was published very recently by Matusiewicz and Krawczyk.30 They successfully employed an integrated atom trap with flame AAS for volatile Ni species. The aim of our work was to investigate the feasibility of volatile Ag species collection in the QTA in a similar way as reported earlier for hydrides18–23 and consequently to decrease LOD substantially. Basic requirements were to reach as high εcol as possible and to develop a convenient collection procedure. A new modular QTA construction that is analytically useful for both in situ collection and atomization of Ag species is described. The potential of modular atomizer constructions for fundamental studies was recently shown by Řezáčová and Dědina when a special L-design configuration was employed to test the optical tubes made of different materials and evaluate their atomization performance.31
2. Experimental
2.1. Instrumentation
The detection was carried out by the atomic absorption spectrometer (Perkin-Elmer 503) with a Ag hollow cathode lamp (Perkin-Elmer, 328.1 nm line, 0.7 nm spectral bandpass) operated at 12 mA. Background correction was not employed. Signals from the spectrometer outputs for a strip chart recorder were AD converted, collected and processed in a PC. Averages from at least 3 replicate measurements of peak areas are shown in the figures, error bars represent standard deviation (SD) or combined SD where results are relative.2.2. Standards and reagents
Deionized water (<0.2 μS cm−1, Ultrapure, Watrex, Czech Republic) was employed for preparation of all solutions. Working Ag standards were prepared by serial dilution of 1000 μg ml−1 stock solution (BDH, UK) in 0.6 M HNO3 (ultrapure, Ventron, Germany) that was also used as a carrier liquid. A reductant solution containing 2.4% (m/v) NaBH4 (Fluka, Germany) and 133 μg ml−1 of Antifoam B emulsion (Sigma, USA) in 0.1% (m/v) KOH (semiconductor grade, Sigma-Aldrich, Germany) was prepared fresh daily. 20 μg ml−1 of Triton X-100 (Aldrich Chemical Co., USA) in 0.1 M HNO3 was used as a reaction modifier.7,32 A groundwater reference material VKI RM Grumo-K (Eurofins, Denmark) and a proficiency testing sample of drinking water SAR C from PT#V-8-2008 test produced by National Institute for Public Health (Prague, Czech Republic) with defined Ag contents were employed to verify the accuracy of the analytical method.2.3. VSG system
A scheme of the generator in the FI mode is displayed in Fig. 1 and it is described in detail elsewhere.7 The reagents were pumped by peristaltic pump 1 at the flow rate of 0.5 ml min−1. Bubbles of H2 evolving from THB decomposition were removed from the reductant channel on-line just before the reaction by means of a PTFE degassing chamber (0.5 ml).33 The reduction takes place inside a glass gas–liquid separator (GLS, 3 ml volume) at the tip of three concentric capillaries. The innermost capillary (non-polar fused silica, 0.25 mm i.d.) leads the carrier with the sample plug and the chemical modifier, the middle one (non-polar fused silica, 0.53 mm i.d.) the reductant solution and the outermost (PTFE, 1 mm i.d.) the carrier Ar (see ref. 7 for detailed capillaries arrangement). The capillaries end in approximately 1 mm distances in a sequence from the innermost to the outermost tube. The mixing with THB thus takes place at the tip of the capillaries and the reaction mixture is immediately dispersed to the whole GLS volume by the carrier Ar. The waste liquid was pumped out at an arbitrary rate by peristaltic pump 2 to maintain a stable level of the reacting mixture inside the GLS (approximately 0.5 ml). The clean generator was pre-conditioned by means of VSG from 4 ml of 10 μg ml−1 Pd solution (BDH, UK) in 0.1 M HNO3 and 20 μg ml−1 Triton X-100 to form a permanent modification of the GLS surface by reduced Pd for enhanced rate of THB decomposition, as described earlier.34 The flow rate of the carrier Ar was 50 ml min−1 for all experiments. The flow rate of H2 released from THB decomposition in the GLS was always 26 ml min−1. Where stated, an additional flow of H2 was used downstream the GLS to support the release of trapped Ag species. All gas flows were controlled by mass flow controllers (FMA-2400 or 2600 Series, Omega Engineering, USA).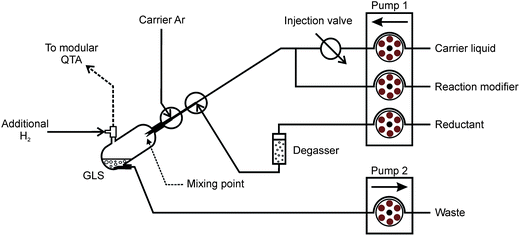 | ||
| Fig. 1 Scheme of the FI-VSG system with a 250 μl sample loop. | ||
2.4. Atomizers
In preliminary tests collection of volatile Ag species on the quartz surface was tested using a special multiatomizer design with a modified inlet arm (see Fig. S1 in the ESI†). This design is described in detail in ref. 9 and was also employed in radiotracer experiments (see Section 2.6). The optical part of this atomizer was made of two concentric tubes. The inner tube (optical, 145 mm long, 7 mm i.d.) was evenly perforated with 14 holes of approximately 0.5 to 1 mm diameter. The outer tube of the multiatomizer was heated electrically to 900 °C by an in-house made furnace controlled by the REX-C100 controller (Syscon, Indiana, USA) with the K-type thermocouple sensor (Omega Engineering, USA). 20 ml min−1 of air as the outer gas was introduced from the sides to the cavity between the optical and the outer concentric tube of the horizontal arm.7 The inlet arm (170 mm long, 2 mm i.d., 6 mm o.d.) was modified in order to connect it directly to the GLS outlet and was wrapped with the Ni80–Cr20 wire (0.6 mm o.d., 5.275 Ω m−1, 10.7 Ω total resistance, Omega Engineering, USA) for constant heating by laboratory power supply (type EA-PS 3065-10 B, Viersen, Germany). The appropriate current value for heating the inlet arm to 300 °C (ref. 9) was calibrated by means of the K-type thermocouple sensor (Omega Engineering, USA) when its tip was placed inside the inlet arm in a position 6.5 cm from the optical tube axis. The auxiliary inlet (35 mm long, 1 mm i.d., 3 mm o.d.) prolonging the horizontal part of the heated inlet arm was used so that a silica capillary (i.d. 0.53 mm) could be axially centered inside the inlet arm in arbitrary positions from the optical tube. This capillary served for delivery of O2 necessary for trapping of volatile Ag species in the inlet arm as well as for H2 introduction for efficient revolatilization.The new modular QTA for in situ collection of volatile Ag species is depicted in Fig. 2. It is composed of three main parts: inlet arm, outer tube and inner exchangeable optical tube with a central orifice. The quartz inlet arm (165 mm long, 2 mm i.d., 4 mm o.d.) was connected directly to the GLS outlet and was wrapped with the Ni80–Cr20 wire (6.4 Ω total resistance) again for constant heating. The appropriate current value for heating the inlet arm to 300 °C (ref. 9) was obtained by means of the K-type thermocouple sensor again when its tip was placed inside the inlet arm in a position 8 cm from the optical tube axis. The outer tube consisted of the main quartz tube (165 mm, 13.3 mm i.d., 16.3 mm o.d.) with three quartz inlets. Both side inlets served for introduction of the outer gas (O2) to the cavity between the optical and the outer tube. PTFE rings were used to seal the optical tube inside the outer tube on both sides. The central inlet served as a mechanical support for the inlet arm that was sealed by a silicone rubber ring in combination with ceramic wool fiber. The inner optical tube was made of quartz (165 mm, 7.2 mm i.d., 11.0 mm o.d.). The central orifice (4.2 mm i.d.) was 0.2 mm broader than the o.d. of the inlet arm (4 mm). Due to this leakage, outer O2 could enter the optical tube around the end of the inlet arm as shown in the inset in Fig. 2. If not stated otherwise, the outer tube of the modular QTA was heated electrically to 1000 °C. A 2-position switching valve (10-port, Valco Instruments Co. Inc., USA) was utilized for the quick change of outer O2 and additional H2 flows at the beginning of the revolatilization stage.
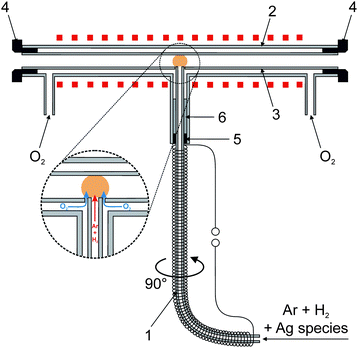 | ||
| Fig. 2 New modular QTA design with a detailed view of O2 introduction to the optical tube; 1 – inlet arm, 2 – outer tube, 3 – optical tube, 4 – PTFE rings, 5 – silicon rubber ring and 6 – ceramic wool fiber. | ||
2.5. Procedure
Measurements were performed in the FI mode either with on-line atomization or in the collection mode. The collection procedure consisted of the following two stages – trapping and revolatilization.The trapping stage starts by switching the 2-position valve into the trapping position when a stoichiometric excess of O2 over H2 is introduced into the optical tube. At the same time the sample is injected via a manual injection vent with the 250 μl loop to the stream of the carrier liquid. The total trapping time was 150 s since such a period was necessary for the complete generation of volatile Ag species and their introduction from the GLS to the QTA, as observed previously.7,9 If explicitly stated, a “breakthrough” signal was recorded. It corresponded to the on-line atomization signal measured in the course of the trapping stage under various trapping conditions. This signal indicated the analyte portion that broke through the trap to the remaining optical tube volume or the analyte portion that could be still atomized in the flame due to some unburnt H2 remaining at the downstream end of the inlet arm.
The revolatilization stage followed after the trapping stage. Without stopping the VSG pumps, signal reading was switched on and subsequently, after a 5 s delay, revolatilization was carried out by the change of the gas composition as follows: the 2-position valve was switched to the revolatilization position in which outer O2 flow was decreased to an appropriate value and additional H2 was introduced to the inlet arm. This change in the gas composition was immediately followed by the release of Ag species into the volume of the optical tube and their atomization/detection.
Each collection procedure for the standard or sample was always followed by the measurement of blank and the trapping signal was corrected for the blank value. The on-line atomization mode was used as a reference signal for estimation of εcol, defined as a product of εtrap and εrev. Additional H2 and outer O2 flow rates were strictly kept the same in the revolatilization stage of the collection mode as in the on-line atomization mode to enable estimation of εcol by comparing the respective peak areas.
2.6. Radiotracer experiments
The preparation, chemical separation and isolation of a 111Ag radioactive indicator (half-life 7.45 days) of high specific activity were completely described elsewhere.7 300 μl of 111Ag radiolabeled solution (ca. 15 kBq per experiment) with addition of non-active Ag standard solution (50 ng ml−1) was introduced to the system in 3 replicates in the same way as in ref. 9. The radiotracer experiments were focused on visualization and quantification of the analyte in the multiatomizer after the trapping stage and after the whole collection procedure (trapping and revolatilization). Four columns in series, each 40 mm long, filled with activated charcoal granules and separated by polyurethane foam plugs were connected to both outlets from the optical tube. Activity in the columns or in HF and HNO3 leaches of the multiatomizer inlet arm and its optical tube was quantified using the automatic gamma counting system equipped with the scintillation NaI(Tl) well-type detector (Minaxi 5000, Packard) with 2 min counting time. The range of the measured energy was 200–400 keV. The obtained count values were corrected for blank counts and radioactive decay. The spatial distribution of the activity in the multiatomizer was investigated by image plate autoradiography (exposure time of 120 min) and the radiograms were obtained by the laser scanner (Fuji BAS 5000).3. Results and discussion
3.1. Preliminary observations
In order to test trapping of volatile Ag species on a quartz surface in excess of O2 over H2, the multiatomizer design previously described in ref. 9 and depicted in Fig. S1 (ESI†) was utilized together with the capillary for O2 delivery. The capillary position was chosen as 2.5 cm from the optical tube axis during the first trapping experiments. The temperature at the tip of the capillary was around 500 °C. A clearly visible small oxygen–hydrogen flame was formed at the capillary tip during VSG. The breakthrough signal was monitored during the trapping stage (see Section 2.5). At first, O2 flow rate through the capillary was altered. While the highest response was measured at 5 ml min−1, 50% and 98% decrease in the signal were observed at 12.5 ml min−1 and 15 ml min−1 of O2, respectively. This observation exactly corresponds with the fact that 13 ml min−1 of O2 is the minimum flow necessary to remove completely 26 ml min−1 of H2 originating from the VSG reaction. At 20 ml min−1 of O2 going through the capillary, no Ag free atoms were detected in the multiatomizer.An independent proof that Ag species were efficiently trapped in the inlet arm under an O2 rich atmosphere was made by means of the experiment with 111Ag radioactive isotope. In this case γ-radiation in the multiatomizer was scanned after 3 replicates with 111Ag (see Section 2.6). It is clearly seen in the image plate autoradiogram (Fig. 3a) that all the analyte was trapped in a narrow zone near the capillary tip. Thus, all the generated volatile Ag species were efficiently prevented from entering the optical arm in excess of O2 over H2. As discussed in the Introduction, revolatilization of hydride forming elements is achieved by the change in the gas composition when O2 flow is decreased under the stoichiometric amount (or totally switched off) and the trapped analyte is revolatilized by H2 addition. With this experimental arrangement H2 was introduced through the capillary when the flow of O2 was switched to H2. This approach was tested with 111Ag in order to visualize potential changes in the analyte spatial distribution in the multiatomizer (Fig. 3b) as well as to estimate εcol. After 3 replicates with the whole collection procedure (trapping followed by revolatilization), most of the γ-radiation was detected at both ends of the optical tube, i.e. in the regions of the multiatomizer that were not covered by the body of the furnace. It means that the trapped analyte was released from the trapping point to the optical tube and it was deposited again in the colder parts of the optical tube (∼600 to 650 °C). Hardly any 111Ag (only 2.1% of the gaseous phase) was detected in the columns connected to both outlets from the optical tube. It should be noted that in the standard use without columns, the analyte deposition pattern might be influenced by possible hydrogen radical formation due to O2 diffusing from the open tube ends.35 Assuming 100% εtrap in the inlet arm according to Fig. 3a, the resultant εcol was estimated to be around 87% from the HF and HNO3 leaches of the multiatomizer optical tube. The remaining activity was found in the inlet arm (11% of the gaseous phase) or deposited on the quartz capillary (2%). These 111Ag experiments demonstrated the functionality of this approach even for volatile Ag species.
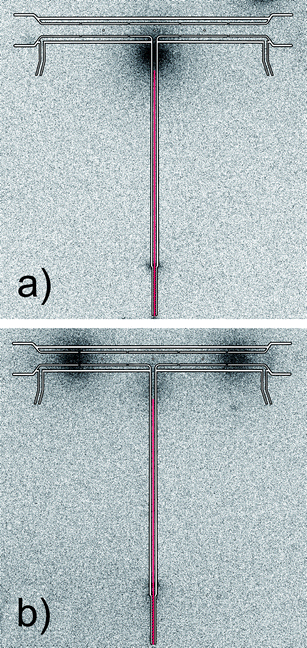 | ||
| Fig. 3 Autoradiograms of the multiatomizer after 3 replicates with 111Ag: (a) after the trapping stage and (b) after the whole collection procedure; exposure time 120 min, Tat = 900 °C, capillary position 2.5 cm from the optical axis ∼500 °C; trapping conditions: 20 ml min−1 of O2 through the capillary; revolatilization conditions: 100 ml min−1 of H2 through the capillary. | ||
The first collection attempts with AAS detection were reflected in too broad trapping signals due to pronounced tailing. Subsequently, it was found out that the width of the measured trapping peaks (and mainly their tailing) was dependent on the applied revolatilization temperature and especially on the distance between the trapping point and the optical tube. The best peak profiles were obtained when the capillary tip was aligned with the optical tube axis (capillary position 0 cm, ∼900 °C). The disadvantage of this design was that the capillary tip serving for O2 delivery was broken very easily after a few measurements. This was attributed to very corrosive conditions with so many aerosol droplets produced in the generator.7,9 The capillary was also responsible for some losses of Ag volatile species on its surface, as indicated above in 111Ag experiments. As a consequence, the apparatus design was changed and a new modular QTA design (see Fig. 2 and Section 2.4) was constructed to overcome these problems.
3.2. New modular design of QTA
This design allows trapping of the analyte without the need of the capillary for O2 delivery. The use of the heated inlet arm, well established for efficient transport previously,9 was found to be essential in the new modular design since the analyte was efficiently transported from the generator, focused at the trapping point and not dispersed along the way. Simultaneously, very fast revolatilization and 100% εctr are achieved because the analyte is trapped and revolatilized directly in the optical tube (in situ). Quartz was tested as the first material of the optical tube without any perforation as Ag was earlier reported to be atomized in the QTA with the same efficiency as in the multiatomizer.7 The only centered orifice (i.d. 4.2 mm) was drilled for the entrance of the inlet arm (o.d. 4 mm). Due to this annular gap between the optical tube and the inlet arm, outer O2 could enter the optical tube and react with H2 leaving the inlet arm. The modular atomizer design also permits very simple exchange of devitrificated quartz parts as discussed in Section 3.6.1. Moreover, the novel construction without the use of the capillary, which is easy to break, prolongs the lifetime of the whole device.The following conditions had to be optimized for this QTA design: the outer O2 flow rate for trapping Ag species, the flow rate of H2 for revolatilization and atomizer temperature. For simplicity, the carrier Ar flow rate (50 ml min−1) that was optimal in the on-line atomization mode9 was chosen for both steps of the collection mode.
3.3. Optimization of O2 flow rate in the trapping stage
The dependence of the breakthrough signal (in the trapping stage) and the resulting trapping signal measured after revolatilization on the outer O2 supply is displayed in Fig. 4. No additional H2 was used throughout the collection procedure. The highest breakthrough signal was obtained at 4 ml min−1 of O2 when obviously no trapping occurred. The same O2 amount added to the carrier Ar was also found to be optimal for on-line atomization in the QTAs earlier.7,9 A small signal decrease was observed for zero outer O2 flow rate. This was attributed to the leakage of the analyte from the optical tube volume through the annular gap rather than to the influence of the presence of O2 on the atomization process because the signal was not significantly impaired when Ar was used as the outer gas at the same flow rate instead of O2. This was why 4 ml min−1 of outer O2 was maintained also during the revolatilization stage. As seen in Fig. 4, a significant signal decrease was observed at outer O2 flow rates higher than 12 ml min−1. A relatively higher flow rate of O2 was necessary for complete trapping (40–50 ml min−1) in comparison to the multiatomizer design with the capillary (see Section 3.1). There was always some breakthrough signal detectable, namely 4 and 3% for 40 and 50 ml min−1 of outer O2. It was probably caused by some H2 fraction remaining unburnt in the optical axis. Therefore, a small portion of Ag free atoms had been always detected before the Ag species were trapped on the surface of the optical tube. The resulting trapping signals were complementary to the breakthrough signals (see Fig. 4). To calculate the accurate values of εcol at given outer O2 flow rates, the trapping signals were related to the highest breakthrough signal (4 ml min−1 of outer O2) represented by the upper dotted line. At the optimized outer O2 flow rate for trapping (50 ml min−1) εcol reached 99.8 ± 6.5%.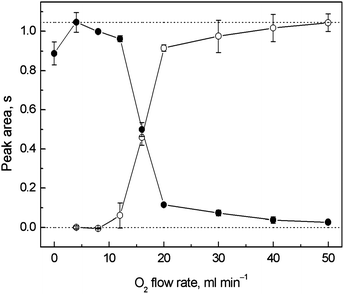 | ||
| Fig. 4 Dependence of the breakthrough and trapping signals on the outer O2 flow rate for in situ collection in the modular QTA; ● – breakthrough signal, ○ – trapping signal; Tat = 1000 °C, 5 ng ml−1 (1.25 ng) of Ag; trapping conditions: variable outer O2 flow rate; revolatilization conditions: 4 ml min−1 of outer O2; the lower dotted line corresponds to a zero signal level, the upper dotted line corresponds to the highest breakthrough signal measured at 4 ml min−1 of outer O2. | ||
3.4. Influence of H2 flow rate on revolatilization
When Ag species were revolatilized only by H2 from THB decomposition, a long time was necessary for signals to return to the baseline (60 s) and repeatability of the peak areas was rather worse (4–9%). Additional H2 was found to support the release of Ag species from the trapping place and as a result more symmetric peaks with less tailing were obtained, permitting shorter integration times. On the other hand, it was earlier reported that additional H2 as well as its fraction in the carrier gas decreased the measured sensitivity in the multiatomizer9 due to two effects – the dilution of free analyte atoms at higher total gas flow rates and decrease of atomic absorption coefficient in H2 owing to absorption line broadening as known for Se.36 Using an additional H2 stream introduced downstream of the the GLS in the on-line atomization mode, the signal of 10 ng ml−1 Ag decreased from the value 2.22 ± 0.07 s for zero additional H2 flow rate (corresponding to the 34% H2 fraction in the Ar stream) to the value 1.06 ± 0.06 s for 100 ml min−1 of additional H2 (72% H2 fraction). 40 ml min−1 of additional H2 was chosen as a compromise between reduced sensitivity and peak tailing. Such a flow rate was found to reduce sensitivity by around 35% compared to the lowest H2 setting.To decide what the real impulse for revolatilization of Ag species was, a special experiment was performed. After trapping by means of 50 ml min−1 of outer O2, VSG was switched off and the rest of the reacting mixture was removed. Another 60 s was allowed to flush out remaining H2 (originating from THB decomposition) from the system. After that, the outer O2 flow was switched off too and Ar remained the only gas coming to the QTA. No peaks had been observed by this time indicating that no Ag species had been revolatilized. The trapping signal was observed only when H2 was introduced to the carrier Ar. This experiment clearly demonstrated that the release and revolatilization of Ag species were activated by H2 coming to the atomizer under a substoichiometric amount of O2 and not by a simple thermal process.
To simplify the collection procedure maintaining additional H2 (40 ml min−1) throughout the whole collection procedure was tried out. 10 ml min−1 of the outer O2 was found to be optimal for on-line atomization under such conditions and subsequently it was also applied during the revolatilization stage. At outer O2 flow rates higher than 30 ml min−1 a significant decrease in the breakthrough signal was observed. The highest εcol was obtained at 140 ml min−1 of outer O2 applied during the trapping stage but it reached only 91.3 ± 4.4%. It was indicated also by the relatively higher breakthrough signal (7%). The lower value of εcol was ascribed to the different flame profile at the high flow rate of gaseous mixture and to some fraction of H2 that was difficult to burn immediately at the desired trapping point, i.e. at the entrance to the optical tube and in the center of the optical tube. Despite the slightly worse εcol, revolatilization of Ag species was favorably affected by additional H2 and peak tailing was reduced. This was why the collection procedure was modified – during the trapping stage additional H2 was not applied and it was introduced only during the revolatilization stage. The optimized conditions for the gaseous flows were thus the following: 50 ml min−1 of outer O2 and no additional H2 in the trapping stage and 10 ml min−1 of outer O2 and 40 ml min−1 of additional H2 in the revolatilization stage.
3.5. Temperature of trapping, revolatilization and atomization
Since collection was carried out in situ, inside the optical tube, temperatures of trapping, revolatilization and atomization could not be independently optimized and εcol was determined only by one parameter, i.e. atomizer temperature. This is not favorable when the optimal temperatures for trapping, revolatilization and atomization differ significantly. However, as is clear from Fig. 5, the values of trapping signals did not significantly differ from those obtained in the on-line atomization mode for all the tested atomizer temperatures measured at the same gas composition. No losses due to incomplete trapping or revolatilization at a given temperature were observed and εcol ranged between 90 and 96% for temperatures of the QTA between 650 and 1000 °C (see the inset graph of Fig. 5). It can be concluded that trapping and revolatilization of Ag species are highly efficient in a wide range of temperatures making the trapping mode fairly robust and that temperature has an influence only on the atomization process in terms of sensitivity. This is in good agreement with the observation that revolatilization is not controlled by thermal processes (see Section 3.4). 1000 °C was chosen as optimal not only due to the highest sensitivity but also due to the best peak profile and time necessary to reach the baseline. Under the optimized conditions for in situ collection (Table 1) εcol reached 94.1 ± 3.2%.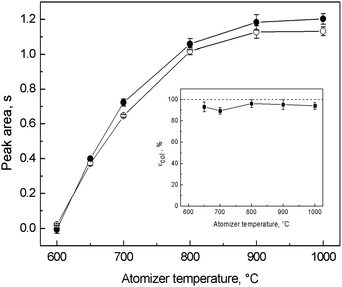 | ||
| Fig. 5 Optimization of atomizer temperature for in situ collection in the modular QTA and dependence of εcol; ● – on-line atomization signal, ○ – trapping signal; Tat = variable, 8 ng ml−1 (2 ng) of Ag; on-line atomization conditions: 10 ml min−1 of outer O2, 40 ml min−1 of additional H2; trapping conditions: 50 ml min−1 of outer O2; revolatilization conditions: 10 ml min−1 of outer O2, 40 ml min−1 of additional H2. | ||
Fig. 5 also suggests that volatile Ag species entering the optical tube in the on-line atomization mode and those Ag species that are revolatilized from the quartz surface by H2 in the collection mode are similar in nature and/or atomized by a similar mechanism because they are atomized in the QTA with the same efficiency. We do not believe that Ag nanoparticles are formed after revolatilization within the optical tube. As proven previously in the on-line atomization mode,9 the generated nanoparticles are relatively stable during the passage through the inlet arm heated to 300 °C but they start decomposing either at slightly higher temperatures or in the oxygen–hydrogen flame. Owing to the inevitable temperature gradient at the downstream end of the inlet arm, Ag nanoparticles are most probably decomposed (likely to free atoms) before entering the optical tube both in the on-line atomization and collection modes. Revealing the identity of revolatilized species would be extremely useful for clarification of the mechanism of Ag species collection. Unfortunately, sampling of these species on the grids for a transmission electron microscope as in ref. 7 and 9 is not possible at such a high temperature.
3.6. Analytical performance
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, v/v) but their lifetime was limited. The total lifetime of the quartz optical tubes and of the inlet arms was around 200–250 runs.
:7, v/v) but their lifetime was limited. The total lifetime of the quartz optical tubes and of the inlet arms was around 200–250 runs.
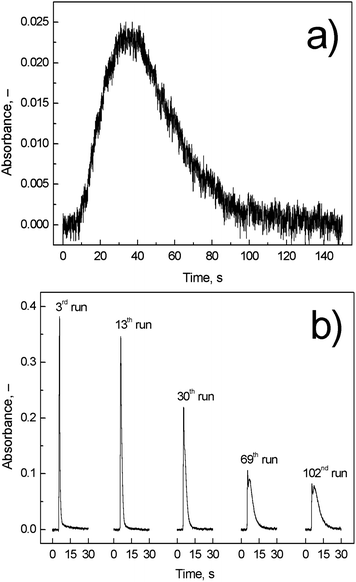 | ||
| Fig. 6 Comparison of (a) on-line atomization signal and (b) trapping signal; Tat = 1000 °C; on-line atomization conditions: 10 ml min−1 of outer O2, 40 ml min−1 of additional H2, and 8 ng ml−1 (2 ng) of Ag; trapping conditions: 50 ml min−1 of outer O2; revolatilization conditions: 10 ml min−1 of outer O2, 40 ml min−1 of additional H2, and 4 ng ml−1 (1 ng) of Ag. | ||
4. Conclusion
The collection method, hitherto employed for hydrides, applying excess of O2 over H2 to trapping of volatile species was successfully extended also for VSG of Ag. A new modular design of the quartz tube atomizer was constructed for this purpose. Revolatilization of Ag species was attributed to the change of the gas composition when O2 flow was decreased under stoichiometry and additional amount of H2 released Ag species. Under optimized conditions of trapping and revolatilization the collection efficiency was found to be nearly 100%. The improvement of an order of magnitude in the LOD was achieved with the use of still inexpensive equipment surpassing significantly the original on-line atomization approach. A good feature of this modular QTA construction is that it enables us to test various materials of the optical tube in future, e.g. ceramics or modified quartz, and thus to avoid devitrification of the optical tube that decreases its lifetime and impairs the method performance. As found in our preliminary observations, ceramic tubes could be a suitable choice. The other possibility to prevent devitrification is to reduce the amount of alkali metals going to the atomizer along with aerosol droplets by using a milder concentration of THB or to use borane ammonia complexes instead.38 Our future investigation will be also focused on reagent contamination and decrease of the blank signals because the LOD could be, in principle, substantially improved by using higher sample volume and trapping time. The same atomizer construction could be also beneficial for in situ collection of hydride forming elements that can be, after revolatilization, efficiently atomized in the QTA, namely Bi and Sb.18–20Acknowledgements
This project was supported by the Czech Science Foundation (no. 203/09/1783) and by the Academy of Sciences of the Czech Republic, v.v.i. (Institutional research plan no. AV0Z40310501 and RV0: 68081715). The authors are much obliged to Dr Jiří Dědina for valuable comments on the manuscript.References
- J. Dědina and D. L. Tsalev, Hydride Generation Atomic Absorption Spectrometry, John Wiley & Sons, Inc., Chichester, 1995 Search PubMed.
- J. Dědina, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyers, John Wiley & Sons, Ltd., 2010 Search PubMed.
- R. E. Sturgeon, J. Liu, V. J. Boyko and V. T. Luong, Anal. Chem., 1996, 68, 1883–1887 CrossRef CAS.
- P. Pohl, TrAC, Trends Anal. Chem., 2004, 23, 21–27 CrossRef CAS.
- P. Pohl and B. Prusisz, Anal. Bioanal. Chem., 2007, 388, 753–762 CrossRef CAS.
- A. D'Ulivo, Spectrochim. Acta, Part B, 2010, 65, 360–375 CrossRef.
- S. Musil, J. Kratzer, M. Vobecký, J. Hovorka, O. Benada and T. Matoušek, Spectrochim. Acta, Part B, 2009, 64, 1240–1247 CrossRef.
- Y. Arslan, T. Matoušek, J. Kratzer, S. Musil, O. Benada, M. Vobecký, O. Y. Ataman and J. Dědina, J. Anal. At. Spectrom., 2011, 26, 828–837 RSC.
- S. Musil, J. Kratzer, M. Vobecký, O. Benada and T. Matoušek, J. Anal. At. Spectrom., 2010, 25, 1618–1626 RSC.
- J. Dědina, Spectrochim. Acta, Part B, 2007, 62, 846–872 CrossRef.
- O. Y. Ataman, Spectrochim. Acta, Part B, 2008, 63, 825–834 CrossRef.
- T. Matoušek, Anal. Bioanal. Chem., 2007, 388, 763–767 CrossRef.
- H. Matusiewicz and R. E. Sturgeon, Spectrochim. Acta, Part B, 1996, 51, 377–397 CrossRef.
- D. K. Korkmaz, N. Ertas and O. Y. Ataman, Spectrochim. Acta, Part B, 2002, 57, 571–580 CrossRef.
- D. Korkmaz, J. Dědina and O. Y. Ataman, J. Anal. At. Spectrom., 2004, 19, 255–259 RSC.
- I. Menemenlioglu, D. Korkmaz and O. Y. Ataman, Spectrochim. Acta, Part B, 2007, 62, 40–47 CrossRef.
- D. Korkmaz, C. Demir, F. Aydin and O. Y. Ataman, J. Anal. At. Spectrom., 2005, 20, 46–52 RSC.
- J. Kratzer and J. Dědina, Spectrochim. Acta, Part B, 2005, 60, 859–864 CrossRef.
- J. Kratzer and J. Dědina, Spectrochim. Acta, Part B, 2008, 63, 843–849 CrossRef.
- J. Kratzer and J. Dědina, J. Anal. At. Spectrom., 2006, 21, 208–210 RSC.
- J. Kratzer and J. Dědina, Anal. Bioanal. Chem., 2007, 388, 793–800 CrossRef CAS.
- J. Kratzer, Spectrochim. Acta, Part B, 2012 DOI:10.1016/j.sab.2012.04.005.
- J. Kratzer, M. Vobecký and J. Dědina, J. Anal. At. Spectrom., 2009, 24, 1222–1228 RSC.
- G. Ertas and O. Y. Ataman, Appl. Spectrosc., 2004, 58, 1243–1250 CrossRef CAS.
- G. Ertas and O. Y. Ataman, Appl. Spectrosc., 2006, 60, 423–429 CrossRef CAS.
- H. W. Sun and R. Suo, Int. J. Environ. Anal. Chem., 2008, 88, 791–801 CrossRef CAS.
- H. B. Ma, X. F. Fan, H. Y. Zhou and S. K. Xu, Spectrochim. Acta, Part B, 2003, 58, 33–41 CrossRef.
- A. S. Luna, H. B. Pereira, I. Takase, R. A. Goncalves, R. E. Sturgeon and R. C. de Campos, Spectrochim. Acta, Part B, 2002, 57, 2047–2056 CrossRef.
- C. B. Zheng, R. E. Sturgeon and X. D. Hou, J. Anal. At. Spectrom., 2010, 25, 1159–1165 RSC.
- H. Matusiewicz and M. Krawczyk, Cent. Eur. J. Chem., 2011, 9, 648–659 CrossRef CAS.
- O. Řezáčová and J. Dědina, Spectrochim. Acta, Part B, 2009, 64, 717–720 CrossRef.
- T. Matoušek and R. E. Sturgeon, J. Anal. At. Spectrom., 2003, 18, 487–494 RSC.
- T. Matoušek, J. Dědina and M. Vobecký, J. Anal. At. Spectrom., 2002, 17, 52–56 RSC.
- T. Matoušek and R. E. Sturgeon, J. Anal. At. Spectrom., 2004, 19, 1014–1016 RSC.
- T. Matoušek and J. Dědina, Spectrochim. Acta, Part B, 2000, 55, 545–557 CrossRef.
- J. Dědina, Spectrochim. Acta, Part B, 1991, 46, 379–391 CrossRef.
- S. Suda, Y. M. Sun, B. H. Liu, Y. Zhou, S. Morimitsu, K. Arai, N. Tsukamoto, M. Uchida, Y. Candra and Z. P. Li, Appl. Phys. A: Mater. Sci. Process., 2001, 72, 209–212 CrossRef CAS.
- A. D'Ulivo, C. Baiocchi, E. Pitzalis, M. Onor and R. Zamboni, Spectrochim. Acta, Part B, 2004, 59, 471–486 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ja30112a |
| This journal is © The Royal Society of Chemistry 2012 |