DOI:
10.1039/C1NJ20671K
(Paper)
New J. Chem., 2012,
36, 125-129
Synthesis of hollow BaSO4 nanospheres templated by core–shell–corona type polymeric micelles†
Received
(in Montpellier, France)
3rd August 2011
, Accepted 31st October 2011
First published on 14th November 2011
Abstract
Hollow barium sulfate (BaSO4) nanospheres were synthesized by templating a polymeric micelle of a triblock copolymer poly(styrene-b-acrylic acid-b-ethylene glycol) (PS-b-PAA-b-PEG). This polymer is known to form a micelle with a PS core, a PAA shell and a PEG corona in aqueous solutions. Barium chloride and sodium sulfate were used as precursors of BaSO4. In the synthesis, the PS core acts as a template for cavities of the hollow particles, the PAA shell is beneficial for arresting Ba2+ ions to produce BaSO4, and the PEG corona stabilizes the BaSO4/polymer nanocomposite to prevent secondary aggregate formation. Hollow BaSO4 nanospheres were obtained by removing the polymeric template from the BaSO4/polymer nanocomposite by calcination. The hollow BaSO4 nanospheres thus fabricated were characterized by various techniques including transmission electron microscopy and X-ray diffraction analysis. The average diameter of the spheres is around 25 nm and the average cavity diameter is around 16 nm. The significance of the present method is that it can avoid formation of large crystals which is generally unavoidable in other methods.
Introduction
Barium sulfate (BaSO4) has industrial and medical relevance due to its inertness and opacity to UV and X-ray radiation.1 It is frequently used clinically as a radio-contrasting agent for X-ray imaging, a radio-opaque filler in commercial bone cement, and in other diagnostic procedures.1d The synthesis of morphologically controlled BaSO4 nanoparticles has been the object of numerous studies.2 Several strategies have been suggested for the controlled growth of BaSO4 nanocrystals. Surfactants,3 organic(polymeric) inhibitors,4 organic additives,5 inorganic stabilizers6 and different reaction media7 were used. BaSO4 nanoparticles can also be synthesized by using double hydrophilic block copolymers with different functional groups such as carboxylic acids, aspartic acids, sulfonates, and phosphonates.8 Mann et al. synthesized a variety of well defined morphogenesis of BaSO4 such as bundled fibres, brush-like and cone-shaped structures, using a block copolymer as a nucleation and stabilizing agent.9 They also used a mixture of two double hydrophilic block copolymers to control the crystallization and organization of BaSO4 microstructures.10 These structures are of several hundred micrometres in length. Such large (microsized) BaSO4 particles have a disadvantage that they limit the stain penetration during micro-computed tomography.1d
Recently, there has been a great deal of interest in the synthesis of hollow nanoparticles because of their potential applications in controlled drug delivery, catalysis, sensors, light weight fillers, photonic crystals, biomedical diagnosis and therapy, and so on.11 Their capacity for encapsulating sensitive materials such as therapeutics, fluorescent markers, and field-responsive agents has been exploited by many groups for drug delivery and biomedical imaging.11d,12
Various methods, such as templating, sonochemical, and hydrothermal, have been reported as the procedures for the preparation of inorganic materials with hollow spherical structures. In recent years, the use of a polymeric micelles template to synthesize inorganic hollow nanoparticles has proven to be successful.13 Lee and Char demonstrated a morphological change of silica from tubules to large hollow spheres to small hollow spheres by adjusting the intermolecular interactions within micelles of a poly(styrene-b-vinylpyridine) block copolymer.14 This method can improve the quality of the product in terms of controlling the size and morphology of hollow particles.15
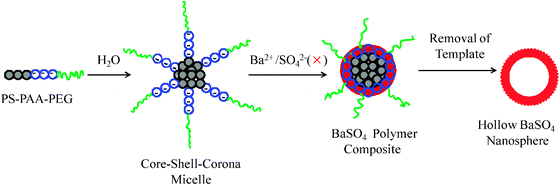
Herein, we present a facile synthesis of hollow BaSO4 nanospheres with a size of several tens of nanometres. We employed a template for polymeric micelles of poly(styrene-b-acrylic acid-b-ethylene glycol) (PS-b-PAA-b-PEG), which has a PS core, a PAA shell and a PEG corona in aqueous solutions. We incorporated Ba2+ ions into PS-b-PAA-b-PEG micelles by interaction between Ba2+ ions and the anionic PAA block of the polymer. The interaction of Ba2+ ions with the PAA shell of the polymeric micelles was elucidated by dynamic light scattering (DLS), zeta-potential, and transmission electron microscopy (TEM). Ba2+/PS-b-PAA-b-PEG chelated particles were then used as a template for the fabrication of hollow BaSO4 nanospheres. In this synthesis, the PS core acts as a template for hollow cavities, the PAA shell works as a nanoreactor and nanocontainer for the precursors of BaSO4, and the PEG corona prevents the polymer–BaSO4 composite nanospheres from forming secondary aggregates by steric repulsion between PEG chains in aqueous solutions. The hollow BaSO4 nanospheres were obtained after removing the template polymer by calcination. The hollow nanospheres thus obtained were characterized by various techniques including TEM and X-ray diffraction (XRD). The fabrication of nanometre-sized hollow BaSO4 spheres is generally difficult because, after initial mixing of Ba2+ and SO42− ions the growth of BaSO4 is uncontrollably rapid to give large (micrometre-sized) crystals. However, the use of a PS-b-PAA-b-PEG micelle template overcomes this problem of crystal overgrowth, allowing the formation of nanometre-sized hollow spheres with uniform void space.
Experimental
Materials
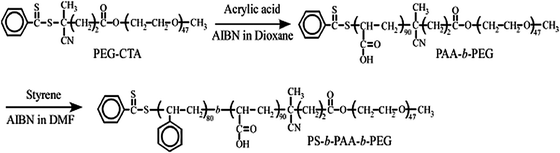
A polyethylene glycol-based chain transfer agent (PEG-CTA) was prepared as previously reported.16 We first prepared the diblock copolymer poly(ethylene glycol-b-acrylic acid) (PEG-b-PAA) by reversible addition–fragmentation chain transfer polymerization using PEG-CTA. Styrene, 2,2′-azobis(isobutyronitrile), and PEG-b-PAA were dissolved in DMF. The solution was deoxygenated by purging with Ar gas for 30 min. The polymerization was carried out at 60 °C for 24 h. The polymerization mixture was dialyzed against acetone for 3 days and pure water for one day. The obtained PS-b-PAA-b-PEG was recovered by a freeze-drying technique. The details of synthesis were reported elsewhere.17 The number-average degrees of polymerization of the PS, PAA, and PEG blocks are 80, 90, and, 47, respectively, as estimated by 1H NMR (Fig. S1, ESI†). The synthetic procedure is shown in Scheme 1. BaCl2 (Katayama, 99%), Na2SO4 (Katayama, 99%), and phosphotungstic acid hydrate (Alfa Aesar) were used without purification.
Preparation of polymeric micelles
A known amount of PS-b-PAA-b-PEG was dissolved in water and gently agitated with a magnetic stirrer at room temperature till a clear solution was obtained. The solution was then transferred to a volumetric flask to obtain a stock solution. The final concentration of polymeric micelles is 1 g L−1.
Incorporation of Ba2+ ions into micelles
A known amount of PS-b-PAA-b-PEG micelle solution was titrated with Ba2+ ions. The amount of added Ba2+ ions is expressed in terms of the apparent degree of charge neutralization (DN), which is defined as| | 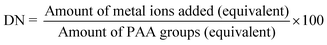 | (1) |
All the experiments were carried out under pH 7–8, where almost all carboxylic groups of the PAA block have an anionic form.
Synthesis of hollow BaSO4 nanospheres
The hollow BaSO4 nanospheres were synthesized using the micelle of PS-b-PAA-b-PEG as a template (Scheme 2). To induce BaSO4 mineralization in polymeric micelles, an aqueous BaCl2 solution (50 μL, 0.94 M) was first added to a stirred solution of polymeric micelles at pH 7–8 and equilibrated for 2 h under stirring. An aqueous solution of Na2SO4 (50 μL, 0.94 M) was slowly added drop-wise to the reaction mixture, and the solution was stirred magnetically at room temperature. The BaSO4/PS-b-PAA-b-PEG composite particles were recovered by centrifugation. The composite particles were thoroughly washed with de-ionized water and dried in an oven at 50 °C. The polymeric template was removed by calcination at 500 °C for 4 hours.
 |
| | Scheme 2 Synthesis of hollow BaSO4 nanospheres from a PS-b-PAA-b-PEG micelle template. | |
DLS measurements
The DLS measurements were carried out using an Otsuka ELS Z zeta-potential and particle analyzer. All the measurements were carried out at 25 °C. All the solutions were passed through a 0.2 μm cellulose filter before the measurements. The scattered light of a vertically polarized He–Ne laser (632.8 nm) was measured at an angle of 90° and was collected by an autocorrelator. The correlation functions were analyzed by the contin method and used to determine the diffusion coefficient (D) of the particles. The hydrodynamic diameter (Dh) was calculated from D using the Stokes–Einstein equation: where kB is the Boltzmann constant, T is the absolute temperature and η is the solvent viscosity. The polydispersity factor of micelles is represented as μ2/Γ2, where μ2 is the second cumulant of the decay function and Γ2 is the average characteristic line width.
Zeta-potential measurements
The electrophoretic mobility (EPM) was measured by an Otsuka ELS Z zeta-potential and particle analyzer. The zeta-potential (ζ) was calculated from the EPM using the Smoluchowski equation: where μE is the EPM, ε is the permittivity of the solvent, and η is the solvent viscosity.
The TEM measurements were carried out using a JEOL JEM-1210 electron microscope at an accelerating voltage of 80 kV. To prepare the samples for the TEM measurements, the nanospheres were dispersed in water followed by casting on a copper grid. The excess solution was removed by soaking with a blotting paper. The samples were then dried in air for one day. For the preparation of a polymeric micelles sample, 0.1 wt% phosphotungstic acid solution is used to improve the contrast.18
XRD analysis
Phase identification of the hollow nanospheres was carried out using a Shimadzu-7000 X-ray diffractometer at 40 kV, 30 mA, and with CuKα radiation (1.5406 Å). Data were collected in the scan range of 20–70° in the continuous scan mode at a scanning speed of 2° min−1 using 0.15 mm receiving slits.
Thermal analysis
A simultaneous thermogravimetric analysis (TGA) and a differential thermal analysis (DTA) were carried out using a SEIKO-6300 TG/DTA instrument at the heating rate of 10 °C min−1 in air.
The Fourier transform infrared (FTIR) absorption spectra were measured by a JASCO FT/IR-7300 spectrometer at room temperature. The samples were prepared by the KBr method.
Results and discussion
Characterization of polymeric micelles
The polymeric micelles obtained were characterized by DLS and TEM measurements. The Dh of the polymeric micelles is about 70 nm (Fig. S2, ESI†) at pH 8. The Dh ranges from 50 to 70 nm as the pH is increased from 3.5 to 6.0 and becomes almost constant after that. This is explained by the conformational change of the PAA block. At low pH (pH < 4) the PAA block is protonated, and has a shrunken conformation resulting in the minimum value of Dh. At high pH (pH > 5.5), the PAA block swells considerably in water because the carboxylic acid groups are converted to carboxylate ions19 leading to a larger Dh value. The polymeric micelles were stained with phosphotungstic acid and observed by TEM. The TEM image shown in Fig. 1 revealed that the micelle has a spherical structure. The white sphere of around 25 nm average diameter corresponds to the hard PS core, because the phosphotungstic acid preferentially stains the PS core. The TEM image provides the clear evidence for the micelles formation.
 |
| | Fig. 1
TEM image of the PS-b-PAA-b-PEG micelles. The micelles were stained with phosphotungstic acid. | |
Interaction of Ba2+ ions with micelles
When Ba2+ ions are introduced into PS-b-PAA-b-PEG solutions at pH 8, the PAA block will be bound to the Ba2+ ions, which leads to the formation of bidentate metal–polymer chelated particles.20 In these chelated particles, metal ions are bound to PAA segments via electrostatic interaction, forming an insoluble hydrophobic shell that is stabilized by a PEG corona. In order to confirm the interaction of Ba2+ ions with the PAA shell, we titrated PS-b-PAA-b-PEG with BaCl2 solutions, and examined the particle size, zeta-potential, and shape of the particles. Fig. 2a shows the dependence of Dh on DN. In the absence of metal ions almost all carboxylic acid groups are deprotonated at pH 9 as their pKa value is 4.521 and the PAA chains are fully extended to produce the maximum size of the micelles. The average Dh of the particles varies from 73 to 43 nm depending on DN. There is a sharp decrease in Dh above 60% DN and the limiting value above 100%. The polydispersity factor, μ2/Γ2, of Dh lies between 0.07 and 0.14 over all DN, indicating that the size distribution of the Ba2+/PS-b-PAA-b-PEG chelated particles is fairly mono-disperse. The added Ba2+ ions cancel the charges of the carboxylate anions. The electrostatic repulsion among the anionic shells, therefore, is weakened and the blocks are shrunken, resulting in the decrease of the total micellar size. The binding of Ba2+ ions to PS-b-PAA-b-PEG was checked by zeta-potential measurement. Fig. 2b shows the dependence of zeta-potential of Ba2+/PS-b-PAA-b-PEG chelated particles on DN. The successive addition of Ba2+ ions increases the zeta-potential. Above 100% DN the value of zeta-potential is not increased. This fact shows that the complex formation is triggered essentially by electrostatic interactions. But the zeta-potential values do not reach zero when DN exceeds 100%. This implies that all the carboxylate groups in PAA are not available for binding of Ba2+ ions; it is due to the complicated structure of the polymer and steric hinderance.22
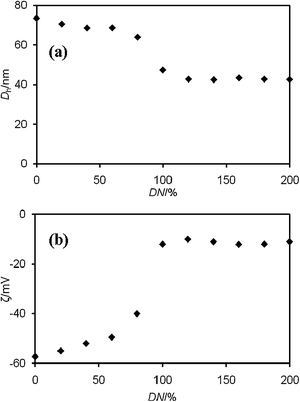 |
| | Fig. 2 (a) Hydrodynamic diameter (Dh) and (b) zeta-potential (ζ) of Ba2+/PS-b-PAA-b-PEG chelated particles as a function of DN. | |
The TEM image of the Ba2+/PS-b-PAA-b-PEG nanocomposites is shown in Fig. 3a. The image clearly shows the effective binding of Ba2+ ions to the polymer. The dark periphery is the PAA block of PS-b-PAA-b-PEG stained with Ba2+ ions. The spheres observed by TEM basically correspond to a glassy PS core surrounded by a compact Ba2+/PAA layer, because the PEG corona seems to be less stained compared to the PAA shell. Therefore, the observed diameter (35 nm) is smaller than the Dh (ca. 40 nm) in DLS measurements.
 |
| | Fig. 3
TEM pictures of (a) Ba2+/PS-b-PAA-b-PEG chelated particles and (b) hollow BaSO4 nanospheres. | |
Fabrication of hollow BaSO4 nanospheres
The attractive interaction between the anionic PAA block and Ba2+ ions induces the formation of chelated particles of the copolymer and the metal ions, which act as a soft template for the synthesis of hollow BaSO4 nanospheres. The Ba2+/PS-b-PAA-b-PEG chelated particles were used for mineralization of BaSO4 by adding sulfate ions in the form of Na2SO4. Hollow BaSO4 nanospheres were obtained after removing the polymeric template by calcination at 500 °C. Fig. 3b illustrates a TEM image of hollow BaSO4 nanospheres. The hollow BaSO4 nanospheres have a uniform size. The outer diameter is ∼25 nm, and the void space diameter is ∼16 nm. In our method the PS core acts as a template to form cavities of hollow BaSO4 nanospheres. But the void space diameter is less than the core size of the polymeric template shown in Fig. 1. It has been reported by several groups23 that the cavity size of the hollow inorganic particles becomes smaller than the size of the template polymers, which is ascribed to the shrinking of the particles during the calcination. Therefore, the difference between the cavity size of hollow BaSO4 nanospheres and the core size of the polymeric template seems to be due to shrinking during the calcination. The nanospheres are somewhat aggregated as seen in Fig. 3b. It is probably due to calcination.24 The BaSO4/PS-b-PAA-b-PEG nanocomposite particles, prior to calcination, are well dispersed and no agglomeration is seen in aqueous solution (Fig. S3, ESI†). Removal of the polymeric template was confirmed by FTIR measurements. Fig. S4 (ESI†) shows the FTIR spectra of the polymer and hollow BaSO4 nanospheres after calcination of the BaSO4/PS-b-PAA-b-PEG composite particles. The disappearance of the PEG band (around 2900 cm−1) and the carbonyl stretching band (around 1720 cm−1) after calcination indicates the removal of the polymeric template.
The crystalline characteristics of the obtained hollow BaSO4 nanospheres were investigated by XRD analysis. The XRD pattern is shown in Fig. S5 (ESI†). It is clearly shown that the nanospheres are crystallized in an orthorhombic structure (ICDD No. 001-1229).
The thermal behavior of BaSO4/PS-b-PAA-b-PEG composite particles was studied by TG-DTA in the temperature range 20–1000 °C. The TG-DTA curves for the BaSO4/PS-b-PAA-b-PEG composites are shown in Fig. 4. The initial parts of the TG curves slightly decline. This implies desorption of absorbed water molecules. The decomposition of the polymer is the main step of the weight loss. The strong exothermic peak on DTA curves around 350 °C and 450 °C shows decomposition of the template polymer. The thermogravimetric analysis of the dried powder samples shows about a 16% weight loss in air for the BaSO4/PS-b-PAA-b-PEG composites. This weight loss corresponds to decomposition of the polymer.
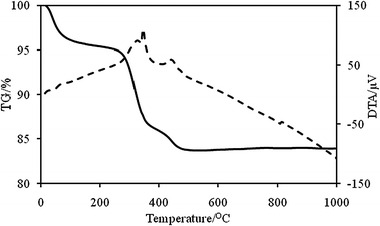 |
| | Fig. 4
TG (—)/DTA (⋯) graphs of BaSO4/PS-b-PAA-b-PEG nanocomposites. | |
Conclusions
Complexation of Ba2+ ions with the PS-b-PAA-b-PEG micelles was induced under basic conditions. This is based on the electrostatic interaction between cationic Ba2+ ions and an anionic (deprotonated) PAA block. The mono-dispersed Ba2+/PS-b-PAA-b-PEG chelated particles were successfully used as a template for the synthesis of well-defined hollow BaSO4 nanospheres. The obtained nanospheres have a uniform cavity size and wall thickness. This synthesis strategy is potentially expandable to other inorganic materials which undergo crystal growth too fast to be obtained by other methods.
Acknowledgements
The authors thank Mr Toshimi Tabata for his help in the TEM measurements, and Professor Takanori Watari and Dr Hom Nath Luitel for the XRD and TG-DTA measurements. The present study was supported by a Grant-in-Aid for Scientific Research (20310054) from the Japan Society for the Promotion of Science (JSPS).
References
-
(a) H. Bala, W. Fu, Y. Guo, J. Zhao, Y. Jiang, X. Ding, K. Li, M. Yu and Z. Wang, Colloids Surf., A, 2006, 274, 71–76 CrossRef CAS;
(b) B. M. Nagaraja, H. Abimanyu, K. D. Jung and K. S. Yoo, J. Colloid Interface Sci., 2007, 316, 645–651 CrossRef CAS;
(c)
J. P. Barraud, Encyclopedia of Diagnostic Imaging, ed. A. A. Baert, Springer-Verlag, vol. 2, pp. 2485–2488 Search PubMed;
(d)
H. Leng, Micro-computed Tomography of Microdamage in Cortical Bone, PhD dissertation, University of Notredum, 2006 Search PubMed.
-
(a) J. Xiao, A. T. Kan and M. B. Tomson, Langmuir, 2001, 17, 4668–4673 CrossRef CAS;
(b) A. E. Murdaugh and S. Mann, Langmuir, 2009, 25, 9792–9796 CrossRef CAS;
(c) A. W. Xu, Y. Ma and H. Colfen, J. Mater. Chem., 2007, 17, 415–449 RSC;
(d) M. Uchida, A. Sue, T. Yoshioka and A. Okuwaki, CrystEngComm, 2001, 3, 21–26 RSC.
- H. Zhang, Y. Liu, J. Zhang, H. Sun, J. Wu and B. Yang, Langmuir, 2008, 24, 12730–12733 CrossRef CAS.
- S. N. Black, L. A. Bromley, D. Cottier, R. J. Davey, B. Dobbs and J. E. Rout, J. Chem. Soc., Faraday Trans., 1991, 87, 3409–3414 RSC.
-
(a) M. Li, H. Schnablegger and S. Mann, Nature, 1999, 402, 393–395 CrossRef CAS;
(b) G. Wu, H. Zhou and S. Zhu, Mater. Lett., 2007, 61, 168–170 CrossRef CAS.
-
(a) R. I. C. Ibara, G. R. Gattorno, M. F. G. Sanchez, A. S. Solis and O. Manero, Langmuir, 2010, 26, 6954–6959 CrossRef;
(b) A. Gupta, P. Singh and C. Shivakumara, Solid State Commun., 2010, 150, 386–388 CrossRef CAS.
- M. Li and S. Mann, Langmuir, 2000, 16, 7088–7094 CrossRef CAS.
- L. Qi, H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2000, 39, 604–607 CrossRef CAS.
- L. Qi, H. Cölfen, M. Antonietti, M. Li, J. D. Hopwood, A. J. Ashley and S. Mann, Chem.–Eur. J., 2001, 7, 3526–3532 CrossRef CAS.
- M. Li, H. Colfen and S. Mann, J. Mater. Chem., 2004, 14, 2269–2276 RSC.
-
(a) X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS;
(b) Y. Wang, A. S. Angelatos and F. Caruso, Chem. Mater., 2008, 20, 848–858 CrossRef CAS;
(c) W. J. Tong and C. Y. Gao, J. Mater. Chem., 2008, 18, 3799–3812 RSC;
(d) S. J. Son, X. Bai and S. B. Lee, Drug Discovery Today, 2007, 12, 650–656 CrossRef CAS.
-
(a) Y. Chen, H. Chen, D. Zeng, Y. Tian, F. Chen, J. Feng and J. Shi, ACS Nano, 2010, 4, 6001–6013 CrossRef CAS;
(b) H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
-
(a) A. Khanal, Y. Inoue, M. Yada and K. Nakashima, J. Am. Chem. Soc., 2007, 129, 1534–1535 CrossRef CAS;
(b) J. Yang, J. U. Lind and W. C. Trogler, Chem. Mater., 2008, 20, 2875–2877 CrossRef CAS;
(c) K. K. Perkin, J. L. Turner, K. L. Wooley and S. Mann, Nano Lett., 2005, 5, 1457–1461 CrossRef CAS.
- H. Lee and K. Char, ACS Appl. Mater. Interfaces, 2009, 1, 913–920 CAS.
- D. Liu, A. Khanal, Y. Inoue, M. Yada and K. Nakashima, Chem. Lett., 2009, 38, 130–131 CrossRef CAS.
- S. Yusa, Y. Yokoyama and Y. Morishima, Macromolecules, 2009, 42, 376–383 CrossRef CAS.
- B. P. Bastakoti, S. Guragain, Y. Yokoyama, S. Yusa and K. Nakashima, Langmuir, 2011, 27, 379–384 CrossRef CAS.
- S. O. Obare, N. R. Jana and C. J. Murphy, Nano Lett., 2001, 1, 601–603 CrossRef CAS.
- S. Ludwigs, K. Schmidt and G. Krausch, Macromolecules, 2005, 38, 2376–2382 CrossRef CAS.
- I. Pochard, P. Couchot and A. Foissy, Colloid Polym. Sci., 1998, 276, 1088–1097 CAS.
- D. G. Leaist, J. Solution Chem., 1989, 18, 421–435 CrossRef CAS.
- Y. An, T. Ushida, M. Susuki, T. Koyama, K. Hanabusa and H. Shirai, Polymer, 1996, 37, 3097–3100 CrossRef CAS.
-
(a) A. Syoufian, Y. Inoue, M. Yada and K. Nakashima, Mater. Lett., 2007, 61, 1572–1575 CrossRef CAS;
(b) K. G. Li and Z. C. Zhang, Mater. Lett., 2004, 58, 2768–2771 CrossRef.
- N. L. Wu, S. Y. Wang and I. A. Rusakova, Science, 1999, 285, 1375–1377 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.