Research highlights
Šeila
Selimović
ab and
Ali
Khademhosseini
*abcd
aCenter for Biomedical Engineering, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Cambridge, Massachusetts 02139, U. S. A. E-mail: alik@rics.bwh.harvard.edu
bHarvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, U. S. A.
cWyss Institute for Biologically Inspired Engineering, Harvard University, Boston, Massachusetts 02115, U. S. A.
dWorld Premier International–Advanced Institute for Materials Research (WPI-AIMR), Tohoku University, Sendai 980-8577, Japan
First published on 24th November 2011
Rapid self-healing materials
Self-healing properties are important in a variety of artificial materials, from bio-inspired sutures and bandages to protective clothing items to large-scale objects such as aeroplane wings.1 Since microcracks can lead to functional failure in these materials, it is not only crucial that they self-repair, but also that this healing process occurs rapidly. Recent research on self-healing properties has produced synthetic materials with built-in vascular structures, designed to hold and, when needed, release epoxy solutions. When a crack is formed, these solutions flow out, mix with each other and harden to form a new, solid and healed material surface.A major shortcoming of these materials is that the healing process often takes days. Lewis and colleagues have now addressed this problem and designed a polymeric microvasculature that enables self-healing of cracks in the order of a few hours.
Hansen et al.2 fabricated a vascularized architecture by alternately direct-writing wax and liquid Pluronic F127 inks. The fluidic channels were embedded in the wax layers and separated by the Pluronic ink. Two sets of these interconnected yet separate channels were filled with an epoxy resin and a curing agent, respectively. They were oriented perpendicularly with respect to the ink layers, such that the channels penetrated both the wax and Pluronic sheets. In addition to this vascular structure, the researchers also embedded a third set of channels that cycled heated water (Fig. 1). The placement of these heating channels was chosen such that they interdigitated with the epoxy channels, thereby heating them locally.
 | ||
| Fig. 1 Schematic (a) and photograph (b) of a self-healing structure with three interpenetrating sets of channels. Blue designates the epoxy resin, red the curing agent, and green the heating channels, which carry warm water and heat the device locally. Scale: 5 mm. Reprinted with permission from Advanced Functional Materials from Hansen et al.2 | ||
When a microcrack occurred in the device, epoxy resin and hardener flowed out to mix and seal the crack. As the epoxy was heated and thus became less viscous, however, the hardening process was reduced from a few days to below 10 h, depending on the water temperature. The elastic and viscous moduli, as well as the complex modulus of the hardening epoxy were recorded as a measure of the stiffness of the crack sealant. It was shown that when heated with 70 °C warm water, the epoxy was crosslinked to 80% (in terms of the normalized complex modulus) in 15 h. The epoxy heated to only 30 °C did not reach this level of crosslinking even after 2.5 days. Similarly, after cooling down to room temperature the healed materials reached the same heal stress (3 × 106 Pa) in 4 h when originally heated to 70 °C, vs. more than 50 h (when heated to 30 °C).
The proposed ternary network of microchannels was thus shown to greatly reduce the self-healing time of the synthetic material. In addition, comparison of the rheological properties of the room-temperature and heated epoxies confirmed that the mechanical properties of the sealed fissures did not suffer from the heating process. Finally, the embedded heating element is superior to electrically controlled elements, as it offers highly localized heating and is compatible with large thermal loads. It is unclear how an element that requires a continuous flow of water could be incorporated in portable materials, but incorporation of non-toxic chemicals in an exothermic reaction could be a potential answer. Still, the observations presented by the researchers make this ternary structure appealing to further exploration and potential biological or engineering applications.
Single-cell and single-enzyme microfluidics
Single-cell detection in large cell populations, such as detection of circulating tumor cells (CTCs), has recently become a focus in the development of microfluidic platforms for diagnostic and therapeutic purposes. Similarly, the evidence and direct observation of single molecules and enzymatic reactions has also been a goal in the design of new microfluidics-based experiments for basic research.3 To improve the detection sensitivity in enzyme assay screens, Knudsen and co-workers have now employed a microfluidic approach bridging single-cell and single-enzyme studies.In their paper, Juul et al.4 introduce a poly(dimethyl siloxane) flow-focusing setup for encapsulating individual cells in picolitre-size aqueous droplets (Fig. 2 top). The droplets, with a lysis buffer as the carrier solution, were sheared off by an oil stream (FC-40) and contained cells and a DNA sensor solution. The droplets were then stored in vials and transferred to the 2D array of droplet traps when needed (Fig. 2 bottom).
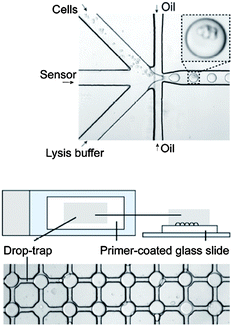 | ||
| Fig. 2 Top: encapsulation of cells and the DNA sensor molecules in picolitre lysis buffer droplets, separated by fluorocarbon oil, in a nozzle structure. Bottom: array of droplet traps, with each trap holding a single droplet with encapsulated cells. Reprinted and adapted with permission from ACS Nano from Juul et al.4 | ||
Two processes took place during the experiment. First, the DNA sensor molecules entered the cells and recorded individual enzyme-based DNA cleavage-ligation events. Then, the DNA sensors were converted into circular products, which enabled rolling circle amplification (RCA). Contrary to the exponential PCR amplification method, RCA multiplies stretches of DNA linearly, such that each RCA signal (observed via a fluorescent probe) correlates with a single DNA cleavage-ligation event. Furthermore, the RCA process is completed isothermally, and it does not require a temperature cycler like PCR does. The RCA process was thus easily combined with microfluidics, as the signal amplification chiefly took place in the droplet storage array and could be immediately visualized.
To test the sensitivity of the RCA and readout method, the researchers observed enzyme activity in untransfected or control cells (human embryonic kidney cells HEK293) and in cell populations containing 0.025%, 0.25% and 2.5% cells expressing an immunomodulatory protein FIp. Another protein that was tested for was hTOPl, a topoisomerase involved in the torsional alignment of DNA strands. Finally, signals from single cells (0.5 million ml−1), cells encapsulated in picolitre droplets, and cells in bulk solutions were compared (5 million cells ml−1). Cells expressing FIp emitted a red fluorescent signal, and cells expressing hTOP1 glowed green. The number of observed red signals decreased linearly with the concentration of FIp-active cells, and the number of hTOP1 events increased.
The bulk solution experiment did not yield any results beyond the 2.5% concentration of active cells, as the signal was overwhelmed by the background. This was in stark contrast to the droplet based assays, which gave measurable results even at 0.025% concentration of active cells. Due to the uneven number of cells encapsulated in different droplets, however, the variation in the average signal was large: the number of fractional signals was 13 + −7 at some conditions. Nonetheless, this issue could likely be resolved in the future by sorting droplets with the desired number of cells. Single cell experiments gave the most precise measurements of FIp activity (fractional number of recorded FIp events was 23 + −2). Finally, the control solutions did not yield any observations of FIp-activity, as expected.
Storing the reaction pods or droplets in a 2D array during this process enabled the researchers to directly and in real-time visualize the enzymatic activity, and reducing the droplet size to the picolitre scale vastly increased the sensitivity of the experiment and shortened the reaction times. Future modifications to the platform could include storage pods for femtolitre size droplets, with each droplet carrying a single cell, as well as a mechanism for controlled access to individual droplets and cells. The choice of the enzymes tested in this research also directly exhibits the value of this platform. Namely, hTOP1 is a protein of great interest in chemotherapeutics, in particular in cancers of the colon and ovaries. It would be straightforward, however, to use this platform in detection of enzymatic activity in CTCs or rare, abnormal cells, chiefly for the promise of high measurement accuracy and precision.
Complex, 3D hydrogel patterning
Basic research involving stem cells always requires mimicking some features of the native external cell microenvironment, including physical properties (stiffness and topography of the substrate) and chemical characteristics (substrate functionalization, growth and differentiation factors). Thus, a 3D synthetic environment for stem cell culture, such as hydrogel particles, generally simulates the native conditions better than a 2D surface, but a planar surface is easier to modify and selectively functionalize than a 3D object.5 Progress in stem cell research requires that this gap between function and convenience be bridged, and Shoichet and colleagues have recently succeeded in doing just that: in their paper, Wylie et al.6 detail a novel method for simultaneous 3D patterning of various proteins in a hydrogel particle for cell culture.In this approach, agarose gel containing thiol groups was first crosslinked, which activated the thiols. Then, a solution of maleimide–barnase proteins permeated during a soak into the hydrogel. When selectively exposed to two-photon irradiation, the thiols were activated and available for bonding and the proteins were immobilized. After a buffer wash, this process was repeated with a maleimide–streptavidin solution in a spatially different volume of the 3D hydrogel. The maleimide was anchored to the thiols, and the functional proteins were added to serve as a bonding agent for future growth factors. Finally, the fusion molecules barstar–SHH and biotin–CNTF were simultaneously introduced into the hydrogel via diffusion and attached to the already present bonding agents. The barstar proteins specifically bound to barnase, and biotin formed bonds with streptavidin, as expected. The final wash removed uncrosslinked SHH and CNTF, leaving only spatially controlled patches of the growth factors in the 3D gel. Patches in the order of 20–40 μm in any of the three dimensions were successfully patterned with either protein, with concentrations varying from 10 nm to more than 140 nm, depending on the total photon energy. The bound concentration of the proteins was determined by labelling them with fluorescent dye and then measuring its intensity in the crosslinked patches. It should be noted that the bound concentration also depended on the number of binding sites of barnase (1) and streptavidin (4 binding sites).
When retinal precursor cells were encapsulated in the hydrogel, the observed viability in the functionalized volumes of the gel was comparable to or better than the results on pure streptavidin and pure biotin. However, certain biological functions were affected. For example, cells cultured on immobilized CNTF expressed phosphorylated STAT3, a transcription factor active in cell proliferation and angiogenesis, as well as in suppression of the host immune response to tumor cells. When exposed to immobilized CNTF, however, the cells did not express this marker. Further, cells cultured on patches of SHH, a factor in oncogenic pathways, expressed a transcription factor mediating that pathway, while cells grown directly on barstar did not show this marker. Thus, both SHH and CNTF had remained bioactive throughout the gel patterning process.
In the proposed fashion, various 3D arrangements as well as concentration gradients of SSH and CNTF were patterned in the agarose gel, including convex and concave shapes. This method represents an exciting new step in the mimicry of cellular microenvironments, which is crucial for many bioengineering applications, from stem cell differentiation to conducting cancer drug assays. In the future, hybrid technologies could develop that combine the present protein patterning method with layer-by-layer fabrication of hydrogel units with spatially varying porosities or stiffnesses. Such a fabrication strategy would allow us to better understand the physical and chemical characteristics of the complex 3D cell microenvironment.
References
- R. P. Wool, Self-healing materials: a review., Soft Matter, 2008, 4, 400–418 RSC.
- C. J. Hansen, et al., Accelerated Self-Healing Via Ternary Interpenetrating Microvascular Networks, Adv. Funct. Mater., 2011 DOI:10.1002/adfm.201101553.
- W. T. S. Huck, et al., Microdroplets, Angew. Chem., Int. Ed., 2010, 49, 5846–5868 Search PubMed.
- S. Juul, et al., Detection of Single Enzymatic Events in Rare or Single Cells Using Microfluidics, ACS Nano, 2011, 5, 8305, DOI:10.1021/nn203012q.
- A. W. Lund, et al., The natural and engineered 3D microenvironment as a regulatory cue during stem cell fate determination, Tissue Eng., Part B: Rev., 2009, 15, 371–380 CrossRef.
- R. G. Wylie, et al., Spatially controlled simultaneous patterning of multiple growth factors in three-dimensional hydrogels, Nat. Mater., 2011, 10, 799, DOI:10.1038/nmat3101.
| This journal is © The Royal Society of Chemistry 2012 |