Bio-inspired synthesis: understanding and exploitation of the crystallization process from amorphous precursors
Junwu
Xiao
and
Shihe
Yang
*
Department of Chemistry, William Mong Institute of Nano Science and Technology, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong. E-mail: chsyang@ust.hk
First published on 9th November 2011
Abstract
Many biominerals, such as mollusk nacre, sea urchin, bone and teeth, are found to form by an amorphous precursor pathway, and these biominerals have remarkable properties, which are better than their artificial material counterparts that are formed at high temperatures and high pressures. More than ever, synthesizing technologically relevant materials following nature's way with a specific size, shape, orientation, organization, and complex form has been a focus of ongoing interest due to the increasing need for low cost and environmentally friendly approaches to processing advanced materials. Herein, we present recent developments in the crystallization process from amorphous precursors by primarily drawing on results from our own laboratory, and discuss some unique characteristics from the transformation process that can be exploited for the design and synthesis of artificial functional materials.
 Junwu Xiao | Junwu Xiao received his BSc degree in Applied Chemistry from Huazhong University of Science & Technology, China, in 2005. He got his MSc degree in Materials Science in 2008 from Shanghai Institute of Ceramics, Chinese Academy of Sciences, under the supervision of Prof. Yingchun Zhu. He is now a PhD student under the supervision of Prof. Shihe Yang, at the Department of Chemistry, The Hong Kong University of Science & Technology. His research focuses on in vitro studies of biomineralization and bio-inspired design and synthesis of inorganic nanomaterials for energy applications. |
 Shihe Yang | Shihe Yang received his B.S. (1982) in Chemistry from Sun Yat-Sen University in China. Through the US–China CGP program, he was admitted to Rice University in 1983 and obtained a Ph.D. in Physical Chemistry in 1988 (with Prof. Richard E. Smalley). He did his post-doctoral research at Argonne National Laboratory and the University of Toronto (with Prof. John C. Polanyi) before joining the faculty at The Hong Kong University of Science and Technology in 1992, where he is currently a full professor. He was active in understanding the structure, dynamics, and photochemistry of isolated clusters and complexes in the gas phase. His current research interests include the understanding, manipulation and applications of zero-, one-, and two-dimensional nanosystems. |
1. Introduction
The bio-inspired synthesis of technologically relevant materials is a quickly expanding area of research due to its low cost, high efficiency, and low environmental impact. During the biomineralization process, organic matrices exert control over the mineral structure in exquisite crystalline and hierarchical morphologies spanning the nano- to macro-scales, and impart unique mechanical properties to biomineralized tissues,1–3 which outperform traditional engineering composites formed under high temperatures and pressures. Researchers hope to extrapolate the knowledge gained from biomineralization systems and apply it to other inorganic systems, including non-biological materials, to regulate their crystallographic properties for advanced materials applications. Thus, it's important to thoroughly understand the biomineralization mechanisms and, by learning from nature, use the biomineralization mechanisms to guide the design and in vitro synthesis of technologically useful materials.Early on, researchers were eager to know how organic matrices in biomineralized tissues exert their chemical, spatial, structural, morphological, and constructional control over the inorganic constituents. Studies focused on specific organic–inorganic interactions that modulate the crystal morphology.4–13 There has been a shift of attention lately, since it was found that many biominerals may be formed through an amorphous precursor pathway.14–26 Remarkably, crystallization from amorphous precursors can yield nanostructures and metastable phases which are not found in the products of conventional solidification pathways.27–31 By all accounts, such a process and the materials that are obtained from the process are of both scientific and technological interest. It is believed that the first step in the crystallization pathway is the formation and transformation of amorphous precursor nanoparticles.14,20,21,32 This strategy is especially capable of generating complex crystal morphologies, because the amorphous precursors can be moulded and formed into any shape, as in pottery or in glass blowing. Furthermore, crystallization from amorphous materials is also an attractive technique for the synthesis of new materials. Over the past few years, in an effort to understand and exploit the biomineralization processes for advanced synthetic materials, we have focused on the in vitro studies of mineralization by identifying important factors that determine nucleation, and transformation of the amorphous precursors from a supersaturated mother solution. The game-changing factors we have investigated in the in vitromineralization include organic matrices, inorganic impurities, and their combination and specific organizations. Herein, we summarize recent developments, based primarily on our own research work, in the crystallization process of CaCO3 from amorphous precursors. For detailed accounts of many excellent works that have been done by other research groups in this area, we refer readers to some recent reviews.33–37
2. Non-classical crystallization and biomineralization
Fig. 1 categorizes the different possible crystallization routes of minerals known to date.32 Established nucleation-driven crystallization theories mainly dealt with classical crystallization, whereby the primary nanoparticles nucleated from monomers and clusters as the smallest crystalline units grow further by simple ion attachment and unit cell replication to form a macroscopic single crystal, which is more or less an amplification of the initial crystal (pathway a). Later on, researchers also discovered non-classical crystallization pathways. Here, the crystallization proceeds by initially forming crystalline nanostructures or amorphous intermediates, which then assemble among themselves and continually rearrange and densify towards the final crystalline structure.38,39 As the building blocks, the primary nanocrystals are fused into a single crystal by oriented attachment since their surfaces contain face-specific information (pathway b), or are covered by a polymer or other additive before they undergo a mesoscale assembly to form a mesocrystal, or even a single crystal (pathway c). On the other hand, when the amorphous particles are formed as a precursor, they could assemble and transform into complicated morphologies (pathway d) simply because the amorphous precursors can be easily moulded. This type of non-classical crystallization is a general strategy adopted by the biological world and known as biomineralization, which is the central concern of this survey.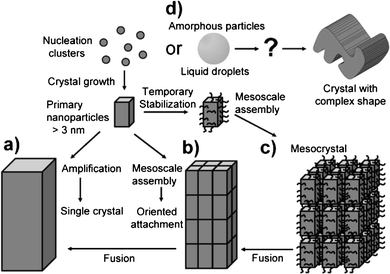 | ||
| Fig. 1 A schematic representation of classical and non-classical crystallization. Pathway (a) represents the classical crystallization pathway, and pathways (b–d) represent the non-classical crystallization processes. Reproduced with permission from ref. 32. Copyright 2003, Wiley-VCH. | ||
For the biomineralization process from amorphous precursors to complex crystals, questions remain about what factors control the crystal size, shape, orientation, phase, texture, and spatial and temporal evolution. Given that biominerals are typically organic–inorganic hybrid materials, a simplistic view about the inorganic and organic constituents would be in regulating and sustaining biomineral formation. In biological systems, organic matrices range from a very small quantity of proteins occluded within the urchin spines (∼0.02 wt%), to an interconnected “foam-like” network within individual tablets of sheet nacre, to a predominant matrix of collagen in bone (roughly 20 wt%).1,40–42 They are generally classified as the insoluble matrix and the soluble additives depending upon their solubility in water after removing the inorganic components. The insoluble matrix provides a scaffolding framework for the deposition of the mineral, such as chitin in some invertebrates or collagen in the case of vertebrate bone and dentin.43,44 The soluble additives are commonly referred to as acidic macromolecules that regulate the mineral formation, which are rich in aspartic and/or glutamic acids, as well as phosphorylated serine and threonine residues, which are rich in carboxyl groups and sometimes sulfates as well.43,45–50 Besides the organic matrices and the obvious requirement of mineral reactants, inorganic impurities found in the physiological environment (such as Mg2+ and Sr2+ ions) may also have a pronounced impact on the mineralization process.21,51–54
3. Template-directed crystallization pathways of CaCO3 under a Langmuir monolayer
Although the amorphous calcium carbonate (ACC) precursor pathway to the final crystalline phases is widespread in biology, it has been overlooked in biomimetic growth studies due to its relatively high solubility and difficulty in its detection, especially when it is intimately associated with a crystalline phase. A minimalist approach to mimic the biomineralization process is to use a Langmuir monolayer to study the crystallization process at well-defined organic/inorganic interfaces. Indeed, the mineralization process of CaCO3 was expansively investigated under Langmuir monolayers of many different lipids,12,55–58 especially stearic acid (SA).59–61 Loste et al.60 studied CaCO3 precipitation under a series of fatty acid Langmuir monolayers using Brewster angle microscopy, and suggested that the polar heads and chain lengths of the surfactants strongly affected crystallization, resulting in changes in the crystal polymorphs and morphologies. Maas et al.61 described the growth of thin films of CaCO3 beneath Langmuir monolayers of stearic acid, and inferred that a crystallization process was induced by a precursor phase with a thin, film-like structure. Surprisingly, direct observation of any amorphous calcium carbonate precursor phase under the SA monolayers was not achieved, not to mention the transformation processes from ACC to stable crystalline phases.By combining in situ (Brewster angle microscopy) and ex-situ (transmission electron microscopy) microscopic studies, we revisited the mineralization process of CaCO3 under a stearic acid monolayer.62 The systematic and careful investigation allowed us to observe that amorphous precursor nanoparticles, 20–80 nm in diameter, were firstly nucleated from a supersaturated Ca(HCO3)2 solution under a stearic acid monolayer (Fig. 2). The direct observation of ACC under the SA monolayer provides evidence for the effect the SA molecules have in mediating ACC formation, which is akin to the role of carboxylic-rich biomacromolecules in biomineralization. As we know, the supersaturation for an amorphous phase (Samorph) should be lower than that for a thermodynamically stable crystalline phase (Scrystal) at a given value of the activity product. Then, according to the nucleation theory,63 the nucleation rate of the crystalline phase should be higher than that of the amorphous phase without any stabilizer. However, the lower supersaturation level of the amorphous phase could be offset and even overtaken by a significantly reduced value for the interfacial energy (δ) due to the amorphous phase itself and its interaction with the carboxyl groups of stearic acid. Thus, the nucleation rate of the amorphous phase would be higher than that of the thermodynamically stable crystalline nuclei under a stearic acid monolayer, allowing the ACC precursor phase to be observed. Of course, after a sufficient reaction time, the ACC precursor could be further transformed into more stable polymorphs, such as calcite, since the ACC precursor is the most thermodynamically unstable phase and has the highest solubility among all the CaCO3 polymorphs.
 | ||
| Fig. 2 TEM images of ACC nanoparticles nucleated from a supersaturated Ca(HCO3)2 solution under a stearic acid monolayer. The inset of (A) is the size distribution of the ACC particles and the inset of (B) is the corresponding electron diffraction pattern. Reproduced with permission from ref. 62. Copyright 2009, the American Chemical Society. | ||
As for what precedes the ACC precursor, Cölfen et al.64 first demonstrated by means of equilibrium thermodynamics that in a dissolved calcium carbonate solution prenucleation clusters formed even when the solution was undersaturated in calcium carbonate. Sommerdijk et al.65 actually observed the prenucleation clusters with dimensions of 0.6 to 1.1 nm in a Ca(HCO3)2 solution by TEM, which are the smallest stable agglomerates of CaCO3 present from the beginning of the reaction and important intermediates between the dissolved calcium carbonate monomer species and the amorphous precursor. By further monitoring the crystallization process under the stearic acid monolayer by in situTEM, the authors were able to propose a mineralization mechanism as follows (Fig. 3). The prenucleation clusters are firstly formed from the dissolved calcium carbonate solution (step 0). Then, the aggregation of these clusters leads to the nucleation of ACC (step 1). The ACC phase at the template surface continues to grow using the neighboring ACC nanoparticles as feedstock (step 2). Next, randomly oriented nanocrystalline domains are formed inside the ACC (steps 3 and 4). Reorientation of the domains occurs in such a way that the orientation that is stabilized through the interaction with the monolayer becomes dominant (step 5) and develops into a single crystal (step 6), and probably grows by further addition and incorporation of ions and clusters from solution.
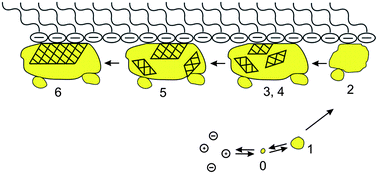 | ||
| Fig. 3 A schematic pathway of the mineralization under a stearic acid monolayer. Step 0: formation of prenucleation clusters. Step 1: aggregation of the clusters to form ACC nanoparticles. Step 2: clustering and growth of ACC particles at the interface of the monolayer. Step 3: the start of the crystallization: formation of poorly crystalline particles. Step 4: formation of nanocrystalline domains inside the amorphous particle. Step 5: oriented growth of the crystalline domain stabilized by the template. Step 6: formation and growth of oriented single crystals. Reproduced with permission from ref. 65. Copyright 2009, the American Association for the Advancement of Science. | ||
Phospholipids, as the key membrane constituents of biological systems, are commonly involved in delineating reaction compartments for the crystallization of biominerals, such as the coccolith of marine alga Emiliania huxleyi,66 the mineralizing tissues of vertebrates,67 and magnetosomes in magnetotactic bacteria.68–70 In pursuing in vitro models, Mann et al.71 used phospholipid unilamellar vesicles to study membrane-mediated growth of iron oxide crystals. It was found that lipid vesicles not only acted as passive hosts to enclose mineralization reactions but also strongly influenced the growing inorganic phase through the molecular recognition of chemical, electrostatic, and chiral complementarity. However, with the mineral formation occurring inside vesicles, it was difficult to compellingly demonstrate that the nucleation actually occurs on the membrane itself. To shed light on this issue, we studied the crystallization process under a zwitterionic phospholipid (1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine, DPPC) Langmuir monolayer.72Fig. 4 shows the particles formed through the mineralization pathways of calcium carbonate under a DPPC monolayer, which is proposed as follows: firstly, the ACC precursor particles nucleate at the air–water interface (Fig. 4A and B), and quickly transform into vaterite nanocrystals (Fig. 4C and D). Driven by the trend to decrease the surface energy, the vaterite crystal nuclei self-aggregate into the loose-packed hollow ellipsoidal particles, resulting in a polycrystalline structure (Fig. 4E and F), and then gradually evolve into the single crystal-like, tightly packed ellipsoidal particles by orientational rearrangement and consolidation, with the (001) crystal faces being stabilized by the DPPC molecules (Fig. 4G and H). Fig. 5 schematically illustrates our observations. Taken together with the results from the crystallization processes under stearic acid and the phospholipid monolayers,62,64,65,72 we conclude that the template-directed crystallization of CaCO3 proceeds as follows: the dissolved calcium carbonate → prenucleation clusters → ACC → nanocrystals in ACC → polycrystals → mesocrystals → single crystals.
 | ||
| Fig. 4 The formation of calcium carbonate particles under a DPPC monolayer at a surface pressure of 40 mN m−1 after reaction for various times: (A) TEM image and Raman spectrum (inset) of ACC particles formed after 0 h; (B) SAED pattern of the ACC particles in (A); (C) TEM image of the vaterite particles formed after 0 h; (D) HRTEM image of the vaterite particles in (C); (E) TEM image of the loosely-packed ellipsoidal particles formed after 0.5 h; (F) HRTEM image of the loose-packed ellipsoidal particles in (E); (G) a top-view TEM image and SAED pattern (inset) of the tightly-packed ellipsoidal particles formed after 1 h; (H) a side-view TEM image and SAED pattern (inset) of the tightly-packed ellipsoidal particles formed after 1 h. In taking the SAED patterns shown in the inset of Fig. 4B, G and H, the electron beam was 110 nm, 1 μm and 1 μm in diameter, respectively, and directed to the whole particle under examination, which are marked by white hollow circles in A, G and H. Reproduced with permission from ref. 72. Copyright 2010, the American Chemical Society. | ||
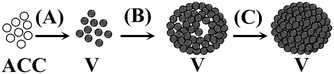 | ||
| Fig. 5 A schematic representation of the mineralization pathways of calcium carbonate under a DPPC monolayer. The formed ACC particles were transformed into vaterite crystals (A), self-aggregated into loosely-packed ellipsoidal, polycrystalline particles (B), and evolved into tightly-packed ellipsoidal, mesocrystals (C). (ACC: amorphous calcium carbonate; V: vaterite). Reproduced with permission from ref. 72. Copyright 2010, the American Chemical Society. | ||
4. Polymorph selection controlled by the surface functional group density of the Langmuir monolayer
Biomineralizing organisms selectively form multiple polymorphs of calcium carbonate (e.g., vaterite, aragonite, and calcite) from an amorphous precursor.1,73 Polymorph selection can be chemically controlled by kinetic effects involving additives or through transformation processes that proceed along a series of structures with decreasing solubility and increasing thermodynamic stability. During our study of the crystallization process under a phospholipid monolayer, we found that vaterite mesocrystals crystallized from ACC precursors could further transform into the most thermodynamically stable rhombohedral calcite crystals when the monolayer was not compressed due to the thermodynamic effect, whereas more and more of them radially grew into floret-like vaterite crystals with an increase in the surface pressure owing to the kinetic effects.72 Moreover, the relative contents of vaterite and calcite at various surface pressures were quantitatively analyzed according to XRD results on the basis of the standard reference intensity ratio (RIR) method, as listed in Table 1. The results show that the high surface pressure inhibits the transformation from vaterite to the most thermodynamically stable calcite phase. Generally, vaterite doesn't expose the (001) crystal face because of its high surface energy in the absence of growth modifiers.74,75 Conceivably, under a higher surface pressure, more phospholipid molecules could be more readily adsorbed onto the high energy (001) face of the vaterite crystals. This would then decrease the surface energy and thus create and/or increase the kinetic barrier for the transformation into the more stable calcite phase. In another study, Volkmer et al.76 proposed that a monolayer with a sufficiently high surface charge density is able to arrest a metastable crystal phase at the air–water interface (see Fig. 6). For example, vaterite crystals are preferentially formed at a surface charge density corresponding to 6.7–7.2 COO− nm−2.58 The surface charge densities in the range of 4.65–5.00 COO− nm−2 lead to a selective crystallization of aragonite.77,78 The uniformly oriented calcite crystals with the highly polar {012} face oriented toward the monolayer are formed at charge densities of 2.0–2.4 COO− nm−2.57,79 The switching between the polymorphs of CaCO3 possessing different thermodynamic stabilities occurs above a critical surface charge density of the monolayer molecules and is primarily a result of a kinetically controlled nucleation process. Thus, organic functional groups, be they carboxylic, hydroxyl, or phosphate groups, can capture a particular metastable polymorph at a given surface density, driven by a reduced surface energy and thus an increased kinetic hindrance to the conversion into a more stable polymorph.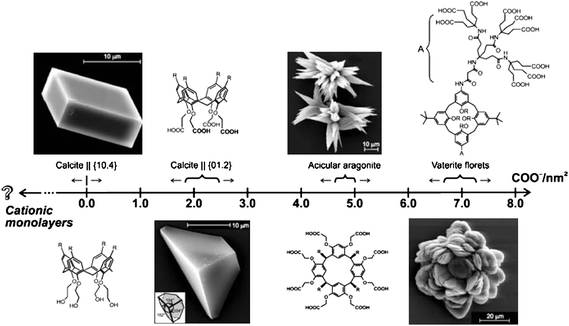 | ||
| Fig. 6 The effect of the charge densities of the macrocyclic polyacid monolayers on the growth of calcium carbonate. Polyacids are arranged in the direction of increasing (negative) charge density, which is expressed here as the number of carboxylate residues per unit area. (Charge density values were directly obtained from the Langmuir isotherms.) Reproduced with permission from ref. 76. Copyright 2007, Springer Science and Business Media. | ||
5. Morphological control of the anhydrous polymorph transformed from amorphous precursors by a template structure
In fact, biominerals are often formed in a preformed three-dimensional (3D) matrix, soaked in fluids consisting of ions of defined concentrations and organic macromolecules, which are mainly acidic macromolecules,48,80,81 rather than under a Langmuir film. For example, in the biomineralization process of shellfish, the insoluble chitin matrix is postulated to provide a scaffold for mineralization,9,82,83 and the soluble biomacromolecules (proteins and acidic polysaccharides) control the formation and morphology of the biominerals.45,73 Most previous investigations were focused on the derivatives of chitin, such as chitosan, since chitin is water insoluble and intractable. Chitosan was normally cast on to a flat substrate (essentially a two-dimensional (2D) matrix)84–88 or made into a 3D scaffold by freeze-drying89,90 or self-assembly91 for mineralization studies. These studies have not yet focused on 3D matrix-mediated mineralization from amorphous precursors, which is more akin to the real biomineralization processes.In mimicking biomineralization, which commonly involves carboxyl-rich molecules, we utilized trisodium citrate molecules to tempt the nucleation and stabilization of amorphous precursors, and to control the subsequent crystallization under an insoluble chitosan scaffold. Fig. 7 shows the SEM images of crystals formed during the crystallization process under an insoluble chitosan scaffold.92 Firstly, a gel-like hydrated ACC precursor with complete disorder was nucleated from the citrate–Ca2+ ion pairs (Fig. 7A and A2). The strong interaction between citrate and the hydrated ACC precursor with complete disorder presumably obstructed its fast reorganization into one of the stable crystalline anhydrous phases. Thus, as a compromise, stable ACC nanoparticles with a short-range order of calcite were formed (Fig. 7B and B2). Secondly, with an increase in the reaction time, the stable ACC nanoparticles in the diameter range of 70–80 nm were transformed and grew into larger calcite nanocrystals ∼150 nm in diameter (Fig. 7C and C2). Afterward, the calcite nanocrystals kept growing into the spherically shaped, polycrystalline calcite microparticles by aggregation, accompanied and sustained by the continuous formation and, thus, supply of ACC precursor (Fig. 7D and D2), which is in keeping with the increasing calcite content with the reaction time. With more freedom and being in close proximity to water, the building block calcite nanocrystals in the outer shell of the spherical shaped polycrystalline calcite microparticles gradually oriented themselves into the rough but faceted rhombohedral calcite mesocrystals through electrostatic interactions (Fig. 7E and E2). Finally, the rough rhombohedral calcite mesocrystals gradually evolved together to form nearly perfect, smooth rhombohedral calcite mesocrystals (Fig. 7F and F2). This result, when compared with that presented in Section 3, reveals that the mineralization process from amorphous precursors in an insoluble scaffold is parallel to the template-directed crystallization process.
 | ||
| Fig. 7 SEM images of the CaCO3 crystals formed from carboxyl-rich molecules stabilized by amorphous precursors under an insoluble chitosan scaffold at various reaction stages: (A, A2) firstly, the gel-like hydrated ACC precursor are nucleated; (B, B2) they are then transformed into the stable ACC nanoparticles; (C, C2) the stable ACC nanoparticles are transformed into calcite nanocrystals; (D, D2) they gradually grow into calcite polycrystals; (E, E2) the calcite polycrystals are oriented into the rough but faceted rhombohedral calcite mesocrystals; and (F, F2) finally formed into smooth rhombohedral calcite mesocrystals. Reproduced with permission from ref. 92. Copyright 2011, Wiley-VCH. | ||
Biomineralization yields single crystals whose morphologies bear no relation to their crystallographic structure, as in the skeletal plates of echinoderms, or the curved and rounded surfaces in sponge spicules.1 To exploit this trait of biomineralization, we studied the crystallization processes starting from an amorphous calcium carbonate precursor stabilized by citric acid molecules under cetyltrimethylammonium bromide (CTAB) micellar templates.27Citrate molecules were utilized for tempting the nucleation of amorphous precursors and their subsequent stabilization. Then, the ACC precursors were crystallized into CaCO3 crystals with various novel morphologies, including hollow radiating cluster-like particles, hollow sheaf-like crystals, and hollow rods, regulated by the CTAB concentration, as seen in Fig. 8. This strategy has also been used by other groups. Aizenberg and co-workers prepared a specially designed self-assembled monolayer with quasi-two dimensional (q2D) micropatterns for hosting oriented nucleation of calcite from ACC precursors.28 In a work by Qiet al., monodisperse colloidal spheres were assembled into colloidal crystals as sacrificial templates to fabricate 2D honeycomb-patterned CaCO3 crystals93 and “3D ordered macroporous materials (3DOM)” of single crystalline calcite from ACC precursors.29 Conceivably, soluble organic matrices, such as carboxyl-rich molecules, acted to tempt the nucleation and temporary stabilization of the amorphous precursors, allowing ACC to fill the voids of the insoluble organic frameworks. The amorphous-to-crystalline transformation process occurred within the restricted insoluble organic frameworks leading to the pre-defined morphologies.
 | ||
| Fig. 8 SEM images of CaCO3 crystals crystallized from citrate stabilized amorphous precursors in the presence of various CTAB concentrations: (A) 1 mM; (B) 5 mM; (C) 10 mM; (D) 100 mM; and (E) 277 mM. Reproduced with permission from ref. 27. Copyright 2011, the Royal Society of Chemistry. | ||
6. Cooperative influence of organic matrices and Mg2+ on the morphology and polymorph of the minerals
Besides the organic matrices mentioned above, inorganic impurities, such as Mg2+ ions, also exist in the carbonate skeletons of marine organisms with widely varying concentrations ranging from a near-zero level of magnesium in calcitic portions of mollusk shells, to over 30 mol% in certain skeletons of algae and echinoderms21,51,52 and even 50 mol% in the specialized teeth of sea urchins.94 The Mg content in the crystal lattice is thought to have governed the polymorphic and morphological transitions,53,95,96 but it is still unclear about what factors control the Mg content in the crystal lattice and influence the polymorphic and morphological transitions. Generally, crystalline calcium carbonate is transformed from amorphous calcium carbonate (ACC) as a transient precursor and depot, and almost all known biogenic hydrated ACC (CaCO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 1:1) contains a high percentage of magnesium.52 The acidic proteins, usually rich in aspartate and glutamate, and polysaccharides regulate the Mg content in biogenic ACC97–99 since the carboxyl-rich molecules, with a strong affinity for Ca2+ over Mg2+, promote the formation of Mg-enriched ACC.100
:H2O = 1:1) contains a high percentage of magnesium.52 The acidic proteins, usually rich in aspartate and glutamate, and polysaccharides regulate the Mg content in biogenic ACC97–99 since the carboxyl-rich molecules, with a strong affinity for Ca2+ over Mg2+, promote the formation of Mg-enriched ACC.100
The Mg-ACC precursors would subsequently crystallize into stable polymorphs, such as aragonite and high magnesium calcite. Jiang et al.101 reported that Mg-ACC precursors were transformed into magnesium calcite in a confined crystallization environment without any organic additives, and they suggested that the confined crystallization environment had prevented the escape of Mg2+. We have been interested in how organic matrices in biogenic carbonates modulate the magnesium content of the final calcite or aragonite that comprises a calcified carbonate skeleton. We have employed gelatin, a denatured collagen protein, as an acidic biomacromolecule to study how the Mg contents are regulated in the mineralization and crystallization processes and how, in turn, they influence polymorphs and morphologies of the minerals and crystals therefrom.102 Firstly, we have studied the influence of the gelatin concentration on the polymorphs and morphologies, with the Mg/Ca molar ratio in the mother solution being set as 50/50. Without gelatin, amorphous nanoparticles containing 14.6 mol% magnesium were formed. With an increasing gelatin concentration, we successively obtained CaCO3·H2O spindles, aragonite hemispheres, and magnesium calcite. The molar percentages of Mg changed from 6.8, 2.7 to 14.3 mol% corresponding to the samples of CaCO3·H2O, aragonite, and magnesium calcite, respectively. Thus, the concentration of gelatin not only altered the Mg contents in the samples, but also influenced the polymorphs at a given Mg2+ molar percentage. Secondly, the influence of the Mg2+ concentration in the mother solution on the polymorph and morphology was considered at a fixed gelatin concentration. Upon decreasing the molar percentage of Mg2+ in the mother solution, a proportional decrease in the Mg content in the calcite crystal products was observed (14.3 mol% for Mg:Ca = 50:50; 4.7 mol% for Mg:Ca = 20:80; and 0.0 mol% for Mg:Ca = 100:0). A similar observation was documented by Ma et al.,103 who found that the magnesium content in high magnesium calcite transformed from polymorph-stabilized Mg-ACC precursors via a non-classical crystallization process, which could be tuned from 15 to 40 mol% by changing the Mg/Ca ratio in the mother solutions.
How did the gelatin concentration alter the Mg contents in the final crystals at a given Mg2+ concentration in the mother-liquor? Previously, it was found that acidic amino acids, peptides, and proteins accelerate the calcite growth rate in a way that is correlated with the hydrophilicity of the biomacromolecules,104 which is a measure of their interactions with water. Theoretical simulations have also revealed an enhanced rate of Ba2+ desolvation in the presence of aspartic acid monomers in a barite (BaSO4) system.105 A similar mechanism may be invoked here to explain the Mg uptake into the crystals. Quintessentially, Mg2+ ions are more strongly solvated than Ca2+ ions, hence the dehydration step could limit the crystal growth rate.106,107 Thus, the role of the acidic groups in gelatin, by virtue of their stronger attractive interaction with Mg2+ over Ca2+, is then to lower the desolvation barrier and create a low-energy pathway for facilitating the uptake, desolvation, and transport of the Mg2+ ions from the bulk solution to the mineral surface.
Next, what factors play a role in the transformation processes from Mg-ACC precursors into aragonite and magnesium calcite? Mg-ACC precursors are transformed into aragonite and magnesium calcite depending on the Mg2+ content incorporated in the final crystals. Mg2+ is also known to have no effect on the rate of crystal growth of aragonite but has a strong retarding effect on that of calcite as the denser aragonite structure is much more averse to the incorporation of partially dehydrated magnesium ions. The incorporation of Mg2+ into the calcite structure induces strain in the solid lattice, thereby increasing the internal free energy of the crystal and the crystal solubility.108 In fact, calcite with a magnesium content of about 10 mol% has the same solubility as aragonite.109–111 If no organic molecules were used to decrease the dehydration energy barrier of Mg2+ incorporation into the crystal lattice, an aragonite polymorph would be formed from the Mg-ACC precursors prior to high magnesium calcite. Gelatin influences the polymorph transformation from amorphous precursors to aragonite or magnesium calcite by modulating the Mg incorporation into the crystal lattice.
There have also been reports on the inhibition of the calcite crystal growth by inorganic dopants112–116 added at the growth step.117 De Leeuw et al.54 showed that growth of pure calcite is an endothermic process (on average +1.8 to +35 kJ mol−1 per CaCO3) but the growth could be reversed to an exothermic process when an inorganic impurity is incorporated. Indeed, growth of the impurity carbonate was shown to be highly exothermic for the first row at −25 to −155 kJ mol−1 per MCO3, but much less so for the subsequent row (−5 to −80 kJ mol−1 per MCO3), due to the increasing mismatch between the rows of MCO3 at the surface and the underlying calcite lattice. Thus, although the initial growth of the impurity carbonates of calcite can be energetically very favorable, the subsequent incorporation of these impurities would become more and more endothermic (+15 to +75 kJ mol−1 per CaCO3), culminating in the inhibition of calcite growth.
In previous in vitro biomimetic studies on the selection of polymorphs and morphologies, organic and inorganic additives were usually added into the original reactants making no distinction of the nucleation process of the amorphous precursors from the amorphous-to-crystalline transition process.118–124 Thus, it's unclear precisely in which process organic matrices and inorganic impurities control the polymorphism and morphology, and how the functional groups in the organic matrices influence the polymorph selection in cooperation with the inorganic impurities.
Carboxyl, hydroxyl and amine groups are the most important functional groups of proteins and acidic polysaccharides. We utilized three organic molecules (trisodium citrate, poly(ethylene oxide) (PEO) and poly(ethyleneimine) (PEI)) containing these functional groups to show how they influence the crystallization from amorphous precursors.125 The amorphous phases of calcium carbonate were obtained from the mother solution of Ca2+ and Mg2+ (Ca2+:Mg2+ = 1:1) in the absence of any organic molecules or in the presence of trisodium citrate, PEO and PEI, which were designated as ACC, SC-ACC, PEO-ACC and PEI-ACC, respectively. Fig. 9 summarizes the whole portfolio of the mineral transformation processes from the four different amorphous precursors. Evidently, the polymorphic transformations from PEO-ACC and PEI-ACC precursors are in reference to those from ACC precursors in various re-dispersion media. Interestingly, with the ACC, PEO-ACC and PEI-ACC precursors, the CaCO3·H2O polymorph crystallized in the aqueous solutions of carboxyl-rich molecules (trisodium citrate and sodium alginate) and remained unchanged even after 21 days of aging, whereas aragonite crystals were formed in deionized (DI) water, PEO and PEI aqueous solutions. PEO and PEI were used for this study because polymeric molecules commonly act as scaffolds for mineralization; however, they are essentially ethers and secondary amines, and have a limited number of the hydroxyl and primary amine groups needed for a truthful comparison. Nevertheless, our study using small molecules, such as ethylene glycol and hydrazine, added in the nucleation process of the amorphous calcium carbonatein lieu of the PEO and PEI macromolecules, respectively, also found their negligible influence on the polymorphic transformations.
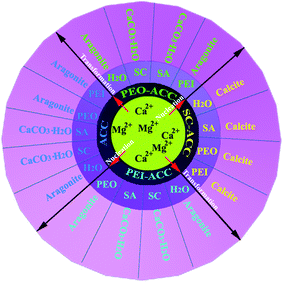 | ||
| Fig. 9 A schematic illustration of the transformation processes from amorphous calcium carbonate (ACC), trisodium citrate stabilized amorphous calcium carbonate (SC-ACC), poly(ethylene oxide) stabilized amorphous calcium carbonate (PEO-ACC), and poly(ethyleneimine) stabilized amorphous calcium carbonate (PEI-ACC) precursors to crystalline phases in various re-dispersion media. (SC: trisodium citrate; SA: sodium alginate; PEO: poly(ethylene oxide); PEI: poly(ethyleneimine). Reproduced with permission from ref. 125. Copyright 2011, the Royal Society of Chemistry. | ||
Understandably, for the re-dispersion media containing trisodium citrate and sodium alginate, the carboxyl groups have strong chemical bonding, as opposed to the weak interactions of the hydroxyl groups of PEO and the amine groups of PEI,126 with the CaCO3·H2O crystals through the Mg2+ ions without leaching. Thus, they hold back their transformation into the aragonite polymorph. Hence in this case, the final polymorph is the crystalline CaCO3·H2O in the carboxyl-rich re-dispersion media. With the SC-ACC precursors, however, the formed samples are all in the calcite polymorph regardless of whether organic molecules are added to the re-dispersion medium or not. Taken together, we can conclude that it is the carboxyl-rich molecules added in the nucleation process of the amorphous precursors, instead of those added during the amorphous-to-crystalline transition process, that really dictate the polymorph transformation into magnesium calcite. Hydroxyl and amine-rich molecules induced a preferential transformation from the amorphous to the thermodynamically metastable aragonite, in a way similar to the case in the absence of organic matrices.
How did the carboxyl-rich molecules added into the nucleation process of the amorphous precursors, instead of added during the amorphous-to-crystallization transition process, influence the polymorph transition? The organic matrices modulate the Mg incorporation into the crystal lattice and thus influence the amorphous-to-crystalline transformation by altering the dehydration energy barrier of Mg2+. As previously stated, the enthalpy of dehydration for Mg2+ (1882 kJ mol−1) is much higher than that of Ca2+ (1569 kJ mol−1) at 25 °C. Thus, the dehydration of the Mg2+ ions prior to incorporation into the calcite lattice presents a sizable barrier to the growth of calcite nuclei. When trisodium citrate molecules were added into the nucleation process of the amorphous precursors, they decreased the dehydration energy barrier for the Mg2+ incorporation into the crystalline lattices, and thus led to the selective formation of magnesium calcite crystals with ∼12 mol% of magnesium. The added PEO and PEI molecules couldn't significantly decrease the dehydration energy for the Mg2+ incorporation into the crystal lattice. Consequently, Mg2+, together with its hydrated water, can't be incorporated into the crystal lattice, which gives rise to the aragonite crystals with less than 5 mol% of magnesium.
7. Mechanical properties of the organic–inorganic composites with hierarchical structures
At present, the main industrial use of CaCO3 in the present day is as a filler, which is simply blended with other components for paper-making and the manufacture of composite materials, paints and coating dispersions. Nature has evolved a way to produce light-weight and strong materials with exceptional properties and functionalities by synergistically combining organic and inorganic components, which are imperative to life.127–129 Tremendous value can be added to the traditional use of CaCO3 by emulating biominerals considering that biominerals can display comparable and frequently superior mechanical robustness to those exhibited by many engineering materials, with similar chemical compositions, but formed at high temperatures and pressures. For example, in sea urchin spines, magnesium-rich calcite crystals highly aligned in a three-dimensional fenestrated mineral network surrounding micrometer-sized spaces occupied by living cellular tissue,130,131 play key functional roles; e.g., protecting the body from predators and they are involved in locomotion and in burrowing. We have synthesized the high magnesium calcite microspheres (HMCMs) containing 14.3 mol% magnesium from amorphous precursors under the cooperation of a soluble organic matrix (gelatin) and inorganic impurities (Mg2+), in which the soft gelatin matrix serves to adhere and align the mineral nanorod building blocks (Fig. 10).102 Remarkably, such HMCMs exhibit high mechanical performance (E = 28.91 ± 2.97 GPa; H = 1.37 ± 0.07 GPa), which are comparable to that of the central stereo of the Paracentrotus lividus spine skeleton (E = 32.20 ± 3.26 GPa, H = 1.76 ± 0.70 GPa).132 The excellent mechanical properties of the HMCMs can be ascribed to their microstructure with the incorporation of the organic matrix among inorganic crystal building blocks, like the brick-and-mortar structure of a nacreous layer. Clearly, this organic–inorganic microstructure has imparted a pronounced enhancement of the fracture toughness, while largely preserving the hardness of the inorganic component.63,133 In comparison with the dense CaCO3 crystal, the reduction of hardness is presumably caused by having a relatively high content of gelatin (7.3 wt%) in the composites. This can be improved by decreasing the organic matrix content in future experiments. | ||
| Fig. 10 (A) SEM image and (B) schematic graphic of the high magnesium calcite microspheres in which nanorod crystals (black part in B) are vertically aligned within the cohesive gelatin matrices (blue part in B). Such a bio-inspired structure gives rise to remarkable mechanical properties (E = 28.91 ± 2.97 GPa; H = 1.37 ± 0.07 GPa) rivalling the central stereo of the Paracentrotus. lividus spine skeleton. Reproduced with permission from ref. 102. Copyright 2011, the Royal Society of Chemistry. | ||
8. Applying the bio-inspired approach to the synthesis of non-biological minerals for energy storage
Besides understanding the biomineralization mechanisms more clearly, it is more important to know whether we can apply it to synthesize non biological materials for advanced technology applications, such as energy storage. Electrochemical capacitors (ECs), also called supercapacitors or ultracapacitors, provide transient but extremely high power densities and are probably one of the most important next generation energy storage devices.134,135 They are divided into two types according to the fundamental mechanisms that govern their capacitance. One is the electrical double layer capacitors (EDLCs), where the capacitance is attributed by the accumulation of charge at the electrode/electrolyte interfaces. The typical electrode materials of EDLCs are carbonaceous materials with high specific surface areas (SSAs) and good electric conductivities, such as CNTs, graphene and so on. Another is pseudocapacitors, where an actual battery-type oxidation–reduction reaction occurs leading to pseudocapacitance, and these are mainly based on transition metal oxides and hydroxides, nitrides, sulfides, and conducting polymers (polyaniline (PANI), polypyrrole (PPy), etc.).Pseudocapacitive materials, especially those for transition metal compounds, have high theoretical specific capacitances, well-defined electrochemical redox activities, and are low cost and, thus, ideal replacements for rare and expensive RuO2. However, the measured capacitances are relatively low when these electroactive materials are grown via the classical crystallization, i.e., simple ion attachment and unit cell replication of nucleation clusters, which is due to the poor electrical conductivity and large size. The electrochemical performance of an EC mainly depends on three factors. One is electric conductivity, which can be enhanced by decreasing the size of the electroactive material and/or incorporating carbonaceous materials; another is the specific surface area, which can be increased by synthesizing nanosized and/or micro- and mesoporous electroactive materials with numerous electroactive sites; and the final is ion transportation, which can be improved by preparing hierarchical (meso- and macro-) porous electroactive materials for easy access of the ions. Thus, there are two ways to improve the performance of pseudocapacitive materials. One is to decrease the crystal size, and another is to introduce carbonaceous materials into the pseudocapactive materials.
Our recent work on electrode materials for supercapacitors by sequential crystallization coupled with morphology-conserved conversion has led to the successful synthesis of sea urchin-like, porous NiCo2O4 spinel.136Nickel cobaltite spinel was chosen for the supercapacitor electrode material as it displays an electronic conductivity which is two orders of magnitude higher than those of the monometallic nickel oxides and cobalt oxides.137 Key to the synthesis was the formation of a bimetallic carbonate hydroxide precursor, albeit not a bona fide amorphous precursor, via a sequential crystallization process, which was found to start with the nucleation of monometallic nickel carbonate hydroxide evolving into flower-like microspheres. This was followed by the nucleation and growth of the bimetallic carbonate hydroxide nanorods from and on the nanoplates in the flower-like microspheres by localized dissolution–recrystallization, leading finally to the sea urchin structure. The crystallization process is similar to biomineralization, in which amorphous precursors or polymorphs with less stable crystalline structures are firstly nucleated and then transformed into a more stable crystalline phase. The morphology-conserved NiCo2O4 spinel nanostructure, after calcination. uniquely comprises hierarchical, interconnected pores with high specific surface areas (198.9 m2 g−1) suitable for fast electron and electrolyte transport. Indeed, the porous sea urchin-like NiCo2O4 nanostructure has exhibited a higher specific capacitance (658 F g−1 at 1 A g−1) than the monometallic cobalt oxides (60 F g−1 at 1 A g−1) and nickel oxides (194 F g−1 at 1 A g−) with similar porous nanostructures. Significantly, even at a high current density of 10 A g−1, the pseudocapacitor made of the NiCo2O4 porous materials retained its high specific capacitance of 530 F g−1 with excellent cycling stability.
We have now started to integrate the amorphous precursor based on the non-classical crystallization to the hybridization strategy to overcome the low specific capacitance of electroactive materials by dispersing them on the surfaces of carbonaceous materials with large specific surface areas, hierarchical porosity and good electrical conductivities. Graphene is a two-dimensional carbonaceous material with a high specific surface area (∼2620 m2 g−1) and electrical conductivity, and it is also a good capacitive material itself. Importantly, its dense functional groups, including the epoxide, hydroxyl, carbonyl and carboxyl groups, can be used as nucleation sites to adjust the crystallization of inorganic compounds,138 which is akin to the carboxylic-rich organic matrices for biomineralization. The pseudocapacitive materials crystallized from amorphous precursors via a non-classical crystallization process can decrease the sizes to the nanoscale, and yield hierarchical nanostructures, which bear no relation to their crystallographic structures but depend only on the template structure. Such an extrapolation of the biomineralization mechanism to the synthesis of pseudocapacitive materials using graphene oxide as a support is expected to improve the electrochemical capacitance performance of these materials compared to those obtained by a classical crystallization. We have successfully prepared Ni(OH)2 nanocrystals/graphene composites crystallized from amorphous nuclei, a strategy illustrated in Fig. 11. The nickel hydroxide nanocrystals, which are less than 20 nm in size, are uniformly grown on and completely cover the graphene sheets, as can be seen from the SEM and TEM images in Fig. 12. This is in contrast to the crystals obtained from the classical crystallization process, which are hexagonal with a lateral size of several hundred nanometers and a thickness of ∼10 nm.138 Comparatively, the Ni(OH)2 nanocrystals formed via the amorphous nuclei not only increase the electroactive sites by decreasing the size of Ni(OH)2, but also facilitate electron transport. Thus, the specific capacitance of the Ni(OH)2/graphene composite has reached 1804 F g−1 at a current density of 1 A g−1. This is a significant improvement over that of the hexagonal Ni(OH)2 plate/graphene composite,138 and represents a stride toward the theoretical specific capacitance of Ni(OH)2 (2602.5 F g−1).
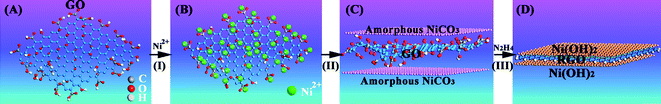 | ||
| Fig. 11 A schematic representation of the formation process of Ni(OH)2/reduced graphene oxide (RGO) composites. (A) GO sheet, (B) adding GO sheets into a supersaturated Ni(HCO3)2 solution, (C) amorphous NiCO3 particles are gradually grown on the GO sheet with the escape of CO2 from the supersaturated Ni(HCO3)2 solution, and (D) amorphous NiCO3 particles are simultaneously crystallized into Ni(OH)2 to form Ni(OH)2/RGO composites during the reduction process from GO to RGO. | ||
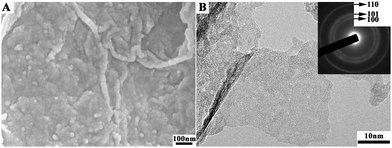 | ||
| Fig. 12 (A) SEM and (B) TEM images of Ni(OH)2/RGO composites transformed from the amorphous precursors. | ||
9. Conclusions and outlook
In this review, we have provided a glimpse of some of the peculiar characteristics of the crystallization processes starting from amorphous precursors, which are recapitulated as follows:1) The picture of the organic template-directed crystallization process of CaCO3 has emerged: dissolved calcium carbonate → prenucleation clusters → ACC → nanocrystals in ACC → polycrystals → mesocrystals → single crystals.
2) During the phase transformation along a given pathway of intermediates from the amorphous to the anhydrous crystalline phase, polymorph selection depends crucially on the surface energy.
3) Amorphous precursors, with a hydrated and disordered structure, can be easily transformed and moulded into anhydrous polymorphs with any shape that conforms to the organic template framework during the amorphous-to-crystalline transformation.
4) Inorganic impurities, such as Mg2+, can mediate the nucleation and stabilization of the amorphous precursors. The transformation from the amorphous precursors into aragonite and magnesium calcite depends on the Mg contents in the final crystals, which are in turn modulated by the organic matrices and depend on the extent to which the organic matrices can decrease the dehydration energy barrier of Mg2+ incorporation into the crystalline lattice.
5) Polymorph selection of Mg2+-containing CaCO3 is primarily determined by adding carboxyl-rich organic molecules in to the nucleation process of Mg2+-containing amorphous precursors, instead of adding them during the amorphous-to-crystalline transition process.
6) The non-classical strategy can be applied to the synthesis of non-biological minerals as advanced materials for device applications.
In the coming years, the use of the biomineralization strategy to synthesize technologically important materials will expand. Firstly, amorphous precursors will be utilized in conjunction with porous scaffolds for constructing materials with novel architectures of regular, ordered, and/or hierarchical natures. Secondly, work will continue to mimic the biomineralization process to synthesize engineering materials with high mechanical performances. Finally, there will be growing interest in the synthesis of non-biological materials inspired by the biomineralization process via amorphous precursors. One can expect significant developments in this area in the near future.
Acknowledgements
This work was supported by the HK-RGC General Research Funds (GRF No. HKUST 604809) and the NSFC/HK-RGC Joint Research Scheme (N_HKUST609/09).References
- H. A. Lowenstam and S. Weiner, On Biomineralization, Oxford University Press, New York, 1989 Search PubMed.
- S. Mann, J. Webb and J. P. Williams, Biomineralization. Chemical and Biochemical Perspectives, VCH, Weinheim, 1989 Search PubMed.
- E. Bauerlein, Biomineralization, Wiley-VCH, Weinheim, 2000 Search PubMed.
- S. Mann, Nature, 1988, 332, 119 CrossRef CAS.
- S. Mann, Nature, 1993, 365, 499 CrossRef CAS.
- S. Mann, B. R. Heywood, S. Rajam and J. D. Birchall, Nature, 1988, 334, 692 CrossRef CAS.
- J. Aizenberg, A. J. Black and G. H. Whitesides, J. Am. Chem. Soc., 1999, 121, 4500 CrossRef CAS.
- A. M. Travaille, J. Donners, J. W. Gerritsen, N. Sommerdijk, R. J. M. Nolte and H. van Kempen, Adv. Mater., 2002, 14, 492 CrossRef CAS.
- A. M. Belcher, X. H. Wu, R. J. Christensen, P. K. Hansma, G. D. Stucky and D. E. Morse, Nature, 1996, 381, 56 CrossRef CAS.
- L. Addadi, J. Moradian, E. Shay, N. G. Maroudas and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2732 CrossRef CAS.
- D. D. Archibald, S. B. Qadri and B. P. Gaber, Langmuir, 1996, 12, 538 CrossRef CAS.
- B. R. Heywood and S. Mann, Chem. Mater., 1994, 6, 311 CrossRef CAS.
- A. Berman, D. J. Ahn, A. Lio, M. Salmeron, A. Reichert and D. Charych, Science, 1995, 269, 515 CAS.
- J. Aizenberg, G. Lambert, L. Addadi and S. Weiner, Adv. Mater., 1996, 8, 222 CrossRef CAS.
- E. Beniash, J. Aizenberg, L. Addadi and S. Weiner, Proc. R. Soc. London, Ser. B, 1997, 264, 461 CrossRef CAS.
- J. Aizenberg, G. Lambert, S. Weiner and L. Addadi, J. Am. Chem. Soc., 2002, 124, 32 CrossRef CAS.
- Y. Levi-Kalisman, S. Raz, S. Weiner, L. Addadi and I. Sagi, Adv. Funct. Mater., 2002, 12, 43 CrossRef CAS.
- S. Weiner and L. Addadi, Geochim. Cosmochim. Acta, 2002, 66, A827 Search PubMed.
- I. M. Weiss, N. Tuross, L. Addadi and S. Weiner, J. Exp. Zool., 2002, 293, 478 CrossRef CAS.
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959 CrossRef CAS.
- S. Raz, P. C. Hamilton, F. H. Wilt, S. Weiner and L. Addadi, Adv. Funct. Mater., 2003, 13, 480 CrossRef CAS.
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Science, 2004, 306, 1161 CrossRef CAS.
- Y. Politi, Y. Levi-Kalisman, S. Raz, F. Wilt, L. Addadi, S. Weiner and I. Sagi, Adv. Funct. Mater., 2006, 16, 1289 CrossRef CAS.
- T. Y. J. Han and J. Aizenberg, Chem. Mater., 2008, 20, 1064 CrossRef CAS.
- J. Mahamid, A. Sharir, L. Addadi and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12748 CrossRef CAS.
- Y. Politi, R. A. Metzler, M. Abrecht, B. Gilbert, F. H. Wilt, I. Sagi, L. Addadi, S. Weiner and P. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17362 CrossRef CAS.
- J. W. Xiao and S. H. Yang, CrystEngComm, 2010, 12, 3296 RSC.
- J. Aizenberg, D. A. Muller, J. L. Grazul and D. R. Hamann, Science, 2003, 299, 1205 CrossRef CAS.
- C. Li and L. M. Qi, Angew. Chem., Int. Ed., 2008, 47, 2388 CrossRef CAS.
- H. Colfen, Angew. Chem., Int. Ed., 2008, 47, 2351 CrossRef.
- D. Volkmer, M. Harms, L. Gower and A. Ziegler, Angew. Chem., Int. Ed., 2005, 44, 639 CrossRef CAS.
- H. Colfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350 CrossRef.
- L. B. Gower, Chem. Rev., 2008, 108, 4551 CrossRef CAS.
- N. A. J. M. Sommerdijk, A. Dey and G. de With, Chem. Soc. Rev., 2010, 39, 397 RSC.
- A. W. Xu, Y. R. Ma and H. Colfen, J. Mater. Chem., 2007, 17, 415 RSC.
- F. C. Meldrum and H. Colfen, Chem. Rev., 2008, 108, 4332 CrossRef CAS.
- N. A. J. M. Sommerdijk and G. de With, Chem. Rev., 2008, 108, 4499 CrossRef CAS.
- H. Colfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576 CrossRef.
- H. Colfen, Biomineralization: From Paleontology to Materials Science, Editorial Universitaria, Universidad de Chile, Santiago, 2006 Search PubMed.
- A. Berman, L. Addadi and S. Weiner, Nature, 1988, 331, 546 CrossRef CAS.
- M. Rousseau, E. Lopez, P. Stempfle, M. Brendle, L. Franke, A. Guette, R. Naslain and X. Bourrat, Biomaterials, 2005, 26, 6254 CrossRef CAS.
- S. Weiner and H. D. Wagner, Annu. Rev. Mater. Sci., 1998, 28, 271 CrossRef CAS.
- G. Falini, S. Weiner and L. Addadi, Calcif. Tissue Int., 2003, 72, 548 CrossRef CAS.
- A. J. Salgado, O. P. Coutinho and R. L. Reis, Macromol. Biosci., 2004, 4, 743 CrossRef CAS.
- B. A. Gotliv, L. Addadi and S. Weiner, ChemBioChem, 2003, 4, 522 CrossRef CAS.
- J. L. Arias, A. Neira-Carrillo, J. I. Arias, C. Escobar, M. Bodero, M. David and M. S. Fernandez, J. Mater. Chem., 2004, 14, 2154 RSC.
- A. Oldberg, A. Franzen and D. Heinegard, J. Biol. Chem., 1988, 263, 19430 CAS.
- S. Weiner, Calcif. Tissue Int., 1979, 29, 163 CrossRef CAS.
- S. Weiner and L. Hood, Science, 1975, 190, 987 CAS.
- M. E. Marsh, J. Exp. Zool., 1986, 239, 207 CrossRef CAS.
- J. N. Weber, Am. J. Sci., 1969, 267, 537 CrossRef CAS.
- S. Weiner, Y. Levi-Kalisman, S. Raz and L. Addadi, Connect Tissue Res, 2003, 44, 214 CAS.
- K. D. Groot and E. M. Duyvis, Nature, 1966, 212, 183 CrossRef.
- N. H. de Leeuw, J. Phys. Chem. B, 2002, 106, 5241 CrossRef CAS.
- S. Mann, B. R. Heywood, S. Rajam and J. B. A. Walker, J. Phys. D: Appl. Phys., 1991, 24, 154 CrossRef CAS.
- J. B. A. Walker, B. R. Heywood and S. Mann, J. Mater. Chem., 1991, 1, 889 RSC.
- D. Volkmer, M. Fricke, C. Agena and J. Mattay, CrystEngComm, 2002, 4, 288 RSC.
- M. Fricke, D. Volkmer, C. E. Krill, M. Kellermann and A. Hirsch, Cryst. Growth Des., 2006, 6, 1120 CAS.
- D. M. Duffy and J. H. Harding, J. Mater. Chem., 2002, 12, 3419 RSC.
- E. Loste, E. Diaz-Marti, A. Zarbakhsh and F. C. Meldrum, Langmuir, 2003, 19, 2830 CrossRef CAS.
- M. Maas, H. Rehage, H. Nebel and M. Epple, Colloid Polym. Sci., 2007, 285, 1301 CAS.
- Y. J. Chen, J. W. Xiao, Z. N. Wang and S. H. Yang, Langmuir, 2009, 25, 1054 CrossRef CAS.
- S. Mann, Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry, Oxford University Press, Oxford, 2001 Search PubMed.
- D. Gebauer, A. Volkel and H. Cölfen, Science, 2008, 322, 1819 CrossRef CAS.
- E. M. Pouget, P. H. H. Bomans, J. A. C. M. Goos, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Science, 2009, 323, 1455 CrossRef CAS.
- J. R. Young, J. M. Didymus, P. R. Bown, B. Prins and S. Mann, Nature, 1992, 356, 516 CrossRef.
- H. C. Anderson, H. H. T. Hsu, P. Raval, T. R. Hunt, J. R. Schwappach, D. C. Morris and D. J. Schneider, Clin. Orthop. Rel. Res., 1995, 313, 129 Search PubMed.
- S. Mann, R. B. Frankel and R. P. Blakemore, Nature, 1984, 310, 405 CrossRef.
- S. Mann, N. H. C. Sparks, R. B. Frankel, D. A. Bazylinski and H. W. Jannasch, Nature, 1990, 343, 258 CrossRef CAS.
- D. A. Bazylinski, B. R. Heywood, S. Mann and R. B. Frankel, Nature, 1993, 366, 218 CrossRef.
- S. Mann, J. P. Hannington and R. J. P. Williams, Nature, 1986, 324, 565 CrossRef CAS.
- J. W. Xiao, Z. N. Wang, Y. C. Tang and S. H. Yang, Langmuir, 2010, 26, 4977 CrossRef CAS.
- G. Falini, S. Albeck, S. Weiner and L. Addadi, Science, 1996, 271, 67 Search PubMed.
- N. H. de Leeuw and S. C. Parker, J. Phys. Chem. B, 1998, 102, 2914 CrossRef CAS.
- J. Kuther, R. Seshadri, W. Knoll and W. Tremel, J. Mater. Chem., 1998, 8, 641 RSC.
- M. Fricke and D. Volkmer, Top. Curr. Chem., 2007, 270, 1 CrossRef CAS.
- D. Volkmer, M. Fricke, C. Agena and J. Mattay, J. Mater. Chem., 2004, 14, 2249 RSC.
- A. L. Litvin, S. Valiyaveettil, D. L. Kaplan and S. Mann, Adv. Mater., 1997, 9, 124 CrossRef CAS.
- D. Volkmer, M. Fricke, D. Vollhardt and S. Siegel, J. Chem. Soc., Dalton Trans., 2002,(24), 4547 RSC.
- M. A. Crenshaw, Biol. Bull., 1972, 143, 506 CrossRef CAS.
- H. Nakahara, G. Bevelander and M. Kakei, Venus, 1982, 41, 33 Search PubMed.
- M. A. Cariolou and D. Morse, J. Comp. Physiol., B, 1988, 157, 717 CrossRef CAS.
- M. Fritz, A. M. Belcher, M. Radmacher, D. A. Walters, P. K. Hansma, G. D. Stucky, D. E. Morse and S. Mann, Nature, 1994, 371, 49 CrossRef CAS.
- S. K. Zhang and K. E. Gonsalves, Langmuir, 1998, 14, 6761 CrossRef CAS.
- A. Sugawara and T. Kato, Chem. Commun., 2000, 6, 487 RSC.
- P. Liang, Y. Zhao, Q. Shen, D. J. Wang and D. F. Xu, J. Cryst. Growth, 2004, 261, 571 CrossRef CAS.
- N. Wada, S. Suda, K. Kanamura and T. Umegaki, J. Colloid Interface Sci., 2004, 279, 167 CrossRef CAS.
- A. Neira-Carrillo, M. Yazdani-Pedram, J. Retuert, M. Diaz-Dosque, S. Gallois and J. L. Arias, J. Colloid Interface Sci., 2005, 286, 134 CrossRef CAS.
- I. Manjubala, S. Scheler, J. Bossert and K. D. Jandt, Acta Biomater., 2006, 2, 75 CrossRef CAS.
- I. Manjubala, I. Ponomarev, I. Wilke and K. D. Jandt, J. Biomed. Mater. Res., Part B, 2008, 84B, 7 CrossRef CAS.
- J. M. Gong, Z. J. Zhou, X. L. Hu, M. K. Wong, K. W. Wong and Z. L. Du, ACS Appl. Mater. Interfaces, 2009, 1, 26 CAS.
- J. W. Xiao and S. H. Yang, Adv. Eng. Mater., 2011, 13, B32 CrossRef.
- C. Li, G. S. Hong, H. Yu and L. M. Qi, Chem. Mater., 2010, 22, 3206 CrossRef CAS.
- J. S. Robach, S. R. Stock and A. Veis, J. Struct. Biol., 2006, 155, 87 CrossRef CAS.
- R. L. Folk, J. Sediment.Petrol., 1974, 44, 40 CAS.
- R. W. Lahann, J. Sediment.Petrol., 1978, 48, 337 CAS.
- L. Addadi and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4110 CrossRef CAS.
- L. Addadi, A. Berman, J. Moradianoldak and S. Weiner, Croat. Chem. Acta, 1990, 63, 539 CAS.
- P. W. Carter and R. M. Mitterer, Geochim. Cosmochim. Acta, 1978, 42, 1231 CrossRef CAS.
- D. B. Wang, A. F. Wallace, J. J. De Yoreo and P. M. Dove, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21511 CrossRef CAS.
- J. Jiang, M.-R. Gao, Y.-H. Qiu, G.-S. Wang, L. Liu, G.-B. Cai and S.-H. Yu, CrystEngComm, 2011, 13, 952 RSC.
- J. Xiao and S. Yang, CrystEngComm, 2011, 13, 2472 RSC.
- Y. R. Ma, X. Long and L. M. Qi, Cryst. Growth Des., 2011, 11, 2866 Search PubMed.
- S. Elhadj, J. J. De Yoreo, J. R. Hoyer and P. M. Dove, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19237 CrossRef CAS.
- S. Piana, F. Jones and J. D. Gale, J. Am. Chem. Soc., 2006, 128, 13568 CrossRef CAS.
- M. Kowacz, C. Putnis and A. Putnis, Geochim. Cosmochim. Acta, 2007, 71, 5168 CrossRef CAS.
- A. E. Stephenson, J. J. DeYoreo, L. Wu, K. J. Wu, J. Hoyer and P. M. Dove, Science, 2008, 322, 724 CrossRef CAS.
- W. J. P. van Enckevort and A. C. J. F. van den Berg, J. Cryst. Growth, 1998, 183, 441 CrossRef CAS.
- K. E. Chave, K. S. Deffeyes, P. K. Weyl, R. M. Garrels and M. E. Thompson, Science, 1962, 137, 33 CAS.
- L. M. Walter and J. S. Hanor, Geochim. Cosmochim. Acta, 1979, 43, 1377 CrossRef CAS.
- R. A. Berner, Geochim. Cosmochim. Acta, 1975, 39, 489 CrossRef CAS.
- R. G. Compton and C. A. Brown, J. Colloid Interface Sci., 1994, 165, 445 CrossRef CAS.
- J. L. Katz, M. R. Reick, R. E. Herzog and K. I. Parsiegla, Langmuir, 1993, 9, 1423 CrossRef CAS.
- L. Brecevic, V. NothigLaslo, D. Kralj and S. Popovic, J. Chem. Soc., Faraday Trans., 1996, 92, 1017 RSC.
- N. NassrallahAboukais, A. Boughriet, J. C. Fischer, M. Wartel, H. R. Langelin and A. Aboukais, J. Chem. Soc., Faraday Trans., 1996, 92, 3211 RSC.
- M. Deleuze and S. L. Brantley, Geochim. Cosmochim. Acta, 1997, 61, 1475 CrossRef CAS.
- K. J. Davis, P. M. Dove and J. J. De Yoreo, Science, 2000, 290, 1134 CrossRef CAS.
- J. W. Xiao, Y. C. Zhu, J. H. Yuan, Q. C. Ruan, Y. Zeng, L. F. Cheng, L. Z. Wang and F. F. Xu, Mod. Phys. Lett. B, 2009, 23, 3695 CrossRef CAS.
- J. W. Xiao, Y. C. Zhu, Y. Y. Liu, H. J. Liu, Y. Zeng, F. F. Xu and L. Z. Wang, Cryst. Growth Des., 2008, 8, 2887 CAS.
- S. E. Wolf, N. Loges, B. Mathiasch, M. Panthofer, I. Mey, A. Janshoff and W. Tremel, Angew. Chem., Int. Ed., 2007, 46, 5618 CrossRef CAS.
- A. W. Xu, W. F. Dong, M. Antonietti and H. Colfen, Adv. Funct. Mater., 2008, 18, 1307 CrossRef CAS.
- S. Raz, S. Weiner and L. Addadi, Adv. Mater., 2000, 12, 38 CrossRef CAS.
- F. C. Meldrum and S. T. Hyde, J. Cryst. Growth, 2001, 231, 544 CrossRef CAS.
- E. Loste, R. M. Wilson, R. Seshadri and F. C. Meldrum, J. Cryst. Growth, 2003, 254, 206 CrossRef CAS.
- J. W. Xiao and S. H. Yang, CrystEngComm, 2011, 13, 6223 RSC.
- M. M. Thomas, J. A. Clouse and J. M. Longo, Chem. Geol., 1993, 109, 201 CrossRef CAS.
- K. M. Towe and H. A. Lowenstam, J. Ultrastruct. Res., 1967, 17, 1 CrossRef CAS.
- L. J. Bonderer, A. R. Studart and L. J. Gauckler, Science, 2008, 319, 1069 CrossRef CAS.
- E. Munch, M. E. Launey, D. H. Alsem, E. Saiz, A. P. Tomsia and R. O. Ritchie, Science, 2008, 322, 1516 CrossRef CAS.
- A. Berman, J. Hanson, L. Leiserowitz, T. F. Koetzle, S. Weiner and L. Addadi, Science, 1993, 259, 776 CAS.
- Y. Politi, R. A. Metzler, M. Abrecht, B. Gilbert, F. H. Wilt, I. Sagi, L. Addadi, S. Weiner and P. U. P. A. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17362 CrossRef CAS.
- C. Moureaux, A. Perez-Huerta, P. Compere, W. Zhu, T. Leloup, M. Cusack and P. Dubois, J. Struct. Biol., 2010, 170, 41 CrossRef CAS.
- F. Nudelman, E. Shimoni, E. Klein, M. Rousseau, X. Bourrat, E. Lopez, L. Addadi and S. Weiner, J. Struct. Biol., 2008, 162, 290 CrossRef CAS.
- J. R. Miller and P. Simon, Science, 2008, 321, 651 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS.
- J. W. Xiao and S. H. Yang, RSC Adv., 2011, 1, 588 RSC.
- M. R. Tarasevich and B. N. Efremov, Electrodes of Conductive Metallic oxides Part A, Elsevier, USA, 1982 Search PubMed.
- H. L. Wang, H. S. Casalongue, Y. Y. Liang and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 7472 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |