DOI:
10.1039/C1NJ20662A
(Paper)
New J. Chem., 2012,
36, 64-72
The importance of cellular localisation of probes: synthesis, photophysical properties, DNA interactions and cellular imaging properties of rhenium dppz complexes with known cellular localisation vectors†
Received
(in Montpellier, France)
29th July 2011
, Accepted 19th September 2011
First published on 12th October 2011
Abstract
The synthesis, photophysical properties, DNA binding, DNA cleavage and cellular imaging behaviour of a range of complexes of the type [Re(CO)3(dppz)(PyR)]+ are reported, where PyR represents a range of substituted pyridines which have previously been studied for cellular localisation of related complexes. Confocal imaging experiments confirm that the complexes retain the variety of cellular localisation behaviour associated with the PyR units in other complexes, and suggest applications as probes for oligonucleotides in specific cellular compartments (e.g. mitochondrial DNA). This study illustrates the importance of considering cellular localisation as a prime consideration in the design of probes for in vivo application.
Introduction
The use of large, planar aromatic systems as DNA intercallators is well established in chemical and biological studies of DNA and related systems, and dipyrido[3,2-a:2′,3′-c]phenazine (dppz) 1 has become a standard ligand in the design of metallointercallators. In the field of luminescent dppz complexes for DNA binding studies, cationic ruthenium complexes of the general formula [Ru(bpy)2(dppz)]2+ have been widely applied and studied both experimentally and theoretically due to their strong interaction with DNA, and the remarkable photoswitching upon interaction with DNA.1–5 Due to complex modulation of the nature of the excited states it is possible to achieve up to 104 enhancement of emission intensity upon DNA binding compared to the free aqueous species.6 While there are many studies of in vitro application of such Ru-dppz systems, in vivo application is rarer, due to complications relating to cellular uptake and distribution of such species. For instance, [Ru(phen)3]2+ shows a tendency to be trapped in endocytotic vessels,7 while other related complexes8 have been successfully applied in cell imaging after bioconjugation to cell penetrating units (e.g. polyarginines). In general, while there are many studies of tuning the photophysical response of these probes to DNA/RNA, there has been much less attention paid to the cellular localisation of the probes, although these may be crucial for in vivo applications. In fact, one study9 has shown that rather than accumulating in the nucleus, a variety of Ru-dppz complexes showed accumulation in cytoplasmic organelles, which could limit applicability as in vivo DNA probes. A recent interesting study10 showed that in a series of dialkoxy substituted complexes [Ru(phen)2dppz-(OR)2], where R = C2–C6, all the probes showed good uptake, with C2 staining only the nucleus, excluding the nucleolus, C4 localising in cytoplasm and nucleoli, and C6 giving intense cytoplasmic staining, and weak nucleolar staining. These dramatic differences in localisation with small changes to structure illustrate one of the problems with probe design, that an alteration in probe structure may change not only its response to analytes (the complexes in this study had differing responses to DNA/RNA/mRNA) but also its localisation. In intensity-based detection this can lead to difficulty in distinguishing between areas with high probe concentration in the less emissive form, and those of low concentration in the highly emissive form. In order to allow meaningful in vivo studies a fuller understanding of the cellular localisation of probes must be developed. By comparison to the dominant ruthenium complexes, there has been less work on the related rhenium complexes, although it was shown some years ago that Re-dppz complexes were capable of binding DNA, and the photophysics and electronic basis of the interaction have been studied in some depth.11–23 In particular, it has been shown that complexes of the general structure [Re(CO)3(dppz)(X)] where X represents a wide variety of axial ligands are very weakly emissive in aqueous media, but show dramatic emission enhancements in the presence of DNA or oligonucleotides, ranging from > 10 fold enhancement,12 to a ‘switch on’ from essentially non-emissive,23 depending upon substitution and conditions. There is a growing body of literature24 showing that rhenium polypyridyl complexes are useful in cell imaging experiments, in particular, rhenium fac-tricarbonyl complexes of chelating bipyridines and related species have been shown to be advantageous over other triplet emitters due to the flexibility of having an additional coordination site once the requirement for emissivity of an [Re(bpy)(CO)3] core is satisfied. This site is usually occupied by a substituted pyridine ligand, which can be used as a vector to control cell uptake and localisation, while the photophysical properties are largely dominated by the nature of the bipyridine. This pyridine ligand has been shown to be capable of controlling uptake and directing localisation to organelles such as mitochondria, membranes, vacuoles and nucleoli.25 In order to help in the design of Re-dppz probes, therefore, it was of interest to investigate the effect of systematically varying the axial ligand to generate a series of rhenium dppz complexes which, in principle, could be expected to retain the useful DNA-related properties but show variations in cellular localisation and uptake. This is of interest for three reasons: to determine how/whether variation of the pyridine ligand changes the interaction with DNA; to determine the optimum design for the cellular uptake of rhenium dppz species; and to potentially target DNA in specific cellular compartments.
Results and discussion
Synthesis of complexes
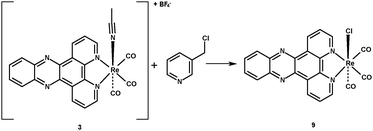
Given the requirement for a range of rhenium dppz complexes with varied axial ligands the common precursor [ReBr(CO)3dppz] was prepared by reaction of [ReBr(CO)5] with dppz 1 (itself prepared by the method of Barton)26 in toluene at sub-reflux for 18 h to give the expected product 2 in excellent yield and high purity. Subsequent activation of the bromide with silver tetrafluoroborate in acetonitrile gave the derived cationic complex 3 containing an acetonitrile ligand, which in analogous rhenium bipyridine chemistry is a common reactive intermediate used to facilitate the formation of the pyridine substituted complexes (Scheme 1). In order to generate a range of pyridine-substituted rhenium dppz complexes 3 was reacted with a number of 3-substituted pyridines under standard conditions (excess ligand, chloroform sub-reflux, 16 h) to give the derived complexes 4–7 in 50–80% yields, and the identity of the complexes was in each case confirmed by NMR, IR and mass spectrometry. The pyridine ligands were chosen as they had previously been shown in the analogous bipyridine series to give a range of degrees of cellular uptake and localisation pattern. One complex which is attractive as in the bipyridine series it shows mitochondrial localisation27 is the chloromethypyridine complex 8, however previous work had indicated that the direct coordination of 3-chloromethypyridine to activated rhenium species is problematical. In the case of the dppz complexes currently under investigation, again, reaction of the acetonitrile complex 3 with 3-chloromethylpyridine proved an unfruitful approach to 8, with only the chloro-complex 9 being isolated as a discrete product indicating that halide abstraction interferes with coordination of the pyridine (Scheme 2).
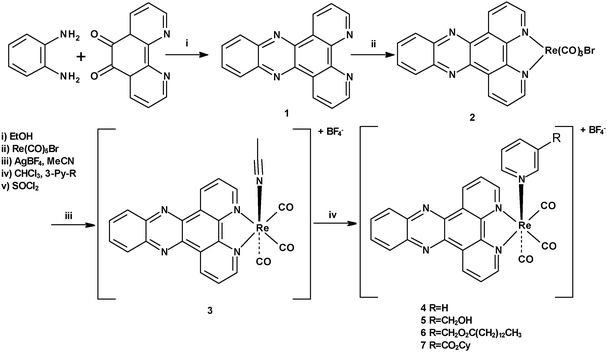 |
| | Scheme 1 Synthesis of complexes 3–7. | |
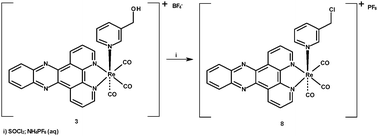
Fortunately it was possible to access the desired chloromethyl complex 8 by treatment of hydroxymethyl complex 5 with thionyl chloride, followed by quenching with saturated aqueous ammonium hexafluorophosphate to isolate the product as the hexafluorophosphate salt (Scheme 3).
Photophysical studies
As the photophysics of rhenium polypyridyl complexes are dominated by the nature of the polypyridine ligands, and the axial ligands typically are less important it was anticipated that the series of complexes 3–8 would show similar photophysical properties to each other, and to the range of previously reported rhenium-dppz complexes, however a brief investigation of these properties was undertaken to confirm that all of the complexes conformed to the expected behaviour. This was deemed especially important in the case of 6 as it has been previously noted that rhenium polypyridines substituted with alkyl chains can show anomalous photophysical behaviour as a result of chain-wrapping events.28 The electronic spectra of complexes 3–8 all show intense absorptions centred around 280 nm, assigned as ligand-centred 1IL π–π* and n–π* transitions, with multiple weaker peaks and shoulders at lower energy centred at around 380 nm assigned as 1MLCT (dπ(Re)–π*(dppz)) absorptions by analogy with previous work (see ESI for more data†).13 It has previously been noted that the luminescence of [Re(dppz)Py] species is moderate, and dominated by 3IL states13 while upon DNA binding12,23 the intensity of emission increases, although the effect is less pronounced than for related ruthenium systems. The nature of any substitution of the axial pyridine ligand typically has little effect upon the emission of such complexes, and so it was to be expected that complexes 4–8 would show similar photophysics, while 3 could have been expected to differ as the replacement of a pyridine ligand by another group in Re dppz complexes has been reported to modulate the luminescence (at least when the axial ligand is a halide). In fact all of the complexes 3–8 showed the typical excitation and emission of [Re(dppz)(Py)] species with excitation maxima around 345 nm and two emission bands centred at 455 ± 10 nm and 550 ± 5 nm except 4 in which the high energy band is shifted to 495 nm (see ESI for more data†). These photophysical attributes suggested that complexes 3–8 had potential as imaging agents and so further studies of their behaviour were undertaken.
While it is well established that simple rhenium dppz complexes are strong DNA binders, it was important to confirm that the inclusion of large units such as in the myristyl or cyclohexyl derived complexes (6 and 7 respectively) had not precluded DNA binding.
Electronic absorption spectroscopy is an effective tool to examine the binding mode of DNA with metal complexes.29 The predominant target of metal-based anticancer complexes is often believed to be binding to DNA through N-7 of guanine.30 Intercalative binding of a complex with DNA is accompanied by both a hypochromic and bathochromic shift in the absorption spectrum.31 Intercalation can occur due to a strong stacking interaction between the aromatic rings present in the ligand framework and the DNA base pairs, and if a π* orbital of the intercalated ligand can couple with a π orbital of the DNA base pairs, the π–π* transition energy decreases inducing a red shift in the observed band, whereas, if a ligand π orbital is partially filled, it decreases the probability and concomitantly results in hypochromism.32 Thus, in order to provide evidence for the possibility of binding of each complex to CT-DNA, spectroscopic titration of solutions of each of the complexes with CT-DNA was performed. In order to compare the binding strength of complexes with CT DNA, the intrinsic binding constant Kb was calculated in each case by monitoring the changes in their absorbance with increasing concentration of DNA. A representative spectrum of complex 6 is shown in Fig. 1 (see ESI for more data†). The value of Kb is obtained as a ratio of slope to intercept obtained from the plot of [DNA]/(εa − εf) vs. [DNA], and are summarised in Table 1. Complexes 3–8 all show values in the region of 0.7 − 6 × 105 M−1 which is comparable with the values previously observed for related species,11,13 and indicative of strong binding to DNA. The Kb values thus obtained varied in the order Kb (6) > Kb (5) ≫ Kb (4) > Kb (3) > Kb (7) > Kb (8), with 5 and 6 being significantly higher than the other complexes. The anomalously strong DNA binding observed with complexes 5 and 6 could be explained by hydrogen bonding from the hydroxymethyl pyridine of 5 to a DNA base and by hydrophobic interaction between the myristyl chain of 6 with DNA. Hydrophobic effects are important in DNA binding33 and the enthalpic gain of removing a C13 chain from the aqueous medium may well be important to the high binding constant observed with this complex. It should be noted that the acetonitrile ligand of 3 is regarded as labile and it is presumed that the species equilibrates between aqua- and halo- complexes in buffered media, with the final species being likely to be coordinated at metal by a group from the DNA fragment itself. That the binding constant of this complex is not significantly higher than species which lack a labile ligand suggest that any such interaction is weak, as may be expected for phosphate oxygens, or even the amidic nitrogens of DNA bases.
![UV-vis absorption spectra of (a) [Complex 3] = 10 μM in the absence and in presence of increasing amounts of DNA = 0–100 μM., (b) [Complex 6] = 10 μM in the absence and in presence of increasing amounts of DNA = 0–100 μM. Arrow shows the absorbance changes upon increasing DNA concentration.](/image/article/2012/NJ/c1nj20662a/c1nj20662a-f1.gif) |
| | Fig. 1 UV-vis absorption spectra of (a) [Complex 3] = 10 μM in the absence and in presence of increasing amounts of DNA = 0–100 μM., (b) [Complex 6] = 10 μM in the absence and in presence of increasing amounts of DNA = 0–100 μM. Arrow shows the absorbance changes upon increasing DNA concentration. | |
Table 1 Kb values for complexes 3–8
| Complex |
3
|
4
|
5
|
6
|
7
|
8
|
| Kb/105 (M−1) |
1.17 |
2.56 |
5.59 |
6.28 |
0.938 |
0.728 |
Competitive binding with ethidium bromide (EB)
The ability of a complex to affect the fluorescence intensity of EB-DNA adduct is a reliable tool for the measurement of its affinity towards DNA, irrespective of its mode of binding. Intense fluorescence is observed from EB in the presence of DNA, owing to its strong intercalation between adjacent DNA base pairs. A complex binds with DNA by the displacement of the EB which is already bound to DNA. Consequently, the intensity of emission is reduced as emission from of free EB is readily quenched by surrounding water molecules.34 Representative emission spectra of the EB-DNA system in the presence and absence of complexes 3 and 6 are shown in Fig. 2 (see ESI for more data). The addition of complexes to DNA pretreated with EB results in appreciable reductions in emission intensity, indicative of DNA binding by the complex, and subsequent aqueous quenching of displaced EB. On the addition of 4–7 μM of complexes to 10 μM of CT DNA pretreated with EB, ∼80% displacement of ethidium bromide was observed. Corresponding quenching plots of Io/I vs. [Complex]/[DNA] (the inset of Fig. 2) are in good agreement with the linear Stern–Volmer equation. The Stern–Volmer constant (Ksv) is evaluated as 1.3, 1.0, 2.0, 5.5, 0.86 & 0.61 for complexes 3, 4, 5, 6, 7, and 8 respectively which gives the order of the observed extent of ethidium bromide displacement by the complexes as 6 > 5 > 3 > 4 > 7 > 8. This is the same gross order as seen in the uv-vis work but with 3 and 4 switching places, presumably as a result of their different interactions with EB complicating the simple DNA affinity relationship seen in the UV-vis titrations.
![Emission spectra of EB bound to DNA in the absence and in the presence of, [complex 3 and 6] 0–5 μM, [EB] 10 μM, [DNA] 10 μM. Arrow shows changes in the emission intensity upon addition of increasing concentration of the complex.](/image/article/2012/NJ/c1nj20662a/c1nj20662a-f2.gif) |
| | Fig. 2 Emission spectra of EB bound to DNA in the absence and in the presence of, [complex 3 and 6] 0–5 μM, [EB] 10 μM, [DNA] 10 μM. Arrow shows changes in the emission intensity upon addition of increasing concentration of the complex. | |
In summary, all complexes 3–8 showed strong DNA binding with KB in the region of 104–105 M−1 which are in the typical range for rhenium dppz complexes and bode well for applications as cellular probes for DNA.
DNA cleavage studies
Binding of transition metal complexes with DNA plays an important role in identification of target structures for chemotherapeutic agents based on DNA cleavage. However, even in cases where binding is strong due to a motif such as dppz, DNA cleavage abilities show additional dependency on the other substituents present in the complexes. The DNA-cleaving ability of complexes 3–8 has been studied by the relaxation of supercoiled pBR322 DNA to the nicked circular DNA. When circular plasmid DNA is subjected to gel electrophoretic mobility assays, relatively fast migration is observed for the intact supercoiled form (S). However, if scission of DNA occurs at one strand (nicking), the supercoiled DNA relaxes to generate a slower-moving open/nicked circular form (NC). If both strands are cleaved, a linear form (L) is generated.35 To assess the DNA cleavage ability of the complexes, supercoiled pBR322 DNA (300 ng μL−1) was incubated separately with 100 μM of the complexes in 5 mM Tris-HCl/50 mM NaCl buffer at pH 8.0 (Fig. 3). As compared to the untreated plasmid DNA sample, there was no change in the intensities of SC and NC forms of DNA when incubated with complexes in aqueous buffered solution up to 24 h, indicating little or no cleavage of DNA by any of the complexes studied.
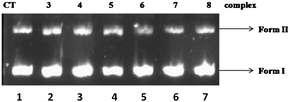 |
| | Fig. 3 Ethidium bromide stained agarose gel (1.5%) of pBR322 plasmid DNA (300 ng μL−1) in the presence of 100 μM complexes after 24 h of incubation, Lane 1: DNA control. Lane 2: pBR322 + 3. Lane 3: pBR322 + 4. Lane 4: pBR322 + 5. Lane 5: pBR322 + 6. Lane 6: pBR322 + 7. Lane 7: pBR322 + 8. | |
As no DNA cleavage was observed with complexes 3–8 under ambient conditions, attempts were made to investigate the photonuclease activity of the complexes. Thus each complex exposed to UV light at 312 nm (64 W) for 10 min in an aqueous solution containing DNA. All complexes except 7 and 8 cause a linear increase in the intensity of NC DNA band accompanied with the similar decline in SC DNA in a concentration dependent manner in the aqueous buffer. Complex 6 converted more than 80% of SC form into NC form at a concentration of 50 μM, whereas 80 μM of 3 and 5 is required for similar conversion (Fig. 4). DNA cleavage by complex 6 is significantly higher than that observed with complex 3, which could be related to the electron releasing substituent in view of an earlier report.36 However, DNA cleavage by complex 3 could be related to the liability of the acetonitrile ligand which provides an opportunity for coordination of rhenium by a DNA base, usually guanine.37 It should be noted that the significantly higher DNA binding ability of complex 6 compared to complexes 3 and 5 may also be significant in their relative cleavage efficiencies.
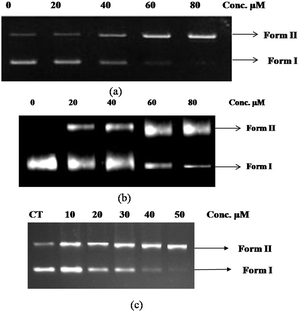 |
| | Fig. 4 UV light induced cleavage of supercoiled pBR322 plasmid DNA in the presence of increasing concentration of complexes in aqueous buffered solution. The plasmid DNA (300 ng μL−1) + complex mix were incubated for 10 min, followed by UV exposure at 312 nm (64 W) for 10 min. The samples were then subjected to 1.5% agarose gel electrophoresis and bands were developed by ethidium bromide staining. (a) Lane 1, DNA control; Lane 2, DNA + 20 μM of complex 3; Lane 3, DNA + 40 μM of complex 3; Lane 4, DNA + 60 μM of complex 3; Lane 5, DNA + 80 μM of complex 3 (b) Lane 1, DNA control; Lane 2, DNA + 20 μM of complex 5; Lane 3, DNA + 40 μM of complex 5; Lane 4, DNA + 60 μM of complex 5; Lane 5, DNA + 80 μM of complex 5 (c) Lane 1, DNA control; Lane 2, DNA + 10 μM of complex 6; Lane 3, DNA + 20 μM of complex 6; Lane 4, DNA + 30 μM of complex 6; Lane 5, DNA + 40 μM of complex 6; Lane 5, DNA + 40 μM of complex 6; Lane 6, DNA + 50 μM of complex 6. | |
In summary, there is some interesting and axial ligand-dependent DNA photocleavage behaviour associated with certain examples of the rhenium dppz complexes studied in this report which will be the focus of further studies. However, none of the complexes studied showed DNA cleavage in the absence of irradiation. Thus, as all complexes bind DNA well, do not cleave DNA under physiological conditions, and show attractive photophysical properties for cell imaging, they were considered potential candidates for live cell imaging studies.
Cell imaging studies
To the best of our knowledge, while their photophysical properties have been widely studied, there are no previous reports of the application of rhenium dppz complexes in fluorescent cell imaging. Complexes 3, 5, 6 and 9 were selected for cell imaging studies due to their favourable binding and fluorescence properties and the known uptake and localisation behaviour of the bipyridine24–28 and (in a recent example involving polylactide chains) phenanthroline38 analogues, which have been examined in detail and confirmed by co-localisation experiments with commercial stains. Given the interest in this case of comparing the known tendency of dppz complexes to show nuclear localisation, and the emission enhancements observed upon binding these complexes with DNA the imaging experiments were undertaken under colocalisation conditions with Hoechst34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 580 a known nuclear and DNA specific stain. By comparing the localization of the new complexes with this material, and by comparing the intensity of rhenium-based emission from the nucleus (which is DNA rich and thus shows intensity-enhanced Re-based emission) with the intensity of other areas the possibility of binding to DNA/RNA present in other organelles can be examined. It should be noted that in addition to the luminescence enhancements reported for rhenium dppz complexes in the presence of DNA, other environments can cause enhanced emission in comparison to aqueous solution. Changing from water to organic solvents can cause many-fold enhancements of emission intensity23 and it is likely that the complexes in question could show interaction with, and subsequent luminescent enhancement in e.g. bilipid membranes. In the event of the complexes localising in aqueous compartments of the cell it is unlikely that they would be detected-the emission of aqueous solutions of such species being barely detectable by fluorimetry, and certainly below the detection limits of the microscope. Thus, it is impossible to be sure of the full localisation of these complexes as in certain areas they will be difficult to detect. However, as they all have the typical photophysics of rhenium dppz complexes and show the same enhancements in the presence of DNA, lipids etc., any differences observed in patterns of emission intensity between the complexes must indicate control of localisation. Thus differences observed between the localisation of these complexes would indicate that using dppz complexes to map cellular DNA/RNA without prior consideration of their localisation preferences may be misleading. Incubation of the complexes with HeLa cells followed by examination by laser scanning confocal microscopy indicated that the complexes were all taken up by cells and showed different patterns of localisation, validating the theory that the choice of axial ligand is vital to the application of rhenium dppz DNA probes in vivo, and could provide probes selective for e.g. mitochondrial DNA. The acetonitrile complex 3 showed (Fig. 5, 3 shown in green) very similar localisation to that of the standard Hoechst stain (shown in blue), indicating localisation in DNA rich sections of the nucleus from the characteristic emission pattern from the centre of the cell (as indicated by overlap with brightfield images) and overlap with the Hoechst dye (turquoise). As has been previously noted this complex is highly reactive and it is likely that the acetonitrile has been displaced by biological material during the incubation, so the localisation is probably dominated by lipophilicity and DNA binding of the dppz ligand, explaining the nuclear localisation. Complex 5, the hydroxymethyl pyridine complex showed (Fig. 6) a slightly different pattern of localisation, with the nuclear localisation observed for the Hoechst stain and 3 being supplemented by some localisation in the perinuclear region of the cytoplasm, apparently concentrated in organelles. As we have previously noted that rhenium hydroxymethylpyridine complexes show some degree of localisation in mitochondria,25 is likely that the extra-nuclear regions in which 5 shows intense staining are mitochondria, and from the similar relative intensity of the emission from these regions and from the nucleus (where 5 is certain to be DNA bound) that in these regions 5 is bound to mitochondrial DNA. Confirmation of this assumption came with the imaging observed with complex 8 which bears a chloromethylpyridine unit, known to strongly direct localisation almost exclusively to mitochondria by reaction of the chloromethyl unit with the high concentration of reduced thiols located in these organelles.27 In this case (Fig. 7) the emission was almost entirely observed in the perinuclear region from localisation in mitochondria, and only a small degree of overlay was observed with the Hoechst nuclear stain, but again the relative intensities of rhenium-derived emission from mitochondrial and nuclear regions indicates that the mitochondrial-localised dye is DNA-bound. Finally, the myristyl derived complex 6 showed (Fig. 8) much more generalised cytoplasmic staining with little nuclear staining and more diffuse patterns of localisation in the cytoplasm than complexes 5 and 8, clearly localising in a range of organelles and membrane structures (as is expected from previous experiments with this ligand). The intensity of the emission from some organelles and regions is clearly much less than that observed in the cases where 6 overlaps with Hoechst, indicating that these regions are not DNA rich so there is no enhancement of emission. We are confident that the alternative explanation, that overall levels of uptake of this material are much lower than the other complexes, is not applicable as previous work demonstrates that the additional lipophilicity associated with the myristyl chain over simple pyridines leads to much higher levels of uptake.25,27
580 a known nuclear and DNA specific stain. By comparing the localization of the new complexes with this material, and by comparing the intensity of rhenium-based emission from the nucleus (which is DNA rich and thus shows intensity-enhanced Re-based emission) with the intensity of other areas the possibility of binding to DNA/RNA present in other organelles can be examined. It should be noted that in addition to the luminescence enhancements reported for rhenium dppz complexes in the presence of DNA, other environments can cause enhanced emission in comparison to aqueous solution. Changing from water to organic solvents can cause many-fold enhancements of emission intensity23 and it is likely that the complexes in question could show interaction with, and subsequent luminescent enhancement in e.g. bilipid membranes. In the event of the complexes localising in aqueous compartments of the cell it is unlikely that they would be detected-the emission of aqueous solutions of such species being barely detectable by fluorimetry, and certainly below the detection limits of the microscope. Thus, it is impossible to be sure of the full localisation of these complexes as in certain areas they will be difficult to detect. However, as they all have the typical photophysics of rhenium dppz complexes and show the same enhancements in the presence of DNA, lipids etc., any differences observed in patterns of emission intensity between the complexes must indicate control of localisation. Thus differences observed between the localisation of these complexes would indicate that using dppz complexes to map cellular DNA/RNA without prior consideration of their localisation preferences may be misleading. Incubation of the complexes with HeLa cells followed by examination by laser scanning confocal microscopy indicated that the complexes were all taken up by cells and showed different patterns of localisation, validating the theory that the choice of axial ligand is vital to the application of rhenium dppz DNA probes in vivo, and could provide probes selective for e.g. mitochondrial DNA. The acetonitrile complex 3 showed (Fig. 5, 3 shown in green) very similar localisation to that of the standard Hoechst stain (shown in blue), indicating localisation in DNA rich sections of the nucleus from the characteristic emission pattern from the centre of the cell (as indicated by overlap with brightfield images) and overlap with the Hoechst dye (turquoise). As has been previously noted this complex is highly reactive and it is likely that the acetonitrile has been displaced by biological material during the incubation, so the localisation is probably dominated by lipophilicity and DNA binding of the dppz ligand, explaining the nuclear localisation. Complex 5, the hydroxymethyl pyridine complex showed (Fig. 6) a slightly different pattern of localisation, with the nuclear localisation observed for the Hoechst stain and 3 being supplemented by some localisation in the perinuclear region of the cytoplasm, apparently concentrated in organelles. As we have previously noted that rhenium hydroxymethylpyridine complexes show some degree of localisation in mitochondria,25 is likely that the extra-nuclear regions in which 5 shows intense staining are mitochondria, and from the similar relative intensity of the emission from these regions and from the nucleus (where 5 is certain to be DNA bound) that in these regions 5 is bound to mitochondrial DNA. Confirmation of this assumption came with the imaging observed with complex 8 which bears a chloromethylpyridine unit, known to strongly direct localisation almost exclusively to mitochondria by reaction of the chloromethyl unit with the high concentration of reduced thiols located in these organelles.27 In this case (Fig. 7) the emission was almost entirely observed in the perinuclear region from localisation in mitochondria, and only a small degree of overlay was observed with the Hoechst nuclear stain, but again the relative intensities of rhenium-derived emission from mitochondrial and nuclear regions indicates that the mitochondrial-localised dye is DNA-bound. Finally, the myristyl derived complex 6 showed (Fig. 8) much more generalised cytoplasmic staining with little nuclear staining and more diffuse patterns of localisation in the cytoplasm than complexes 5 and 8, clearly localising in a range of organelles and membrane structures (as is expected from previous experiments with this ligand). The intensity of the emission from some organelles and regions is clearly much less than that observed in the cases where 6 overlaps with Hoechst, indicating that these regions are not DNA rich so there is no enhancement of emission. We are confident that the alternative explanation, that overall levels of uptake of this material are much lower than the other complexes, is not applicable as previous work demonstrates that the additional lipophilicity associated with the myristyl chain over simple pyridines leads to much higher levels of uptake.25,27
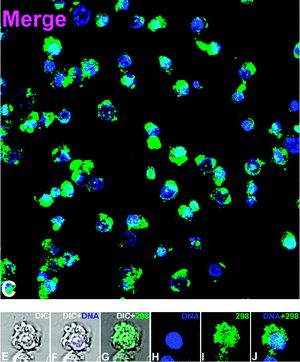 |
| | Fig. 5 Cell imaging with acetonitrile complex 3 (overlaid 3 green Hoechst Blue) (further data in the ESI†). | |
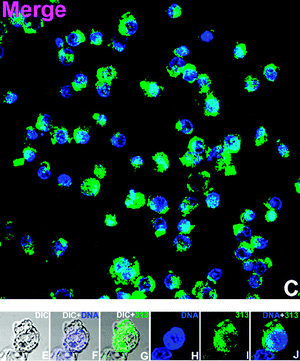 |
| | Fig. 6 Cell imaging with hydroxymethyl complex 5 (overlaid 5 green Hoechst Blue) (further data in the ESI†). | |
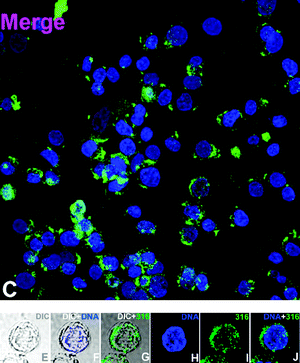 |
| | Fig. 7 Imaging with chloromethyl complex 8 (further data in the ESI†). | |
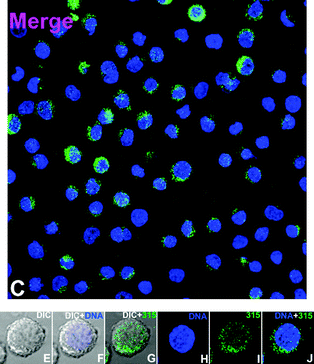 |
| | Fig. 8 Imaging with myristyl complex 6 (further data in the ESI†). | |
Conclusions
From the studies detailed herein, it is clear that rhenium dppz complexes appended through the axial pyridine ligand with groups known to direct cellular localisation retain both their useful DNA binding and photophysical properties, while gaining the organelle specific-localisation desired. The imaging studies suggest that such complexes may be used as probes for e.g. mitochondrial DNA by combining organelle targeting vectors with DNA responsive dppz units. However, crucially this study demonstrates that cellular localisation parameters are a vital consideration when designing probes to be applied in vivo. In the case of probes which rely upon changes in emission intensity in the presence of an analyte for detection, and have no ratiometric aspects,39,40 only a full understanding of the cellular distribution patterns of the probe can allow even qualitative conclusions to be drawn about relative analyte distribution in the organisms studied. The patterns of localisation of metallolumophores are only just becoming understood, as has been highlighted in a recent review,41 and, currently, the examples of vectors for controlling the localisation of rhenium complexes studied in this work represent the only reliable examples in the literature. Until localisation of metallolumophores is better understood and more predictable, therefore, great caution must be applied in the interpretation of the results of imaging experiments relying upon emission intensity to indicate the presence/absence of analytes.
Experimental
Synthesis of complexes
[Re(CO)3(dppz)(MeCN)][BF4] 313.
Complex 238 (400 mg, 0.63 mmol) was dissolved in MeCN (2 ml) and AgBF4 (147 mg, 0.759 mmol) was added. The solution was stirred at sub-reflux in a dark, nitrogen environment overnight. The yellow solution was filtered and evaporated until a minimum amount of solvent remained. The compound was precipitated with ether and was filtered to give the pure product as a yellow solid (288 mg, 42%). 1H NMR (CD3CN) 9.58 (2H, dd, J = 8.3, 1.4) 9.51 (2H, dd, J = 5.3, 1.4) 8.27 (2H dd, J = 6.5, 3.5) 8.15 (2H dd, J = 8.3, 5.3) 8.07 (2H dd, J = 6.6, 3.4) 1.98 (3H, s). 13C NMR (CD3CN) 155.6, 149.0, 142.1, 138.4, 136.0, 132.4, 129.9, 129.1, 127.7, 122.5, 2.6. λmax 361, 380 nm.13
[Re(CO)3(dppz)(Py)][BF4] 4.
Complex 3 (46 mg, 0.068 mmol) was dissolved in CHCl3 and pyridine (0.027 ml, 0.34 mmol) was added. The solution was heated at sub-reflux overnight in a nitrogen environment. The solution was evaporated to a minimum volume and ether was added to precipitate the yellow product (27 mg, 56%).
1H NMR (CHCl3) 9.86 (2H, d, J = 8.3), 9.70 (2H, d, J = 5.2), 9.45–8.38 (4H, m), 8.29 (2H, dd, J = 5.3, 2.9), 8.16-8.12 (2H, m), 7.80 (1H, t, J = 7.0), 7.28 (2H, t, J = 7.0); 13C NMR (CD3CN) 155.4, 151.8, 148.8, 142.2, 139.6, 138.6, 136.4, 132.4, 130.2, 129.2, 128.3, 126.4. λmax 360, 379 nm νmax IR (nujol) 2035 (CO), 1947 (CO), 1909 (CO) cm−1. MS (ES+) [M+H] 632.11; HRMS C26H15N5O3Re calc: 630.0699; observed: 630.0704.
[Re(CO)3(dppz)(3-CH2OH-Py)][BF4] 5.
Complex 3 (150 mg, 0.22 mmol) was dissolved in CHCl3 and 3-hydroxymethylpyridine (26 μL, 0.27 mmol) was added. The solution was heated at sub-reflux overnight in a nitrogen environment. The solution was evaporated to a minimum volume and ether was added to precipitate the yellow product (138 mg, 84%).
1H NMR (MeOD) 9.97 (2H, d), 9.83 (2H, dd, J = 3.9, 1.4), 8.49–8.35 (6H, m), 8.16–8.11 (2H, m), 7.79 (1H, d, J = 8.4), 7.31 (1H, dd, J = 5.5, 2.2) 13C NMR (CD3CN) 155.2, 150.1, 149.5, 148.8, 142.3, 140.6, 138.6, 137.4, 136.6, 132.3, 130.2, 129.2, 128.1, 125.8, 59.8. λmax 362, 380 nm. νmax IR (Nujol) 2032 (CO), 1929 (CO), 1915 (CO) cm−1. MS (ES+) [M+H] 662.08; HRMS C27 H17 N5 O4 Re: calc: 660.0805; observed: 660.0811.
[Re(CO)3(dppz)(Py-3-O2C(CH2)12CH3)][BF4] 6.
Complex 3 (45 mg, 0.0661 mmol) was dissolved in CHCl3 and 3-hydroxymethylpyridyl myristate (20 mg, 0.0627 mmol)28 was added. The solution was heated at sub-reflux overnight in a nitrogen environment. The solution was evaporated to a minimum volume and ether was added to precipitate the yellow product (33 mg, 52%).
1H NMR (CD3CN) 9.77 (2H, d, J = 8.3), 9.59 (2H, d, J = 5.2), 8.35–8.28 (4H, m), 8.18–8.15 (2H, m), 8.05–8.01 (2H, m), 7.66 (1H, d, J = 7.8), 7.16 (1H, pseudo-t) 4.74 (2H, s) 1.95(2H, m), 1.18 (14H, bs), 0.93 (4H, bs), 0.81 (3H, m). 13C NMR (CD3CN) 155.2, 155.1 152.0, 151.2 142.2 140.3 138.6 136.4 135.1 132.2 130.3 129.1 128.9 128.0 125.9 53.8 31.1 28.8–28.7 (multiple peaks) 28.5 28.2 21.8 12.9. λmax 360, 379 nm. νmaxIR (Nujol) 2035 (CO), 1921 (2 × CO) cm−1. MS (ES+) [M+H] 872.29. HRMS calc: 870.2788; observed: 870.2810.
[Re(CO)3(dppz)(Py-3-CO2Cy)][BF4] 7.
Complex 3 (40 mg, 0.0588 mmol) was dissolved in CHCl3 and cyclohexylnicotinate25 (24 mg, 0.118 mmol) was added. The solution was heated at sub-reflux overnight in a nitrogen environment. The solution was evaporated to a minimum volume and ether was added to precipitate the yellow product (26 mg, 52%).
1H NMR (CD3CN) 9.79 (2H, d, J = 7.1), 9.60 (2H, d, J = 4.1), 8.54 (2H, d, J = 4.9), 8.46 (1H, d, J = 1.7), 8.34 (2H, dd, J = 3.5, 3.4), 8.18 (2H, m), 8.04 (2H, dd, J = 3.4, 3.5), 7.30 (1H, dd, J = 5.6, 2.4), 4.73 (1H, bm), 1.66–1.23 (10H, bm). 13C NMR (CD3CN) 180.1, 161.7, 155.5, 155.3, 151.4, 149.1, 142.5, 139.8, 138.8, 136.7, 132.5, 130.7, 129.3, 128.3, 126.4, 74.0, 30.5, 24.5, 22.7. λmax 363, 380 nm. νmaxIR (MeCN) 2031 (CO), 1932 (CO), 1911 (CO), 1718 (COOR) cm−1. MS (ES+) [M]+ 758.14; HRMS C33 H25 N5 O5 Re: calc: 756.1380; observed: 756.1381.
[Re(CO)3(dppz)(3-CH2Cl-Py)][PF6] 8.
Complex 5 (24 mg, 0.0321 mmol) was dissolved in thionyl chloride (1 ml) and the mixture stirred at room temperature for 2 h. The reaction mixture was then evaporated to drynes, redissolved in the minimum volume of acetonitrile, and treated dropwise with a saturated aqueous solution of ammonium hexafluorphosphate. The precipitate was collected by filtration, washed with water and diethyl ether and dried to give the title compound as yellow crystals (17 mg, 64%).
1H NMR (CD3CN) 9.92 (2H, d, J = 7.4) 9.72 (2H, d, J = 4.3) 8.48 (2H, dd, J = 6.6, 3.4) 8.43 (1H br. s) 8.30 (3H, m) 8.16 (2H, dd, J = 6.6, 3.4) 7.83 (1H, d, J = 7.4) 7.26 (1H, dd, J = 7.9, 5.6) 4.50 (2H, s). This complex was insufficiently soluble to obtain meaningful 13C data. λmax 363, 380 nm. νmax IR 2034 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 1923 (CO) cm−1. MS (ES+) [M]+ 680.04; HRMS C27H16ClN5O3Re: calc: 678.0466; observed: 678.0471.
O) 1923 (CO) cm−1. MS (ES+) [M]+ 680.04; HRMS C27H16ClN5O3Re: calc: 678.0466; observed: 678.0471.
Absorption titration.
The binding of the complexes 3–8 with DNA was measured in a Na-phosphate buffer solution (pH 7.2). The absorption ratio at 260 nm and 280 nm of CT DNA solutions was found as 1.9:1. It shows that DNA is sufficiently free from protein. The concentration of DNA was then determined by UV/visible absorbance using the molar absorption coefficient (6600 M−1 cm−1) at 260 nm.42 The absorption titrations of 3–8 (10 μM) in Na-phosphate buffer (pH 7.2) containing 2% acetonitrile against CT DNA were performed by monitoring the changes in their absorption spectra. The titration experiments were performed by maintaining the concentration of metal complexes constant at 10 μM while the concentration of CT DNA was varied within 0–100 μM. An equal quantity of CT DNA was also added to the reference solution to eliminate the absorption by DNA. From the absorption data, the intrinsic binding constant Kb was calculated from a plot of [DNA]/(εa − εf) vs. [DNA] using the equation:
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + [Kb(εb − εf)]−1 |
Where, [DNA] represents the concentration of DNA in base pairs. The apparent absorption coefficients εa, εf and εb correspond to Aobsd./[Re], the extinction coefficient for the free rhenium(I) complex and extinction coefficient for the rhenium(I) complex in the fully bound form, respectively.43Kb is the ratio of slope to the intercept.
Competitive binding of complexes with ethidium bromide.
Relative binding of the rhenium complexes to CT DNA was also studied by fluorescence spectroscopy using ethidium bromide (EB) bound to CT DNA in a Na phosphate buffer solution (pH 7.2). Competitive binding of Re(I) complexes to CT DNA resulted in the displacement of bound EB which subsequently quenches the fluorescence. In a typical experiment, 20 μL of CT-DNA solution (A260 = 2.0) was added to 2.0 mL of EB buffer solution (pH 7.2) and the fluorescence intensity was measured at excitation wavelength 510 nm and maximum emission was observed at 600 nm. Aliquots of a 0.1 mM solution of the complexes were then added separately to DNA-EB solution and resulting fluorescence spectra were measured after each addition until it reached to maximum reduction in fluorescence intensity. Stern–Volmer quenching constants were calculated using the given equation,44
where Io and I are the fluorescence intensities in absence and presence of complexes, respectively, Ksv is a linear Stern–Volmer quenching constant and r is the ratio of the total concentration of complex to that of DNA. The value of Ksv is given by the ratio of slope to intercept in a plot of Io/I versus [Complex]/[DNA].
DNA cleavage.
The nuclease activity of the Re(I) complexes was studied using supercoiled pBR322 plasmid DNA. Electrophoresis in native agarose gel was used to quantify the unwinding induced by Re(I) complexes in pBR322, monitoring the changes in the degree of supercoiling of pBR322. The cleavage reactions were carried out in a total volume of 25 μL containing pBR322 DNA (300 ng μL−1) and different concentrations of complexes prepared in 5 mM Tris–HCl buffer (pH 7.2) containing 50 mM NaCl and 0.02% acetonitrile for 24 h at 37 °C. The samples were analyzed by electrophoresis for 3 h at 50 V on 1.5% agarose gel in 1 × TAE buffer (40 mM Tris acetate and 1 mM EDTA) pH 8.0. The gel was stained with a 0.5 μg ml−1 ethidium bromide and visualized by UV light and then photographed for analysis. The extent of cleavage was measured from the intensities of the bands using the AlphaImager 2200 software.45
Cell imaging.
HeLa cells, obtained from NCCS (National Centre for Cell Science, Pune, India), were grown in culture flask containing DMEM (Delbeco Modified Eagle's Medium) supplemented with 10% FBS (Fetal Bovine Serum) and 5% CO2 at 37 °C. Cells were tripsinized with 0.05% Trypsin-EDTA (GIBCO-25300) for 2 min followed by addition of 1 mL of fresh DMEM medium containing serum to inhibit trypsin activity. Cells were spun at 1200 rpm for 10 min and the sediment was suspended in 1 × PBS (pH 7.4), placed on polylysine coated slides and incubated for one hour at room temperature for adherence. Cells were incubated with the relevant complexes (20 μM) for 30 min, briefly washed twice with 1 × PBS and then incubated with Hoechst34580 (1 μg mL−1) for 5 min followed by washing with PBS. Live cells were imaged by ZEISS LSM-510 Meta Confocal Microscope. Excitation wavelength 488 nm (Argon 488 Laser line, to avoid cell damage at the maximum absorbance in the uv of 380 nm) and emission collected at 520 nm. Excitation wavelength for Hoechst was 405 nm (Diode 405) and emission was recorded at 440 nm. Fluorescence intensities were measured by Zeiss LSM software.
Acknowledgements
We thank the EPSRC Mass Spectrometry National Service Centre, Swansea for invaluable assistance in obtaining mass spectra and high resolution mass spectra on the difficult and labile complexes 4–8.
References
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960 CrossRef CAS.
- Y. Jenkins, A. E. Friedman, N. J. Turro and J. K. Barton, Biochemistry, 1992, 31, 10809 CrossRef CAS.
- E. J. C. Olson, D. Hu, A. Hormann, A. M. Jonkman, M. R. Arkin, E. D. A. Stemp, J. K. Barton and P. F. Barbara, J. Am. Chem. Soc., 1997, 119, 11458 CrossRef CAS.
- J. Fees, W. Kaim, M. Moscherosch, W. Matheis, J. Klima, M. Krejcik and S. Zalis, Inorg. Chem., 1993, 32, 166 CrossRef CAS.
- M. K. Brennaman, T. J. Meyer and J. M. Papanikolas, J. Phys. Chem. A, 2004, 108, 9938 CrossRef CAS.
- R. Hartshorn and J. K. Barton, J. Am. Chem. Soc., 1992, 114, 5915 CrossRef.
- J. W. Dobrucki, J. Photochem. Photobiol., B, 2001, 65, 136 CrossRef CAS.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2009, 131, 8738 CrossRef CAS.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46 CrossRef CAS.
- M. Matson, F. R. Svensson, B. Norden and P. Lincoln, J. Phys. Chem. B, 2011, 115, 1706 CrossRef CAS.
- G. T. Ruiz, M. P. Juliarena, R. O. Lezna, E. Wolcan, M. R. Feliz and G. Ferraudi, Dalton Trans., 2007, 2020 RSC.
- V. W. W. Yam, K. K. W. Lo, K. K. Cheung and R. Y. C. Kong, Chem. Commun., 1995, 1191 RSC.
- K. K. W. Lo and K. H. K. Tsang, Organometallics, 2004, 23, 3062 CrossRef CAS.
- N. J. Lundin, P. J. Walsh, S. L. Howell, J. J. McGarvey, A. G. Blackman and K. C. Gordon, Inorg. Chem., 2005, 44, 3551 CrossRef CAS.
- J. Dyer, C. M. Creely, J. C. Penedo, D. C. Grills, S. Hudson, P. Matousek, A. W. Parker, M. Towrie, J. M. Kelly and M. W. George, Photochem. Photobiol. Sci., 2007, 6, 741 CAS.
- M. K. Kuimova, W. Z. Alsindi, A. J. Blake, E. S. Davies, D. J. Lampus, P. Matousek, J. McMaster, A. W. Parker, M. Towrie, X. Z. Sun, C. Wilson and M. W. George, Inorg. Chem., 2008, 47, 9857 CrossRef CAS.
- M. G. Fraser, A. G. Blackman, G. I. S. Irwin, C. P. Easton and K. C. Gordon, Inorg. Chem., 2010, 49, 5180 CrossRef CAS.
- R. Diaz, A. Francois, A. M. Leiva, B. Loeb, E. Norambuena and M. Yanez, Helv. Chim. Acta, 2006, 89, 1220 CrossRef CAS.
- C. Y. Fu, M. T. Li, Z. M. Su, Z. Hong, W. L. Li and B. Li, Appl. Phys. Lett., 2006, 88, 093507 CrossRef.
- C. Metcalfe, M. Webb and J. A. Thomas, Chem. Commun., 2002, 2026 RSC.
- J. R. Schoonover, W. D. Bates and T. J. Meyer, Inorg. Chem., 1995, 34, 6421 CrossRef CAS.
- V. W. W. Yam, K. K. W. Lo, K. K. Cheung and R. Y. C. Kong, Dalton Trans., 1997, 2067 CAS.
- H. D. Stoeffler, N. B. Thornton, S. L. Temkin and K. S. Schanze, J. Am. Chem. Soc., 1995, 117, 7119 CrossRef CAS.
- V. Fernández-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186 RSC.
- V. Fernández-Moreira, F. L. Thorp-Greenwood, A. J. Amoroso, J. Cable, J. B. Court, V. Gray, A. J. Hayes, R. L. Jenkins, B. M. Kariuki, D. Lloyd, C. O. Millet, C. Ff. Williams and M. P. Coogan, Org. Biomol. Chem., 2010, 8, 3888 Search PubMed.
- C. M. Dupureur and J. K. Barton, Inorg. Chem., 1997, 36, 33 CrossRef CAS.
- A. J. Amoroso, M. P. Coogan, J. E. Dunne, V. Fernández- Moreira, J. B. Hess, A. J. Hayes, D. Lloyd, C. Millet, S. J. A. Pope and C. Williams, New J. Chem., 2008, 32, 1097 RSC.
- M. P. Coogan, V. Fernández-Moreira, J. B. Hess, S. J. A. Pope and C. Williams, New J. Chem., 2009, 33, 1094 RSC.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3053 Search PubMed.
- C. X. Zhang and S. J. Lippard, Curr. Opin. Chem. Biol., 2003, 7, 481 CrossRef CAS.
- B. Lippert, Coord. Chem. Rev., 2000, 200–202, 487 CrossRef CAS.
- H. Chao, W. J. Mei, Q. W. Huang and L. N. Ji, J. Inorg. Biochem., 2002, 92, 165 CrossRef CAS.
- M. M. Patel and T. J. Anchordoquy, Biophys. J., 2005, 88, 2089 CrossRef CAS.
- J.-B. LePecq and C. Paoletti, J. Mol. Biol., 1967, 27, 87 CrossRef CAS.
- B. Selvakumar, V. Rajendiran, P. U. Maheswari, H. S. Evans and M. Palaniandavar, J. Inorg. Biochem., 2006, 100, 316 CrossRef CAS.
- Q.-X. Zhou, F. Yang, W.-H. Lei, J.-R. Chen, C. Li, Y.-J. Hou, X.-C. Ai, J.-P. Zhang, X.-Song Wang and B.-W. Zhang, J. Phys. Chem. B, 2009, 113, 11521 CrossRef CAS.
- Ruchi Gaur, Rais Ahmad Khan, Sartaj Tabassum, Priyanka Shah, Mohammad I. Siddiqi and Lallan Mishra, J. Photochem. Photobiol., A, 2011, 220, 145 CrossRef CAS.
- N. E. Brückmann, S. Kögel, A. Hamacher, M. U. Kassack and P. C. Kunz, Eur. J. Inorg. Chem., 2010, 5063 Search PubMed.
- C. J. Chang, J. Jaworski, E. M. Nolan, M. Sheng and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1129 CrossRef CAS.
- X. Zhao and K. Schanze, Chem. Commun., 2010, 46, 6075 RSC.
- R. G. Balasingham, M. P. Coogan and F. L. Thorp-Greenwood, Dalton Trans., 2011 10.1039/C1DT11219H.
- J. Marmur, J. Mol. Biol., 1961, 3, 208 CrossRef CAS.
- A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392 CrossRef CAS.
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 2nd edn, Plenum Press, New York, 1999 Search PubMed.
- AlphaImager 2200 software, Alpha Innotech Corporation 2401 Merced St. San Leandro, CA 94577.
Footnote |
| † Electronic supplementary information (ESI) available: further details of the photophysical properties of complexes 3–8 and full photophysical data, UV vis and EB titration data, and further cell images are available. See DOI: 10.1039/c1nj20662a |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.