Extending the family of heteroditopic calix[4]diquinone receptors for cooperative AND ion-pair recognition†
Anna J.
McConnell
,
Christopher J.
Serpell
and
Paul D.
Beer
*
Inorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: paul.beer@chem.ox.ac.uk; Fax: +44 1865-272-690
First published on 28th October 2011
Abstract
New heteroditopic calix[4]diquinone receptors with different anion binding units and calix[4]diquinone scaffolds have been synthesised. Ion-pair binding studies using UV/visible and 1H NMR spectroscopies reveal that receptors 1, 2 and 4 are cooperative AND ion-pair receptors, which display little affinity for ‘free’ ions but enhanced binding of the ion-pairs NaCl, NH4Cl and KCl. The more preorganised receptor 2 binds the ion-pairs more weakly than receptor 1. Varying the nature of the calix[4]diquinone scaffold appears to have little effect on ion-pair binding, although the calix[4]diquinone framework is more conformationally flexible than the tert-butylcalix[4]diquinone one and can be synthesised using the milder oxidant chlorine dioxide.
Introduction
Ion-pair recognition1–3 is an emerging field which takes inspiration from both cation and anion coordination chemistry for the design of heteroditopic receptors, which are able to simultaneously bind cations and anions. Given the importance of ions in chemical, biological, medical, environmental and industrial processes, the design of synthetic host systems for ion-pairs is a current area of intense interest for many applications, including the development of membrane transport,4 salt extraction,5 salt solubilisation agents6 and sensors.7–9We have previously reported a series of tert-butylcalix[4]diquinone receptors which display unprecedented cooperative AND ion-pair recognition.10,11 These receptors display little affinity for ‘free’ ions but enhanced binding of contact ion-pairs, such as NH4Cl. It has been proposed that this unique ion-pair recognition behaviour results from intramolecular hydrogen bonding interactions within the receptor which can only be broken in the presence of cations and anions. There is also evidence that a π–π stacking interaction between the calix[4]diquinone motif and the anion binding unit stabilises ion-pair association.
Herein we report an extension of this family of calix[4]diquinone based ion-pair receptors and investigate further the mechanism behind the cooperative AND ion-pair recognition by varying the nature of the anion recognition site and the calix[4]arene framework (Fig. 1). In particular, the anion binding unit tert-butylisophthalamide was incorporated to probe the importance of the π–π stacking interaction for ion-pair recognition. In addition, the 2,6-bis-amide pyridine analogue 2 was prepared in order to study the effect of preorganisation on ion-pair binding, as Chmielewski and Jurczak have reported that macrocyclic 2,6-bis-amide pyridine based receptors bind anions more strongly than their isophthalamide analogues due to preorganisation.12,13 Previously, tert-butylcalix[4]diquinone receptors have been synthesised viathallium trifluoroacetate oxidation.10,11,14–16 However, removal of the tert-butyl groups from the calix[4]diquinone framework allows the use of milder oxidants, such as chlorine dioxide.17–20 We report the preparation of the new heteroditopic calix[4]diquinone receptors 1–5 and their ion-pair binding properties.
![Target tert-butylcalix[4]diquinone receptors 1–2 and calix[4]diquinone receptors 3–5.](/image/article/2012/NJ/c1nj20708c/c1nj20708c-f1.gif) | ||
| Fig. 1 Target tert-butylcalix[4]diquinone receptors 1–2 and calix[4]diquinone receptors 3–5. | ||
Synthesis
The new target tert-butylcalix[4]diquinone receptors 1 and 2 were synthesised according to Scheme 1. The synthesis of 7 and 8 has been reported previously,10 however, microwave syntheses of these compounds have been developed, which significantly reduce the reaction times. Compound 7 was isolated in the improved yield of 75% following recrystallisation from CH2Cl2/EtOH. After quantitative removal of the phthalimide protecting groups using hydrazine monohydrate under microwave irradiation, the resulting bis-amine 8 was treated with tert-butyl isophthaloyl dichloride and 2,6-bis(chlorocarbonyl) pyridine under high dilution conditions to afford the respective macrocycles 9 and 10 in satisfactory yields. Heteroditopic tert-butylcalix[4]diquinone receptors 1 and 2 were synthesised in 74% and 23% yields, respectively, by oxidation of 9 and 10 using thallium(III) trifluoroacetate in a TFA solution.![Synthesis of tert-butylcalix[4]diquinone receptors 1–2.](/image/article/2012/NJ/c1nj20708c/c1nj20708c-s1.gif) | ||
| Scheme 1 Synthesis of tert-butylcalix[4]diquinone receptors 1–2. | ||
New calix[4]diquinone receptors 3–5 were prepared using the multi-step synthetic strategy shown in Scheme 2, where the milder oxidant chlorine dioxide was utilised. Calix[4]arene was synthesised from tert-butylcalix[4]arene by Friedel–Crafts dealkylation21 and reacted with 610,22 or 1123,24 under basic conditions. Following purification by silica gel chromatography, precursors 12 and 13 were isolated in 66 and 48% yields respectively. The phthalimide protecting groups were removed using hydrazine in ethanol to give bis-amines 14 and 15. These were reacted with isophthaloyl dichloride or 5-nitroisophthaloyl dichloride to give the macrocyclic derivatives 16–18 in 35–58% yield. The target calix[4]diquinone receptors 3–5 were prepared in good yields by oxidation with chlorine dioxide using Lin's method.17
![Synthesis of calix[4]diquinone receptors 3–5.](/image/article/2012/NJ/c1nj20708c/c1nj20708c-s2.gif) | ||
| Scheme 2 Synthesis of calix[4]diquinone receptors 3–5. | ||
Crystals of 12, macrocycle 17 and receptors 1 and 3 suitable for single crystal X-ray structural analysis were grown from various solvent mixtures (see ESI†). The structures of 12 and 17 show a number of similarities (Fig. 2); the calix[4]arene unit is in the cone conformation with π-stacking interactions between one of the calixarene rings and the phthalimide and macrocycle substituents respectively. There are intramolecular hydrogen bonds between the phenolic protons and the ether oxygens. A water molecule is bound viahydrogen bonding in the cavity of macrocycle 17, which has been omitted for clarity.
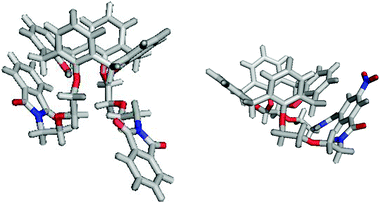 | ||
| Fig. 2 X-Ray crystal structures of 12 (left) and macrocycle 17 (right). | ||
The structures of receptors 1 and 3 show a number of differences to the calix[4]arene precursors (Fig. 3), but are similar to the structure reported for the original cooperative AND ion-pair receptor.10 In both structures, the macrocycle is in an extended conformation with intramolecular hydrogen bonding interactions between the amide protons of the isophthalamide anion binding unit and one of the calix[4]diquinone oxygens with N⋯O bond lengths of 3.1–3.2 Å (Table 1). For receptor 1 the calix[4]diquinone is in the 1,3-alternate conformation with the pairs of tert-phenyl and quinone rings approximately parallel. In contrast, the calix[4]diquinone unit of receptor 3 is in the partial cone conformation with the two quinone rings almost perpendicular to one another. Also, the phenyl rings are no longer parallel but pinched together, most likely due to the greater conformational flexibility of the calix[4]diquinone unit without the tert-butyl groups.
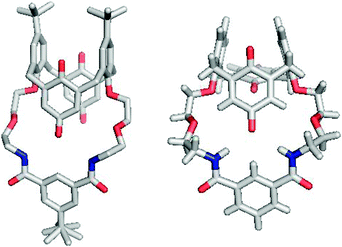 | ||
| Fig. 3 X-Ray crystal structures of receptors 1 (left) and 3 (right). | ||
| Receptor | N⋯O bond lengths/Å |
|---|---|
| 1 | 3.123, 3.152 |
| 3 | 3.216, 3.200 |
With solid state evidence for intramolecular hydrogen bonding interactions in the receptors, 1H NMR experiments were carried out to investigate whether these are also present in solution. There are differences in the 1H NMR spectra of the tert-butylcalix[4]diquinone and calix[4]diquinone receptors in CDCl3 (Fig. 4). Using receptors 1 and 3 as representative examples, the calix[4]diquinone proton signals for receptor 1 are sharp whereas those for receptor 3 are very broad. This broadness could be attributed to the greater conformational flexibility of the calix[4]diquinone unit compared with the tert-butylcalix[4]diquinone unit.
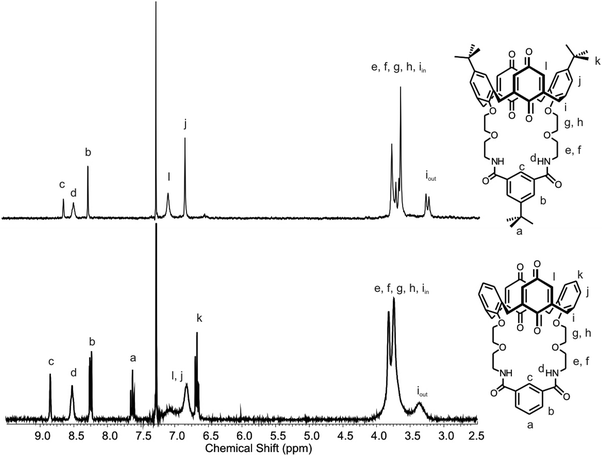 | ||
| Fig. 4 1H NMR spectrum of receptors 1 (top) and 3 (bottom) in CDCl3 at 298 K. | ||
A variable temperature 1H NMR experiment was carried out between 20 °C and 100 °C in DMSO-d6 to investigate this further.25 While the proton spectrum was very broad at 20 °C, heating the solution to 100 °C significantly sharpened the signals and there were a number of chemical shift changes (Fig. 5). For example the amide protond moved upfield by 0.25 ppm and there was also a small upfield shift of the isophthaloyl protonc. In the alkyl region, the methylene proton iin moved downfield and there were significant changes for protonse–h. All of these changes are suggestive of a conformational change of the calix[4]diquinone, possibly due to intramolecular hydrogen bond breaking. The upfield shifts of residual solvent signals for H2O and CHCl3 are also consistent with this.
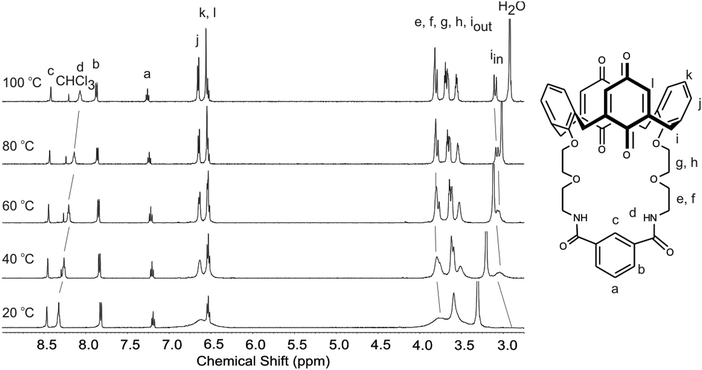 | ||
| Fig. 5 Variable temperature experiment showing the effect of increasing temperature on the 1H NMR spectrum of receptor 3 in DMSO-d6. | ||
For receptor 5 there was evidence of conformational flexibility in solution as a much sharper spectrum was obtained in the solvent CD3CN and ROESY analysis revealed correlations between the calix[4]diquinone protonn and protonsb–d of the anion binding unit (Fig. S1, ESI†). This is not surprising since the macrocycle cavity is larger and intramolecular hydrogen bonding interactions are therefore expected to be weaker.
Ion-pair binding studies
The ion-pair binding properties of the receptors were investigated using UV/visible and 1H NMR spectroscopies in acetonitrile based solvent systems. Ion-pair binding studies with receptor 3 could not be carried out due to insolubility.The cation binding ability of the receptors was investigated using UV/visible spectroscopy by monitoring the characteristic calix[4]diquinone n–π* absorption band. Upon addition of sodium and ammonium cations with non-coordinating counterions, there was little change in the absorption spectra indicating very weak binding (Fig. 6a and Fig. S2a, ESI†). While there was no evidence of potassium cations binding to receptor 4, the addition of potassium ions to receptors 1 and 2 induced small changes to the absorption spectra. However, in the presence of one equivalent of chloride, the absorption spectra changed significantly when increasing amounts of the cation solution were added (Fig. 6b and Fig. S2b, ESI†). Job Plot analysis revealed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding and association constants were obtained from SPECFIT analysis of the titration data (Table 2). Receptors 1–2 and 4 all show enhanced binding of ion-pairs over the ‘free’ ions with a preference for binding NH4Cl over KCl and NaCl. Surprisingly, the more preorganised receptor 2 binds the ion-pairs more weakly than receptor 1. Receptor 4 binds the ion-pairs with a similar magnitude to the previously reported tert-butylcalix[4]diquinone analogue,11 and more strongly than both 1 and 2 due to the electron-withdrawing nitro group increasing the acidity of the amide protons. Disappointingly, there were insufficient changes to infer cation binding to receptor 5, even in the presence of chloride.
:1 binding and association constants were obtained from SPECFIT analysis of the titration data (Table 2). Receptors 1–2 and 4 all show enhanced binding of ion-pairs over the ‘free’ ions with a preference for binding NH4Cl over KCl and NaCl. Surprisingly, the more preorganised receptor 2 binds the ion-pairs more weakly than receptor 1. Receptor 4 binds the ion-pairs with a similar magnitude to the previously reported tert-butylcalix[4]diquinone analogue,11 and more strongly than both 1 and 2 due to the electron-withdrawing nitro group increasing the acidity of the amide protons. Disappointingly, there were insufficient changes to infer cation binding to receptor 5, even in the presence of chloride.
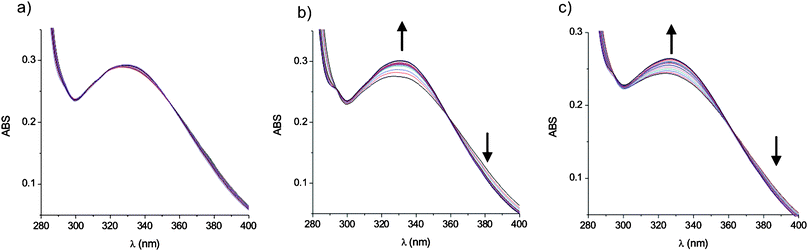 | ||
| Fig. 6 Changes in the absorbance of receptor 1 upon addition of NH4+ cations in the (a) absence and (b) presence of one equivalent of chloride ions in CH3CN and (c) as NH4Cl in 0.5% H2O/CH3CN at 298 K. | ||
| logKM+ |
|||
|---|---|---|---|
| Na+ | K+ | NH4+ | |
| a Association constants derived from SPECFIT analysis of UV/visible spectroscopic titrations in b CH3CN and c 0.5% H2O/CH3CN at 298 K. Errors < 15%. d No evidence of binding. e Evidence of binding but errors > 15%. | |||
| 1 b | —d | 3.26 | —d |
| 1·TBAClb | —e | 3.97 | 4.60 |
| 1·MClc | 3.76 | 3.84 | 4.28 |
| 2 b | —d | 3.25 | —d |
| 2·TBAClb | —e | 3.56 | 3.90 |
| 4 a | —d | —d | —d |
| 4·TBAClb | —e | 4.62 | 5.03 |
| 5 b | —d | —d | —d |
| 5·TBAClb | —d | —d | —d |
Having demonstrated that ion-pair binding occurs by adding the cation to a 1:1 mixture of the receptor and the anion, binding of the ion-pair directly was investigated. This is more difficult to study since ion-pairs of interest are typically insoluble in organic solvents and the receptors are insoluble in water. Therefore, UV/visible titrations were carried out in 0.5% H2O/CH3CN with receptors 1 and 2 adding aqueous solutions of the ion-pairs in small volumes. An increase in the absorbance was observed indicating ion-pair binding to the receptor (Fig. 6c). However, competing sequestration of the ion-pair prevented the determination of association constants for receptor 2. The binding curves for receptor 1 demonstrate that ion-pair sequestration does not appear to be significant over the concentration range investigated (Fig. S3, ESI†). Association constants were determined and these show the same trend as for the previous ion-pair binding studies, although the magnitude of the association constants is lower since a more competitive solvent system was used (Table 2).
The chloride binding ability of the receptors was studied in 98:2 CD3CN/D2O using 1H NMR spectroscopy, monitoring the chemical shifts of the anion binding unit protons upon addition of tetrabutylammonium (TBA) chloride. For receptors 1–2, there was no evidence of chloride binding in the absence of a coordinating cation, as demonstrated by the very small downfield shifts of the amide protonsc and isophthaloyl protonb (Table 3, Fig. 7d). Equally, there were insignificant chemical shift changes upon addition of one equivalent of NH4PF6 (Fig. 7b). For receptors 1–2 and 4, addition of chloride anions to a 1:1 mixture of the receptor and ammonium cations led to significant downfield shifts of protonsb and c (Fig. 7a). Reproducible data for 5 could not be obtained. Job plot analysis of the titration data revealed 1:1 binding and association constants were derived from winEQNMR226 analysis of the titration data. These reveal that chloride recognition is ‘switched on’ in the presence of a coordinating cation for receptors 1–2. Due to the limited solubility of receptor 4, it was only possible to carry out the titration in the presence of a coordinating cation. While there was evidence of NH4Cl binding, an accurate association constant could not be determined due to the anion binding unit proton signals broadening into the baseline for several data points (Fig. S4, ESI†).
| Δδamide/ppm | ΔδisoH/ppm | logKCl− |
|
|---|---|---|---|
|
a Chemical shift changes of the amide and isophthaloyl protons upon addition of one equivalent of TBACl in the presence and absence of NH4PF6 and association constants derived from the amide chemical shift data in 98:2 CD3CN/D2O at 298 K. Errors < 15%.
b No evidence of binding.
c Not applicable.
d Not determined due to insolubility.
e Evidence of binding but an accurate association constant could not be determined.
|
|||
| 1 | 0.051 | 0.017 | —b |
| 1·NH4PF6 | 0.478 | 0.342 | 3.4 |
| 2 | 0.028 | —c | —b |
| 2·NH4PF6 | 0.252 | —c | 3.1 |
| 4 | —d | —d | —d |
| 4·NH4PF6 | —e | 0.592 | —e |
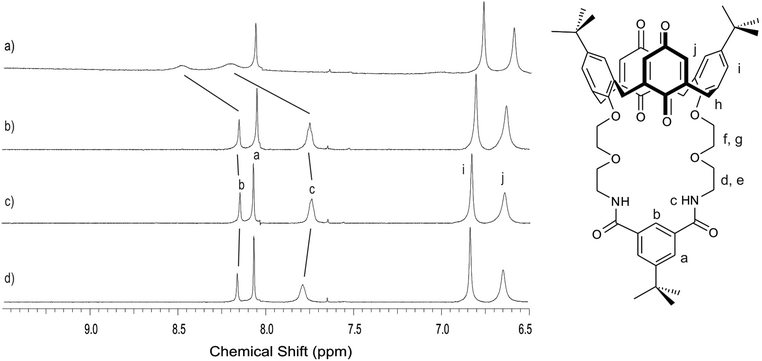 | ||
| Fig. 7 Aromatic region of the 1H NMR spectrum of receptor 1 in the presence of one equivalent of (a) NH4Cl (b) NH4PF6 (c) no guest and (d) TBACl in 98:2 CD3CN/D2O at 298 K. | ||
Direct ion-pair binding was studied by adding aqueous NH4Cl to a solution of receptor 1 in 0.5% H2O/CD3CN. The isophthaloyl and amide proton peaks both shifted significantly downfield upon the addition of up to two equivalents of NH4Cl indicating binding of the ion-pair (Fig. S5, ESI†). However, these peaks began to shift upfield when more than two equivalents of NH4Cl were added, most likely due to sequestration of the ion-pair from the receptor. Therefore, studies with the other receptors were not carried out.
The ion-pair binding studies demonstrate that receptors 1, 2 and 4 are cooperative AND ion-pair receptors. They do not bind free Na+, NH4+ or Cl− but show enhanced binding of ion-pairs NaCl, NH4Cl and KCl. This shows that the tert-butyl substituent on the isophthalamide unit of receptor 1 does not prevent ion-pair recognition. The more preorganised receptor 2 however binds ion-pairs more weakly than receptor 1, most likely due to electrostatic repulsion between the lone pair of the pyridine and the negatively charged anion. It is difficult to compare the ion-pair binding properties of the tert-butylcalix[4]diquinone and calix[4]diquinone receptors since solubility problems limited the scope of the studies. Removal of the tert-butyl substituents on the calix[4]diquinone unit lowers the solubility of the receptors, although it does not appear to significantly alter the ion-pair binding properties, based on the results for receptor 4.
Electrochemistry
The electrochemical properties of calix[4]diquinone receptors in the presence of cations15,16,27 and anions28,29 have been studied previously. The calix[4]diquinone unit can theoretically undergo a four electron reduction to the tetraanionvia two one electron processes, Eo1 and Eo2, and a two electron process Eo3 (Fig. 8). The electrochemistry of receptors 1 and 5 was studied in CH3CN using cyclic voltammetric (CV) and square wave voltammetric (SWV) techniques. Both receptors show two quasi-reversible one electron transfers for redox couples 1/1′ and 2/2′ and a third irreversible two electron transfer for redox wave 3 (Fig. 9 and Fig. S6, ESI†; Table 4). Additionally, receptor 1 has a broad prewave labelled 4, which has also been observed in related tert-butylcalix[4]diquinone systems.15,16![Four electron electrochemical reduction of the neutral calix[4]diquinone, Q–Q, to the tetraanionviaradical anion and radical dianionic species.](/image/article/2012/NJ/c1nj20708c/c1nj20708c-f8.gif) | ||
| Fig. 8 Four electron electrochemical reduction of the neutral calix[4]diquinone, Q–Q, to the tetraanionviaradical anion and radical dianionic species. | ||
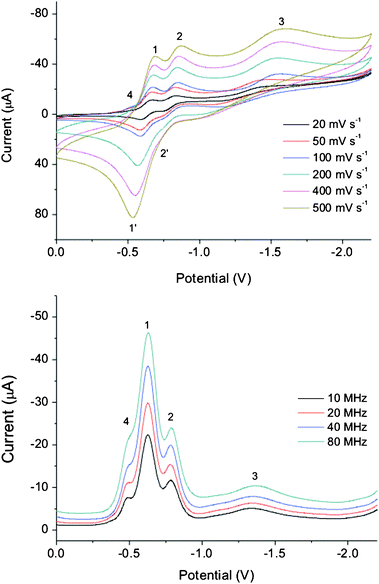 | ||
| Fig. 9 Cyclic and square wave voltammograms of receptor 1 at different scan rates in CH3CN at 298 K. Conditions: 4 mM solution of receptor 1, 0.1 M TBAPF6 supporting electrolyte, glassy carbon working electrode, platinum auxiliary electrode, Ag/AgCl reference electrode. | ||
Disappointingly, electrochemical studies of the receptors in the presence of coordinating cations and anions were hampered by solubility problems and loss of current upon addition of excess ions. Addition of only 0.5 equivalents of cations to receptor 5 led to a complete loss of current and therefore it was not possible to infer cation binding by electrochemistry. Addition of one equivalent of potassium or sodium cations to receptor 1 led to anodic shifts of all three redox waves, while addition of up to 5 equivalents of chloride anions led to no change of the redox potentials but an increase in current (Fig. 10). Addition of one equivalent of potassium to this solution led to anodic shifts of all redox waves, however, addition of excess cations resulted in precipitation of the ion-pair. Unfortunately, repeating the experiment in the presence of one equivalent of chloride, to prevent precipitation of the ion-pair, gave inconsistent results and addition of excess cations led to loss of current. This is suggestive that the mechanism for ion-pair binding is complex. Although it was not possible to obtain any quantitative information about ion-pair binding, the electrochemical studies reveal that receptor 1 binds cations in the presence and absence of anions. Therefore the receptor does not exhibit cooperative AND ion-pair recognition electrochemically, most likely because the background electrolyte disrupts the proposed receptor intramolecular hydrogen bond required for cooperative AND ion-pair recognition.
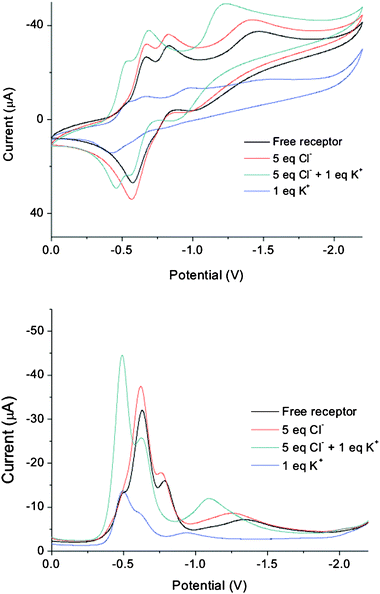 | ||
| Fig. 10 Cyclic voltammograms recorded at 100 mV s−1 and square wave voltammograms recorded at 30 MHz of receptor 1 in the presence of various guest ions in CH3CN at 298 K. | ||
Conclusions
The original family of cooperative AND ion-pair receptors has been expanded to include receptors with different anion binding units and calix[4]diquinone scaffolds in an effort to further elucidate the mechanism behind this unique ion-pair recognition behaviour. A series of calix[4]diquinone receptors have been successfully prepared using the milder oxidant chlorine dioxide.Ion-pair binding studies reveal that receptors 1, 2 and 4 are cooperative AND ion-pair receptors, which display little affinity for ‘free’ Na+, NH4+ and Cl− but enhanced binding of ion-pairs NaCl, NH4Cl and KCl. The calix[4]diquinone receptor 4 displays cooperative AND ion-pair recognition for all of the ion-pairs, whereas the tert-butylcalix[4]diquinone receptors 1 and 2 are cooperative AND ion-pair receptors for NH4Cl and NaCl only, since these receptors bind potassium in the absence of chloride. The more preorganised pyridine based receptor 2 binds ion-pairs relatively more weakly than 1, most likely due to electrostatic repulsion between the pyridine lone pair and the negatively charged anion.
Experimental
Materials and instrumentations
All commercial-grade chemicals and solvents were used without further purification unless otherwise stated. Where dry solvents were used, they were degassed with nitrogen, dried by passing through an MBraun MPSP-800 column and then used immediately. Triethylamine was distilled over and stored over potassium hydroxide. TBA salts were stored prior to use under vacuum in a desiccator containing phosphorus pentoxide and self-indicating silica gel. Deionised water was used in all cases.1H and 13C{1H} NMR spectra were recorded on a Varian Mercury 300, a Varian Unity Plus 500 or a Bruker AVII500 with a cryoprobe spectrometer. Mass spectra were obtained on a Bruker micrOTOF or a MALDI Micro MX spectrometer. Melting points were recorded on a Gallenkamp capillary melting point apparatus and are uncorrected.
Syntheses
Calix[4]arene,21610,22 and 1123,24 were prepared according to the literature procedures.:1 ice–water (50 mL). The orange suspension was extracted with CHCl3 (3 × 50 mL) and the organic layer was dried over MgSO4, filtered and the solution was concentrated in vacuo. Purification by silica gel chromatography (98:2 CHCl3/MeOH) gave 1 as an orange solid (0.075 g, 74%). m.p. 180 °C (dec.). 1H NMR (300 MHz, CDCl3) δ: 1.13 (18H, s, t-Bu), 1.42 (9H, s, t-Bu), 3.29 (4H, d, 2J = 12.9 Hz, ArCHinHoutQu), 3.55–3.99 (20H, m, NCH2, OCH2, ArCHinHoutQu), 6.84 (4H, s, calix ArH), 7.08 (4H, br s, calix QuH), 8.26 (2H, s, isoH), 8.46 (2H, br s, NH), 8.61 (1H, s, isoH). 13C{1H} NMR (125 MHz, CDCl3) δ: 31.21, 31.38, 32.55, 34.04, 35.11, 41.11, 70.14, 72.03, 74.31, 121.51, 126.59, 128.97, 129.07, 133.32, 133.72, 146.44, 147.31, 152.41, 154.44, 168.03, 185.46, 190.50. MS (ES+)m/z: 947.4706 [M + Na]+. Elemental Analysis calcd. for C56H64N2O10·2H2O: C 70.0% H 7.1% N 2.9%. Found: C 69.5% H 6.9% N 2.7%.
:1 ice–water (50 mL). The orange suspension was extracted with CHCl3 (3 × 50 mL) and the organic layer was dried over MgSO4, filtered and the solution was concentrated in vacuo. The crude material was redissolved in CH2Cl2 (20 mL), neutralised with triethylamine and filtered through a plug of silica eluting with CH2Cl2. The band containing 2 was washed with H2O (10 mL), dried over MgSO4, filtered and the solvent was removed in vacuo to give an orange solid (0.05 g, 23%). m.p. 230 °C (dec.). 1H NMR (300 MHz, CDCl3) δ: 1.12 (18H, s, t-Bu), 3.27 (4H, d, 2J = 12.6 Hz, ArCHinCHoutQu), 3.56–4.01 (20H, m, ArCHinHoutQu, NCH2, OCH2), 6.84 (4H, s, calix ArH), 7.02 (4H, br s, calix QuH), 8.03 (1H, t, 3J = 7.9 Hz, pyH4), 8.40 (2H, d, 3J = 7.9 Hz, pyH3 and H5), 9.59 (2H, t, 3J = 5.3 Hz, NH). 13C{1H} NMR (75.5 MHz, CDCl3) δ: 31.38, 32.52, 34.03, 40.98, 70.00, 71.13, 73.80, 125.13, 126.65, 129.10, 133.20, 138.56, 146.33, 147.23, 149.21, 154.17, 164.92, 185.49, 190.09. MS (ES+)m/z: 892.3751 [M + Na]+ (calcd. for C51H55N3O10Na: 892.3785).
:5 CH2Cl2/MeOH), 3 was isolated as a yellow solid (0.057 g, 57%). m.p. 200 °C (dec.). 1H NMR (300 MHz, CDCl3) δ: 3.36 (4H, br s, ArCHinHoutQu), 3.57–3.98 (20H, m, ArCHinHoutQu, OCH2, NCH2), 6.66 (2H, m, calix ArH), 6.82 (4H, br s, calix ArH), 7.07 (4H, br s, calix QuH), 7.62 (1H, t, 3J = 7.8 Hz, isoH), 8.25 (2H, dd, 3J = 7.8 Hz, 4J = 1.5 Hz, isoH), 8.52 (2H, br s, NH), 8.84 (1H, s, isoH). 13C{1H} NMR (125 MHz, CDCl3) δ: 31.13, 41.13, 69.84, 72.28, 73.57, 123.92 124.08, 129.14, 129.81, 130.20, 132.05, 132.58, 133.99, 147.88, 156.03, 167.61, 185.91. MS (ES+)m/z: 757.2729 [M + H]+ (calcd. for C44H41N2O10: 757.2761), 779.2578 [M + Na]+.
:4 CH2Cl2/MeOH), 4 was isolated as a yellow solid (0.12 g, 40%). m.p. 200 °C (dec.). 1H NMR (300 MHz, CDCl3) δ: 3.39 (4H, br s, ArCHinHoutQu), 3.59–3.98 (20H, m, ArCHinHoutQu, OCH2, NCH2), 6.67 (2H, t, 3J = 7.5 Hz, calix ArH), 6.76–6.94 (8H, m, calix ArH, calix QuH), 8.60 (2H, br s, NH), 9.08 (2H, d, 4J = 1.2 Hz, isoH), 9.18 (1H, s, isoH). 13C{1H} NMR (125 MHz, CDCl3) δ: 30.91, 41.41, 69.86, 71.88, 73.49, 124.06, 126.72, 129.15, 130.06, 132.46, 136.04, 147.98, 149.06, 156.03, 165.32, 186.21. MS (ES+)m/z: 824.2421 [M + Na]+ (calcd. for C44H39N3O12Na: 824.2426). Elemental Analysis calcd. for C44H39N3O12·1.3CHCl3: C 56.9% H 4.2% N 4.4%. Found: C 57.0% H 4.4% N 4.3%.
:5 CH2Cl2/MeOH) gave the yellow receptor 5 (0.096 g, 50%). m.p. 200 °C (dec.). 1H NMR (500 MHz, CD3CN) δ: 3.21 (4H, d, 2J = 13.2 Hz, ArCHinHoutQu), 3.52–3.60 (4H, m, NCH2), 3.65–3.75 (20H, m, OCH2), 3.76–3.80 (4H, m, ArCHinHoutQu), 6.63 (2H, t, 3J = 7.7 Hz, calix ArH), 6.67 (4H, s, calix QuH), 6.81 (4H, d, 3J = 7.7 Hz, calix ArH), 7.14 (2H, br s, NH), 7.52 (1H, t, 3J = 7.8 Hz, isoH), 7.91 (2H, dd, 3J = 7.8 Hz, 4J = 2.0 Hz, isoH), 8.00 (1H, s, isoH). 13C{1H} NMR (75.5 MHz, CDCl3) δ: 31.49, 40.36, 69.84, 70.11, 70.52, 73.75, 123.41, 123.70, 129.10, 129.85, 130.23, 131.39, 132.23, 134.25, 147.97, 156.28, 166.65, 186.30, 189.89. MS (ES+)m/z: 845.3275 [M+H]+ (calcd. for C48H49N2O12: 845.3286). Elemental Analysis calcd. for C48H48N2O12·2H2O: C 65.4% H 6.0% N 3.2%. Found: C 65.0% H 5.8% N 3.1%.
:2 EtOAc/petroleum ether) to give 9 as a white solid (0.31 g, 55%). m.p. 176–181°C. 1H NMR (300 MHz, CDCl3) δ: 0.90 (18H, s, t-Bu), 1.31 (18H, s, t-Bu), 1.38 (9H, s, t-Bu), 3.33 (4H, d, 2J = 13.2 Hz, ArCHinHoutAr), 3.71–3.84 (8H, m, NCH2, OCH2), 3.84–3.92 (4H, m, OCH2), 4.18–4.24 (4H, m, OCH2), 4.34 (4H, d, 2J = 13.2 Hz, ArCHinHoutAr), 6.73 (4H, s, calix ArH), 7.04 (2H, s, OH), 7.07 (4H, s, calix ArH), 7.81 (2H, br s, NH), 8.18–8.27 (3H, m, isoH). 13C{1H} NMR (75.5 MHz, CDCl3) δ: 30.91, 31.21, 31.28, 31.68, 33.84, 33.86, 35.11, 40.11, 69.39, 70.13, 75.15, 121.08, 125.17, 125.61, 127.79, 128.57, 132.31, 133.70, 141.90, 147.11, 149.52, 150.02, 152.52, 166.96. MS (ES+)m/z: 1031.6125 [M+Na]+. Elemental analysis calcd. for C64H84N2O8·2H2O: C: 73.5% H 8.5% N 2.7%. Found: C 73.6% H 8.4% N 2.6%.
:5 EtOAc/acetone) gave 10 as a white solid (0.32 g, 55%). m.p. 176–178 °C. 1H NMR (300 MHz, CDCl3) δ: 0.87 (18H, s, t-Bu), 1.27 (18H, s, t-Bu), 3.20 (4H, d, 2J = 13.2 Hz, ArCHinHoutAr), 3.61–3.96 (12H, m, NCH2, OCH2), 4.12–4.32 (8H, m, ArCHinHoutAr, OCH2), 6.68 (4H, s, calix ArH), 6.91 (4H, s, calix ArH), 6.99 (2H, s, OH), 7.73 (2H, t, 3J = 7.6 Hz, pyH4), 8.03 (1H, d, 3J = 7.6 Hz, pyH3 and H6), 9.19 (2H, br s, NH). 13C{1H} NMR (75.5 MHz, CDCl3) δ: 30.88, 30.93, 31.70, 33.75, 33.81, 39.92, 69.43, 69.77, 74.63, 123.94, 124.95, 125.51, 127.59, 132.41, 138.34, 141.43, 146.90, 148.16, 149.01, 149.65, 164.08. MS (ES+)m/z: 976.5576 [M + Na]+.
:5 CHCl3/acetone) eluting 12 as the third band to give a white solid (0.78 g, 66%). m.p. 190 °C. 1H NMR (300 MHz, CDCl3) δ: 3.09 (4H, d, 2J = 12.9 Hz, ArCHinHoutAr), 3.82–4.23 (20H, m, ArCHinHoutAr, OCH2, NCH2), 6.47 (2H, t, 3J = 7.5 Hz, calix ArH), 6.67 (2H, t, 3J = 7.6 Hz, calix ArH), 6.75–6.86 (8H, m, calix ArH), 7.46–7.52 (4H, m, PhthH), 7.52–7.60 (4H, m, PhthH), 7.81 (2H, s, OH). 13C{1H} NMR (75.5 MHz, CDCl3) δ: 30.93, 38.12, 68.54, 70.02, 75.17, 118.82, 122.66, 125.33, 128.01, 128.15, 128.65, 131.78, 133.43, 133.87, 151.35, 152.50, 168.49. MS (ES+)m/z: 881.3537 [M + Na]+.
:5 CHCl3/acetone) to give 13 as a white solid (0.47 g, 48%). m.p. 72–74 °C. 1H NMR (300 MHz, CDCl3) δ: 3.33 (4H, d, 2J = 13.1 Hz, ArCHinHoutAr), 3.68–3.75 (8H, m, OCH2, NCH2), 3.77–3.88 (8H, m, OCH2), 3.94 (4H, t, 3J = 4.7 Hz, OCH2), 4.07–4.15 (4H, m, OCH2), 4.37 (4H, d, 2J = 13.1 Hz, ArCHinHoutAr), 6.61 (2H, t, 3J = 7.6 Hz, calix ArH), 6.71 (2H, t, 3J = 7.4 Hz, calix ArH), 6.86 (4H, d, 3J = 7.6 Hz, calix ArH), 7.03 (4H, d, 3J = 7.4 Hz, calix ArH), 7.68 (4H, dd, 3J = 5.5 Hz, 4J = 3.0 Hz, PhthH), 7.77 (2H, s, OH), 7.82 (4H, dd, 3J = 5.5 Hz, 4J = 3.0 Hz, PhthH). 13C{1H} NMR (75.5 MHz, CDCl3) δ: 31.14, 37.19, 67.96, 70.01, 70.19, 71.03, 75.35, 118.79, 123.17, 125.21, 128.06, 128.37, 128.84, 132.11, 133.35, 133.85, 151.82, 153.20, 168.21. MS (ES+)m/z: 969.4337 [M + Na]+.
:3 EtOAc/acetone) to give 16 as a white solid (0.23 g, 58%). m.p. 285 °C (dec.). 1H NMR (300 MHz, CDCl3) δ: 3.29 (4H, d, 2J = 13.2 Hz, ArCHinHoutAr), 3.70–3.91 (12H, m, NCH2, OCH2), 4.26–4.37 (8H, m, ArCHinHoutAr, OCH2), 6.58 (2H, t, 3J = 7.4 Hz, calix ArH), 6.68 (2H, t, 3J = 7.4 Hz, calix ArH), 6.79 (4H, d, 3J = 7.4 Hz, calix ArH), 6.93 (4H, d, 3J = 7.4 Hz, calix ArH), 7.31–7.41 (3H, m, OH, isoH), 7.52 (2H, br s, NH), 7.93 (2H, dd, 3J = 7.6 Hz, 4J = 1.5 Hz, isoH), 8.10 (1H, s, isoH). 13C{1H} NMR (75.5 MHz, CDCl3) δ: 30.74, 40.03, 68.86, 69.85, 75.28, 119.54, 123.04, 125.40, 127.78, 128.48, 128.98, 129.27, 131.23, 133.04, 133.36, 151.07, 152.08, 166.68. MS (ES+)m/z: 751.3459 [M + Na]+.
:3 EtOAc/acetone) eluting 17 as the third band to give a green solid (0.37 g, 35%). m.p. 290 °C (dec.). 1H NMR (300 MHz, CDCl3) δ: 3.24 (4H, d, 2J = 13.2 Hz, ArCHinHoutAr), 3.69–3.76 (4H, m, NCH2), 3.75–3.83 (8H, m, OCH2), 4.20 (4H, d, 2J = 13.2 Hz, ArCHinHoutAr), 4.23–4.32 (4H, m, OCH2), 6.51 (2H, t, 3J = 7.5 Hz, calix ArH), 6.64 (2H, t, 3J = 7.5 Hz, calix ArH), 6.74 (4H, d, 3J = 7.5 Hz, calix ArH), 6.87 (4H, d, 3J = 7.5 Hz, calix ArH), 7.13 (2H, s, OH), 7.66 (2H, br s, NH), 8.32 (1H, s, isoH), 8.59 (2H, s, isoH). 13C{1H} NMR (75.5 MHz, CDCl3) δ: 30.52, 40.26, 68.86, 69.50, 75.29, 109.99, 119.62, 125.51, 125.97, 127.79, 128.26, 128.57, 129.05, 132.84, 135.12, 150.71, 151.65, 164.58. MS (ES+)m/z: 796.3348 [M+Na]+.
:3 EtOAc/acetone) gave 18 as a white solid (0.31 g, 57%). m.p. 172–174 °C. 1H NMR (300 MHz, CDCl3) δ: 3.33 (4H, d, 2J = 13.2 Hz, ArCHinHoutAr), 3.44–3.58 (4H, m, NCH2), 3.67 (4H, t, 3J = 4.7 Hz, OCH2), 3.71–3.79 (4H, m, OCH2), 3.78–3.89 (8H, m, OCH2), 3.96–4.06 (4H, m, OCH2), 4.33 (4H, d, 2J = 13.2 Hz, ArCHinHoutAr), 6.61–6.75 (4H, m, calix ArH), 6.84 (4H, d, 3J = 7.3 Hz, calix ArH), 7.06 (4H, d, 3J = 7.3 Hz, calix ArH), 7.47–7.56 (4H, m, OH, NH), 7.60 (1H, t, 3J = 7.9 Hz, isoH), 8.11 (2H, dd, 3J = 7.9 Hz, 4J = 1.5 Hz, isoH), 8.29 (1H, s, isoH). 13C{1H} NMR (75.5 MHz, CDCl3) δ: 31.00, 39.93, 69.35, 69.83, 70.54, 75.07, 119.26, 124.51, 125.32, 128.08, 128.51, 128.96, 129.08, 130.96, 133.11, 134.90, 151.63, 152.84, 167.01. MS (ES+)m/z: 839.3873 [M + Na]+.
Acknowledgements
We wish to thank the Woolf Fisher Trust and the Overseas Research Student (ORS) Awards Scheme for a scholarship (A.J.M). C.J.S. thanks the EPSRC and Johnson Matthey for a CASE studentship and the EPSRC for a PhD+ award. We also thank Dr N.H. Rees for valuable NMR advice and the University of Oxford Chemical Crystallography Laboratory for X-ray crystallography.Notes and references
- B. D. Smith, Macrocyclic Chemistry: Current Trends and Future Perspectives, ed. K. Gloe, Springer, Dordrecht, 2005 Search PubMed.
- G. J. Kirkovits, J. A. Shriver, P. A. Gale and J. L. Sessler, J. Inclusion Phenom. Macrocyclic Chem., 2001, 41, 69–75 CrossRef CAS.
- S. K. Kim and J. L. Sessler, Chem. Soc. Rev., 2010, 39, 3784–3809 RSC.
- D. M. Rudkevich, J. D. Mercer-Chalmers, W. Verboom, R. Ungaro, F. de Jong and D. N. Reinhoudt, J. Am. Chem. Soc., 1995, 117, 6124–6125 CrossRef CAS.
- D. J. White, N. Laing, H. Miller, S. Parsons, P. A. Tasker and S. Coles, Chem. Commun., 1999, 2077–2078 RSC.
- N. Pelizzi, A. Casnati, A. Friggeri and R. Ungaro, J. Chem. Soc., Perkin Trans. 2, 1998, 1307–1311 RSC.
- P. D. Beer, Z. Chen and M. I. Ogden, J. Chem. Soc., Faraday Trans., 1995, 91, 295–302 RSC.
- H. Miyaji, S. R. Collinson, I. Prokes and J. H. R. Tucker, Chem. Commun., 2003, 64–65 RSC.
- M. Alfonso, A. Espinosa, A. Tarraga and P. Molina, Org. Lett., 2011, 13, 2078–2081 CrossRef CAS.
- M. D. Lankshear, A. R. Cowley and P. D. Beer, Chem. Commun., 2006, 612–614 RSC.
- M. D. Lankshear, I. M. Dudley, K.-M. Chan, A. R. Cowley, S. M. Santos, V. Felix and P. D. Beer, Chem.–Eur. J., 2008, 14, 2248–2263 CrossRef CAS.
- M. J. Chmielewski and J. Jurczak, Tetrahedron Lett., 2005, 46, 3085–3088 CrossRef CAS.
- M. J. Chmielewski and J. Jurczak, Chem.–Eur. J., 2006, 12, 7652–7667 CrossRef CAS.
- P. A. Reddy, R. P. Kashyap, W. H. Watson and C. D. Gutsche, Isr. J. Chem., 1992, 32, 89–96 CAS.
- P. D. Beer, P. A. Gale, Z. Chen, M. G. B. Drew, J. A. Heath, M. I. Ogden and H. R. Powell, Inorg. Chem., 1997, 36, 5880–5893 CrossRef CAS.
- P. R. A. Webber, P. D. Beer, G. Z. Chen, V. Felix and M. G. B. Drew, J. Am. Chem. Soc., 2003, 125, 5774–5785 CrossRef CAS.
- Y.-L. Lin, T.-S. Yu, W.-Y. Wang and L.-G. Lin, Tetrahedron, 2006, 62, 6082–6089 CrossRef CAS.
- H. M. Chawla and S. N. Sahu, J. Inclusion Phenom. Macrocyclic Chem., 2009, 63, 141–149 CrossRef CAS.
- K.-M. Yang, M.-D. Lee, R.-F. Chen, Y.-L. Chen and L.-G. Lin, Tetrahedron, 2001, 57, 8101–8105 CrossRef CAS.
- M.-D. Lee, K.-M. Yang, C.-Y. Tsoo, C.-M. Shu and L.-G. Lin, Tetrahedron, 2001, 57, 8095–8099 CrossRef CAS.
- V. Percec, T. K. Bera, B. B. De, Y. Sanai, J. Smith, M. N. Holerca, B. Barboiu, R. B. Grubbs and J. M. J. Fréchet, J. Org. Chem., 2001, 66, 2104–2117 CrossRef CAS.
- J. W. Lown, R. R. Koganty and A. V. Joshua, J. Org. Chem., 1982, 47, 2027–2033 CrossRef CAS.
- H. Sato, E. Hayashi, N. Yamada, M. Yatagai and Y. Takahara, Bioconjugate Chem., 2001, 12, 701–710 CrossRef CAS.
- S. Botros, A. W. Lipkowski, A. E. Takemori and P. S. Portoghese, J. Med. Chem., 1986, 29, 874–876 CrossRef CAS.
- A variable temperature experiment was also carried out in CDCl3 between 20–55 °C, which showed similar spectral changes.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- M. Gomez-Kaifer, P. A. Reddy, C. D. Gutsche and L. Echegoyen, J. Am. Chem. Soc., 1994, 116, 3580–3587 CrossRef CAS.
- H. Jeong, E. M. Choi, S. O. Kang, K. C. Nam and S. Jeon, J. Electroanal. Chem., 2000, 485, 154–160 CrossRef CAS.
- S. O. Kang, J. M. Oh, Y. S. Yang, J. C. Chun, S. Jeon and K. C. Nam, Bull. Korean Chem. Soc., 2002, 23, 145–148 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 839213–839216. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1nj20708c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |