Self-assembled amino acids and dipeptides as noncovalent hydrogels for tissue engineering
Derek M.
Ryan
and
Bradley L.
Nilsson
*
University of Rochester, Department of Chemistry, Rochester, NY 14627, USA. E-mail: nilsson@chem.rochester.edu; Tel: +1-585-276-3053
First published on 7th October 2011
Abstract
Noncovalent hydrogels derived from the self-assembly of peptides and proteins have demonstrated advantages over covalent hydrogels for three-dimensional cell scaffolding applications. There is growing interest in exploiting minimal, self-assembling dipeptides and amino acids as hydrogel networks that support cell culture applications, but significant questions persist concerning the mechanism of self-assembly and the relationship between the molecular structure of the assembled materials and their emergent viscoelastic and biochemical properties. This review will critically assess current progress in the use of minimal self-assembling peptides and functionalized amino acids to create hydrogels, with a focus on the challenges of understanding the structure and function of these materials and on the outlook for the use of these modular and dynamic materials as robust networks for tissue engineering.
 Derek M. Ryan | Derek Ryan conducted graduate studies at the University of Rochester working with Professor Bradley Nilsson. His research project focused on the self-assembly of modified amino acids to create hydrogel materials. He will begin postdoctoral studies at the University of North Carolina at Chapel Hill with Professor Marcey Waters in the fall of 2011. |
 Bradley L. Nilsson | Bradley Nilsson conducted PhD studies in organic chemistry at the University of Wisconsin–Madison with Professor Ronald T. Raines studying methodology for the chemoselective ligation of peptides. He subsequently completed postdoctoral research in synthetic organic chemistry with Professor Larry E. Overman at the University of California, Irvine. He joined the faculty of the Department of Chemistry at the University of Rochester in 2006. The Nilsson group is interested in molecular recognition and self-assembly phenomena of peptides and proteins leading to amyloid and in the development of amyloid-inspired materials and therapeutics. |
1. Introduction
The development of hydrogels capable of supporting the growth and proliferation of cells for tissue engineering applications is a rapidly expanding field of study.1–13 There are two general classes of hydrogel defined by the macromolecular architecture of the gel network (Fig. 1). Covalent hydrogels are derived from synthetic polymers that are irreversibly cross-linked through the formation of covalent bonds by chemical or photo-initiated polymerization, disulfide bond formation, catalyzed metathesis, or other chemical reactions and have been extensively reviewed elsewhere.13,14 A second emerging class of hydrogel utilizes noncovalent interactions to build fibril structures by chemical self-assembly; these fibrils form noncovalent fibril-fibril cross-links and entanglements to provide dynamic hydrogel networks.15–21 These two classes of materials have been traditionally referred to as chemically cross-linked and physically cross-linked hydrogels, respectively.1 We have chosen to use the terms covalent and noncovalent hydrogels since these terms more precisely describe the molecular nature of the hydrogel architecture. This article will critically review progress in the development of noncovalent hydrogels derived from the noncovalent self-assembly of simple dipeptide and amino acid motifs in aqueous solution. Our focus on these minimal low molecular weight hydrogelators will serve to illuminate the promise of noncovalent hydrogels and the challenges that exist in understanding and exploiting noncovalent self-assembly in the creation of functional materials.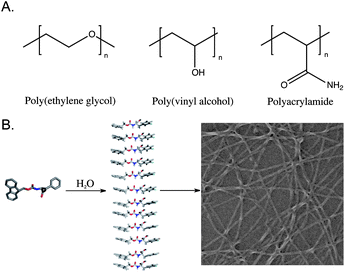 | ||
| Fig. 1 (A) Representative polymers commonly used to form covalent hydrogel networks for cell scaffolding. (B) Schematic representation of the self-assembly process that leads to the formation of a noncovalent hydrogel network. | ||
Hydrogels have distinct advantages over culture media solutions for the growth of cellsex vivo.9 It has become increasingly apparent that traditional two-dimensional cell culture methods are an inaccurate approximation of in vivogrowth conditions.1,9Cells grown in two-dimensional scaffolds experience different microenvironments than are found in vivo; as a result, biological functions including geneactivation and protein expression are significantly perturbed.22–24 It has been demonstrated that hydrogel materials that closely mimic the three-dimensional extracellular matrix are ideal for cell and tissue scaffolding applications.24 Noncovalent hydrogels have been shown to be strong candidates to fill this role since the dynamic nature of the fibril network allows the material to spontaneously adjust to the surrounding environment.1,9 For example, the pore size of the noncovalent material more readily responds to the mechanical forces exerted by cells as they migrate through the matrix, a distinct advantage over covalent hydrogels whose pores are chemically constrained and relatively inflexible.1,25 The ability to tune the mechanical behavior of the noncovalent hydrogel through synthetic customization of the assembling monomer is also highly beneficial, as different cell types require materials of varying rigidity.26–28 These advantages make noncovalent hydrogels attractive candidates as next-generation three-dimensional extracellular matrix mimetics.
In order for noncovalent hydrogels to be useful as three-dimensional scaffolds for tissue engineering they must meet certain criteria. A biocompatible noncovalent hydrogel network must be stable in common cell culture media and support cell viability, adhesion, differentiation, proliferation and migration.1,13,29–31 The components of these gels must be non-toxic and nonimmunogenic in both fibril and monomer states.8,13,31 In addition, the hydrogel network should undergo tunable biodegradation and provide by-products that are neither toxic nor immunogenic.1,10,13,30,31 The formation of an optically transparent hydrogel is highly desirable to enable efficient cell imaging.31 Hydrogels prepared for cell scaffolding applications should require no organic co-solvent and must be solvolytically stable under biological conditions in order to avoid precipitation of the hydrogel network.31 Hydrogels for tissue engineering must exhibit specific viscoelastic properties; a minimum storage modulus (G′) of approximately 100 Pa is necessary to support the mass of a cell, but ideal storage and loss modulus (G′′) values vary depending on cell type.26,31,32 In this regard, the ability to tune the viscoelasticity of the hydrogel is highly desirable. Shear recovery, or the ability to spontaneously heal following shear-thinning by the application of stress, is critical to the design of injection-based delivery systems.31,33 Hydrogelation in response to environmental stimuli including ionic strength, pH, light, and other triggers facilitates spontaneous formation of the supporting network in a manner that enables encapsulation of cells; cell growth can thus occur in a truly three-dimensional matrix and not merely at the surface of the hydrogel.31,34,35 Control over time of gelation upon exposure to the desired environmental trigger is also important.31 Finally, reversible temporal control over sol-gel/gel-sol transitions facilitates growth of cells in three-dimensional hydrogels and allows harvesting of the proliferated cells from an induced solution state.31
2. Peptide and protein-derived noncovalent hydrogels
Noncovalent protein based hydrogels that meet many of the above criteria have been developed and applied to cell culture in three dimensions.2,3,10,15,36Protein and peptide based hydrogelators self-assemble into polymer-like fibers which entangle to form hydrogel networks.2,10,11,17 For example, collagen is comprised of well characterized triple helices and has been isolated from animals for hundreds of years for biomedical applications.11 In the past three decades biological collagen has been used in cell culture, drug delivery, and as artificial skin for wound healing applications.11 Raines et al. have extensively studied the structure of the fibers formed by biological collagen and have developed a synthetic method for the generation of biomimetic collagen-like fibers.37 They demonstrated that the covalent tethering of staggered triple helices viadisulfide bond formation leads to elongation of the helix to yield fibers > 400 nm in diameter, similar to that of natural collagen fibers (>300 nm).37 Prior to this synthesis, attempts to prepare artificial collagen mimetics resulted in the formation of fibers with distinctly different diameters (∼10 nm) and thus, different material properties from biological collagen.11 Hartgerink et al. later increased the sophistication of collagen systems by demonstrating that heterotriple helices could be utilized to assemble collagen-like fibers.38 The ability to chemically synthesize collagen-like fibrils with tunable assembly propensity facilitated the development of collagen-inspired hydrogels for cell scaffolding applications.39,40 Woolfson et al. designed a triggered hydrogelation system based on two-component coiled-coil fiber forming proteins.41–44 When either component was dissolved in biological media no hydrogelation was observed. When the two mixtures were combined in an equimolar ratio, an optically transparent hydrogel with a storage modulus of 1000 Pa was formed.41 This hydrogel was shown to be highly effective for cell culture of rat adrenal pheochromocytoma cells, as differentiation into neural cells was observed for up to 10 days upon introduction of nerve growth factor to the hydrogel.41Amyloid-inspired fibrils have emerged as powerful systems for the preparation of noncovalent hydrogels.45 Biological amyloid, both functional and pathological, forms from the self-assembly of β-sheet peptides into cross-βfibrils.7,46–48 The principles of amyloidself-assembly have been used to engineer noncovalent hydrogels for tissue engineering.46 Zhang et al. have developed several amphipathic, self-assembling β-strand peptides with a general (XZXZ)n sequence in which X and Z are alternating hydrophobic and hydrophilic amino acids; these peptides readily self-assemble in water to form soluble amyloid-like fibrils that entangle to provide mechanically rigid, optically transparent hydrogels.7,49–54 Hydrogels derived from the (RADA)4peptide were shown to be capable of supporting the growth of chondrocytes for cartilage repair, as well as a haemostat for wound healing applications.49,55 Hartgerink et al. designed hydrogels assembled from the amphiphilic peptideK2(SL)nK2, and found them to be capable of supporting the growth and proliferation of mesenchymal stem cells.56,57 In a followup study, Hartgerink demonstrated that inclusion of the metalloproteaseenzyme signal, LRG, into the K(SL)3RG(SL)3Kpeptide facilitated enzymatic degradation of the hydrogel by metalloproteases secreted by mesenchymal stem cells during growth.57 This resulted in enhanced cell viability and proliferation relative to the K2(SL)nK2peptide, and demonstrated that effectively mimicking extracellular matrixex vivo dramatically increases utility of the material for cell culture.57 Others have developed similar amyloid-inspired designs to produce hydrogels that possess external triggers over the sol/gel transitions that are useful for applications ranging from drug delivery agents to immunostimulants.10,58–66
Schneider et al. have designed a class of noncovalent β-hairpin peptides that meet nearly all of the criteria to be ideal hydrogel matrices for cell culture applications.31,67–75 These peptides, based on the general sequence (VK)nVDPPT(VK)n, have been engineered to self-assemble selectively at temperatures, ionic strength, and pH commonly found in cell culture media to form optically transparent hydrogels with G′ values ranging from 1000–10,000 Pa.31,73–75 Dissolution of the peptides into cell culture media triggers folding of the β–hairpin, which aligns the (VK)nβ-strands and promotes self-assembly into fibrils. They found that the kinetics of fibril formation and hydrogelation were sufficiently rapid to allow for homogeneous encapsulation of mesenchymal stem cells in the hydrogel matrix.31Cell viability assays demonstrated that the hydrogel was nontoxic, and cell proliferation was observed throughout the hydrogel network. In addition, these hydrogels exhibited the ability to shear recover following the application and cessation of stress allowing for delivery of the hydrogel and encapsulated cellsvia injection.31 It was demonstrated that encapsulated mesenchymal stem cells survived the injection process with limited loss of viability. These materials have also been shown to capable of supporting the growth of cementoblasts and directing the processes of biomineralization in the development of bone tissue scaffolds.76 The hydrogels developed by Schneider et al. represent significant benchmarks in the development of noncovalent hydrogels for tissue engineering, as they are optically transparent, mechanically robust, shear recoverable, and biocompatible with the cultured cells.31
In the past decade there has been growing interest in further exploration of minimal structures that exhibit equivalent material properties to larger covalent and noncovalent protein based hydrogels.12,21 Stupp and coworkers developed a series of amphiphilic β-strands that were functionalized with a hydrophobic group at the N-terminus and a hydrophilic moiety at the C-terminus.77–83 The β-strands assembled into amyloid-like β-sheets and burial of the N-terminal hydrophobic groups resulted in the formation of cylindrical nanotubes.5 These nanotubes entangle in biological media to form hydrogels that supported the growth of neurons and were also used in the regeneration of nerve cells following injection into spinal chord injury sites.81 Hamley and coworkers observed that a seven amino acid fragment derived from the amyloid-βpeptide, KLVFF, was able to form fibrils that were morphologically similar to the 42-residue parent peptide.84 In addition, this peptide fragment was shown to be capable of forming optically transparent hydrogels (G′ 10 kPa) in PBS buffer (3 wt%).84 Yang et al. investigated the ability of a short peptide fragment bearing a C-terminal collagen inspired motif to form fibrils and hydrogels of similar mechanical properties as long proteins.85 They observed that the naphthalene-functionalized peptide, Nap-GFFYGGKX (X = 4-hydroxyproline), self-assembled (0.3 wt%) to form optically transparent hydrogels (G′ of 30 Pa) that were capable of supporting the growth of mouse embryonic fibroblastcells on the surface.85 They observed cell viability on the hydrogel surface for up to 72 h, and hypothesized that the positively charged Lys residue at the C-terminus may have assisted in binding to the negatively charged cell surfaces.85
3. Development of noncovalent hydrogels from self-assembly of dipeptides and functionalized amino acids
While the structure/function relationship that governs the viscoelastic properties of covalent polymeric materials is reasonably well understood, the dynamic nature of self-assembled, noncovalent materials complicates understanding of these types of hydrogels.1 As recently stated by van Esch, there are numerous reports of noncovalent hydrogels but there is a significant knowledge gap between the discovery of these materials, which often occurs empirically or through serendipity, and understanding of the relationship between the structure of the underlying network and its emergent properties.86 There is a significant need for fundamental insights into the noncovalent forces that drive the self-assembly process and how hydrogelation and self-assembly are related.86 The low cost and ease of preparation of dipeptide and amino acids relative to longer peptides and proteins makes these attractive systems to probe this structure/function relationship.15,16 Low molecular weight self-assembling hydrogelators minimize the available functionality that can participate in stabilizing hydrogen bonding, hydrophobic, and aromatic π-π interactions, providing interesting probes of the scope and limitation of materials that can form noncovalent hydrogels.20,21 By investigating a series of simple, structurally related compounds, fundamental insights into the self-assembly mechanism can be obtained.86 It may then become possible to design from first principles simple materials for cell scaffolding applications. It is important to consider whether minimal self-assembled scaffolds (one or two amino acids) can fundamentally achieve all the hydrogel features associated with larger peptides (hydrogel rigidity, solvolytic stability, external triggers of assembly, etc). We will critically assess progress in the development of hydrogelators derived from the self-assembly of simple one or two amino acid motifs.3.1. Fmoc-protected dipeptides
One of the earliest examples of a self-assembled amino acid-based hydrogel was reported by Gortner and Hoffman in 1921.87 They observed that dissolution of dibenzoylcystine (1a, Fig. 2) in water led to the formation of a self-supporting hydrogel. Nearly 50 years later Menger et al. applied advanced structural characterization methods to investigate the basis of the observed hydrogelation behavior.88,89 They found that 1a self-assembles into fibrils (∼50 nm in diameter) that entangle in solution to form the hydrogel network. Menger et al. synthesized a series of side chain and C-terminal dibenzoylcystine derivatives that included structures 1b–c in order to investigate the driving forces behind the observed self-assembly and hydrogelation.88 It was found that substitution of the C-terminal carboxylic acid with a C-terminal amide, 1b, significantly increased the apparent rates of gelation, as well as the rheological strength of the resulting hydrogel material. The authors hypothesized that the increased intermolecular hydrogen bonding capacity of the C-terminal amide accelerated the rates of assembly and hydrogelation relative to the C-terminal acids.88 It was also observed that incorporation of an electron-donating group on the phenyl side chains of the C-terminal amide, 1c, resulted in improved hydrogelation rates and rheological properties relative to 1a. The most efficient hydrogelator observed in this study, 1c, formed an optically transparent hydrogel within 30 seconds of mixing, and had a G′ of 2000 Pa (at 0.5 mM concentrations).88 This work demonstrated that perturbation of hydrogen bonding and π-π interactions could be used to tune the rheological behavior of the self-assembled hydrogels. It is generally accepted that aromatic interactions are an enthalpically advantageous subset of more general hydrophobic interactions, although the exact role of aromatic effects in self-assembly processes is not well understood.20,90–93 Whether Menger's observations are due to altered π-π interactions or due to changes in hydrophobic/steric effects is not known.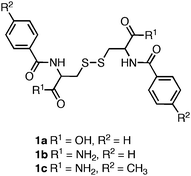 | ||
| Fig. 2 Chemical structure of dibenzoyl cystine derivatives investigated by Menger et al.88 | ||
Gazit and coworkers demonstrated that the Phe-Phe dipeptide self-assembled into nanotubesvia side chain π-π interactions and inter-strand hydrogen bonding between amidegroups.94 The resulting nanotubes did not induce hydrogelation of aqueous media. Gazit and others later discovered that fluorenylmethoxycarbonyl (Fmoc) protection of the N-terminus of Phe-Phe (2, Fig. 3) promoted rapid self-assembly into fibers with average diameters ranging from 10–100 nm.95,96 The formation of fibers instead of nanotubes upon N-terminal protection and the resulting hydrogelation behavior was hypothesized to arise from increased π-π interactions.95 It was found that upon self-assembly, 2 formed an optically transparent hydrogel with a storage modulus of 10 kPa ([2] = 9 mM).95 Hydrogels prepared from 2 supported the viability and growth of Chinese hamster ovary cells. It was observed via live/dead assays that contact with the hydrogel surface did not result in significant cell death (∼90% viability) after a 24 h incubation period.95 In addition, similar proliferation rates as untreated control experiments were observed, indicating that the cells were not negatively affected by exposure to the hydrogel surface. While hydrogels derived from 2 were shown to be capable of gelling biological media and exhibited low cytotoxicity, preparation of the hydrogel materials required dilution of concentrated organic stocks of peptide.95 These conditions limit potential cell scaffolding applications of these materials since some cell types are highly sensitive to organic solvents.13
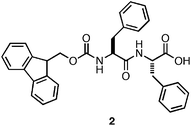 | ||
| Fig. 3 Chemical structure of Fmoc-Phe-Phe. | ||
Ulijn and coworkers reported methodology for self-assembly and hydrogelation of 2 without organic co-solvent.96Dipeptide2 was dissolved in basic water (∼20 mM total peptide, pH > 8) and was titrated with HCl until the formation of a self-supporting hydrogel was observed. At basic pH, the negatively charged C-terminal carboxylates enforce solubility of 2, but prevent self-assembly due to charge repulsion; careful titration to pH < 8 induces hydrogelation by establishing partial protonation at the C-terminus, which reduces charge repulsion and enables hydrogen bond formation, facilitating self-assembly.96 Ulijn et al. investigated the molecular architecture of fibers formed by 2 using fluorescence, CD, and FT-IR spectroscopy.97–99Fluorescence spectra were deemed to be consistent with antiparallel stacking of the Fmocgroups. This was further supported by the observation of an amide I shoulder at 1685 cm−1 in the FT-IR spectra, consistent with the formation of antiparallel β-sheets.97 Modeling these interactions with geometric constraints obtained from fibrilTEM images and X-ray powder diffraction lead to a proposed packing model in which antiparallel β-sheets form with dangling Fmocgroups (Fig. 4).97 Lamination of four of these sheets results in the formation of cylindrical fibrils (∼3 nm) in diameter, which are stabilized by the antiparallel π-π stacking of the Fmocgroups.97,99 These fibrils then bundle side by side, leading to the observation of striated flat ribbons by TEM.99 Further entanglement of these ribbons gives rise to the observed hydrogel behavior of the Fmoc-Phe-Phedipeptides.97,99
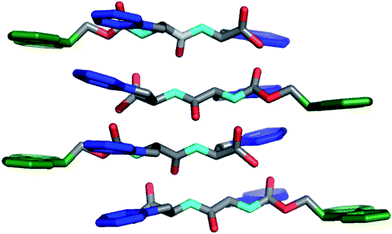 | ||
| Fig. 4 Packing model of Fmoc-Phe-Phedipeptides in self-assembled hydrogels as proposed by Ulijn et al.97,99 Hydrogens omitted for clarity, Fmoc (green), Phe side chain (blue). | ||
While investigating the self-assembly mechanism of Fmoc-Phe-Phe and related Fmoc-protected peptides, Ulijn et al. observed a trend in the pH at which hydrogelation occurred. Several of the dipeptide hydrogelators (3a–e, Fig. 5) formed self-supporting hydrogels at low pH (3–5),96 while Fmoc-Phe-Phe formed a self-supporting hydrogel at neutral pH.96,99 This led the authors to investigate the origin of the apparent shift in the C-terminal pKa of 2. They observed that two apparent pKa transitions occurred at a pH of ∼10 and a pH of ∼6 upon titration of a basic solution of Fmoc-Phe-Phe with HCl.99 The origin of these two transitions were found to be due to two distinct structural transformations that occurred during the course of assembly.99 It was observed that when a solution of 2 (10 mM) was kept at a pH above the apparent pKa 1 (pH 10.2), the dipeptide remained in a monomeric state, as indicated by the absence of β-sheet signal in the FT-IR spectra and lack of higher ordered structure by TEM imaging.99 When the pH of the solution was lowered to 9 (below apparent pKa 1) the formation of fibers (6 nm in diameter) consisting of antiparallel β-sheets was observed by FT-IR (stretches at 1625 and 1687 cm−1) and TEM.99 Lowering of the pH further to 4.6 (below apparent pKa 2) resulted in the formation of larger ribbons (∼100 nm in diameter) that were constructed from the lateral association of fibrils.99 It was also observed that the formation of fibrils at pH 9 coincided with the formation of a weak hydrogel (G′ = 10 Pa) that consisted of flat 6 nm ribbons.99 This G′ value was significantly decreased from the previously observed G′ of 10 kPa for this dipepetide.96,99 The authors hypothesized that self-assembly kinetics account for the different rheological behavior of 2, since the titration-based experiments were conducted at low temperature (4 °C) thus slowing the rate of self-assembly. Schneider et al. and Xu et al. have observed in other self-assembling systems that increasing the rate of fibrillization enhances entangled cross-link formation, increasing G′ values, consistent with Ulijn's hypothesis.73,100
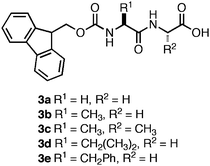 | ||
| Fig. 5 Chemical structures of Fmoc-dipeptides studied by Ulijn et al.96 | ||
Ulijn and coworkers also observed that subtle variations in the method of sample preparation can have a significant influence on the ultimate fibril morphology.98 Exposure to high shear forces during self-assembly, such as those observed with vortex treatment, produced Fmoc-Phe-Phe hydrogels with a G′ of 1000 Pa; however, when hydrogels were prepared using low shear forces (gentle manual agitation), hydrogels with a G′ of 4000 Pa were observed.98 Upon investigation of the resulting fibril structures they observed shear force-dependent differences in fibril morphology.99 High shear forces led to the formation of laminated ribbons, similar to those formed at low pH, while low shear forces lead to the formation of smaller diameter fibrils that more effectively entangled. It was hypothesized that the rheological strength of the hydrogels were also largely dependent on the fibril morphology. It was found that association of fibrils into large ribbons results in decreasing fibril network density, thereby lowering the cross-linking efficiency, resulting in rheologically weaker material.98Assembly into smaller diameter fibrils that can more effectively entangle and cross-link increases the network density, and results in hydrogels with an increased rigidity.31 These studies demonstrate that the kinetics of assembly and fibril morphology can significantly influence the bulk rheological properties of noncovalent hydrogels by mediating the number of effective cross-links.
Based on the strong correlation between solution pH and assembly kinetics on the ultimate rheological properties of the hydrogel, Adams and coworkers investigated alternate methods for adjusting the pH of basic solutions of dipeptides to values at which self-assembly occurs.101,102 They observed that acidification with HCl resulted in rapid local assembly that caused structural defects in the fibril network. It was hypothesized that this was due to the rate of acid diffusion being slower than the rate of fibrilself-assembly.101 The authors introduced an alternate method for the promotion of assembly by treating the basic solution of peptide with glucono-δ-lactone (GdL) (instead of HCl), which diffuses throughout the solution and is subsequently hydrolyzed to the corresponding gluconic acid; the pH of the solution is simultaneously lowered, allowing for homogenous protonation and self-assembly that avoids localized differences in rate leading to defects in the network.
The impact of this methodology on the rheological strength of the resulting materials was readily apparent. Hydrogels of Fmoc-Leu-Gly (4, Fig. 6) were found to be nearly 30 times more rigid when prepared viaGdLacidification (G′ = 6 kPa with HCl; 184 kPa with GdL).101 In addition, variations between measurements of different samples significantly decreased (average error ∼10%).101 This effect was not universal to all the peptides studied, however, highlighting a conceptual gap in our understanding of the structure/function relationship between sequence identity, environment, and bulk rheological properties of the self-assembled material. In addition, hydrogelation induced by GdLacidification required several hours to achieve the maximum G′, decreasing utility of this methodology for the encapsulation of cells, which has been shown to occur most effectively with gelation times that are < 5 min.31
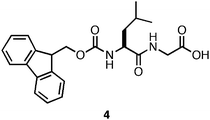 | ||
| Fig. 6 Chemical structure of Fmoc-Leu-Gly. | ||
As stated earlier, Ulijn et al. observed that a series of Fmoc-protected dipeptides (Fig. 5) formed optically transparent, self supporting hydrogels at pH < 4; however, this pH is too acidic for cell culture applications. Xu and coworkers had previously observed that mixtures of Fmoc-Leu and Nε-Fmoc-Lys (5, 6, Fig. 7) could co-assemble and impart the unique characteristics of each component to the ensemble.103,104 Inspired by these results, Ulijn et al. investigated the hydrogelation propensity of an equimolar mixture of 2 with 7.96 They hypothesized that co-incorporation of a basic side chain, such as the amine of lysine, into the fibril network would increase the pH at which hydrogelation occurred, allowing for gelation at biocompatible pH. The 2/7 mixture rapidly formed an optically transparent hydrogel upon titration with HCl to pH 7. They hypothesized that 7 was either being statistically incorporated into the fibril network through co-assembly or was being nonspecifically encapsulated in the hydrogel matrix. The ability of the 2/7 hydrogel to support two and three-dimensional cell scaffolding of bovine chondrocytes was investigated by adding cells to the surface of a preformed hydrogel or encapsulating cells within the gel matrix.96Chondrocytes were viable and proliferated for up to seven days after introduction to either the hydrogel surface (no penetration was observed) or encapsulation within the matrix. These results demonstrated that control over the pH of assembly is critical to hydrogelation of cell culture media and that co-assembly of monomers with complementary pKa values can be used to tune the pH at which hydrogelation occurs.
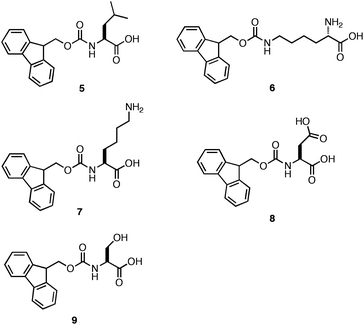 | ||
| Fig. 7 Chemical structures of Fmoc-Leu (5), N∑-Fmoc-Lys (6), Fmoc-Lys (7), Fmoc-Glu (8), Fmoc-Ser (9). | ||
Ulijn et al. subsequently prepared similar equimolar co-assembly mixtures of Fmoc-Phe-Phe with 7, 8, and 9 (Fig. 7) to compare the effect of hydrogel scaffold charge on cell culture applications.105 They hypothesized that forming a positive (2/7), negative (2/8) or neutral (2/9) fibril scaffold would elicit differential effects on cell adhesion and proliferation. Each of the equimolar mixtures of 2 with 7–9 produced fibers ranging in diameter from 30–60 nm. The 2/7, 2/8, and 2/9 hydrogels had respective G′ values of 12.3 kPa, ∼500 Pa, and 3.5 kPa (∼20 mM total peptide/amino acid concentration).105 It was proposed that the origin of these differences in viscoelasticity resulted from variations in the efficiency of the side chain interactions that mediate fibril entanglement.105 The side chain amine of Fmoc-Lys may participate in effective hydrogen bonding and electrostatic interactions with the C-terminal carboxylate of Fmoc-Phe-Phe, resulting in an increase in the number of cross-links and hydrogel rigidity. The carboxylate side chain of Fmoc-Glu may experience electrostatic repulsion from the C-terminal carboxylate of Fmoc-Phe-Phe, thereby inhibiting the efficient entanglement of fibrils. Lastly, the alcohol side chain of Fmoc-Ser could form effective hydrogen bonds although it could not participate in charge interactions resulting in materials with intermediate viscoelasticity.105
Ulijn et al. investigated the utility of each hydrogel mixture on growth of cells in culture.105 It was observed that the 2/7 and 2/8 hydrogels did not support the growth of bovine chondrocytes, mouse embryo fibroblasts, and human dermal fibroblasts as well as the 2/9 hydrogel.105 These observations were unexpected since it was previously reported that fibroblasts and chondrocytes prefer contact with more rigid surfaces in two-dimensional cell culture.22 It was expected that the weaker hydrogels formed from the 2/8 mixture would not possess sufficient rigidity to support the growth of chondrocytes; however, cell adhesion and proliferation were observed to be more favorable in this environment.105 In addition, the most rheologically rigid hydrogel (2/7) did not support cell viability as well as the hydrogels prepared from 2/8 or 2/9. These results indicate that the positively charged 2/7fibrils interact adversely with the cell surface, while the negatively charged 2/8fibrils established beneficial contacts.105 This was counterintuitive since the surfaces of cells are typically negatively charged and it would be expected that the negatively charged fibrils formed by 2/8 would experience electrostatic repulsion with the cell surfaces. The authors hypothesize that the reported cell behavior may be due to the integrin binding at the cell surface.105 These results highlight that binding of cells to soft, three-dimensional hydrogel surfaces does not necessarily correlate with commonly observed behavior in two-dimensional cell culture on hard surfaces.
Ulijn et al. next investigated whether incorporation of the fibronectin-derived RGDcell adhesion motif into the fibril architecture would improve cell adhesion and proliferation.106 They prepared a mixture of Fmoc-Phe-Phe with 0–50 mol% Fmoc-RGD (10, Fig. 8). They observed that hydrogels containing >30 mol% of Fmoc-RGD were significantly less rigid.106 It was hypothesized that the decrease in G′ was due to inhibition of fiber-fiber interactions by the charged RGD motif. At 70% Fmoc-Phe-Phe: 30% Fmoc-RGD, the formation of an optically transparent hydrogel in DMEM cell culture media with a G′ of ∼4 kPa (10 mM total peptide concentration) was observed.106 The cytotoxicity of the hydrogel to HDFacells was investigated using a live/dead assay. It was found that 90% of the encapsulated, cultured cells were viable after a 7-day incubation period.106 In addition, it was observed that the final cell density of the HDFacells was 30% higher than the original cell density indicating effective adhesion and proliferation throughout the gel.106
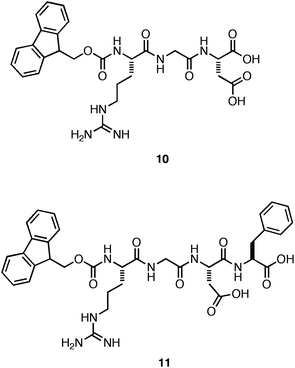 | ||
| Fig. 8 Chemical structures of Fmoc-RGD and Fmoc-RGDF. | ||
Hamley and coworkers subsequently investigated whether 10 could independently self-assemble and form hydrogels that promote cell adhesion and proliferation without Fmoc-Phe-Phe co-peptide.107 They observed that the Fmoc-RGD self-assembled to form rigid (G′ = 10 kPa, ∼35 mM 10), optically transparent hydrogels that were nontoxic, but failed to promote cell proliferation over the time course of the study.107 Gazit and coworkers similarly observed that a hydrogel assembled from a close derivative of RGD, Fmoc-RGDF (11), also failed to support the proliferation of cells on the hydrogel surface; in fact only 50% of the cells were viable after a 7-day exposure.108 These results suggest that the RGDcell adhesion motif was not effectively displayed on the surface of the self-assembled fibers, demonstrating that accessible display of the ligand at appropriate density is necessary for efficient cell adhesion.106 The role of the inherent mechanical properties of these hydrogel materials on cell behavior in culture is unclear in these studies.
Ulijn and coworkers also explored enzymatic methods for triggering assembly and hydrogelation.29,109–112 They observed that in situenzyme-catalyzed ligation could be used to couple Fmoc-Phe to the Phe-Phe dipeptide.109 The resulting Fmoc protected tripeptide (12, Fig. 9) was produced with 55% efficiency, and self-assembled to form fibers with average diameters of 10–20 nm (∼12 mM).109 They also observed the formation of optically transparent self-supporting hydrogel at pH 7 following conversion, although the exact viscoelasticity of these materials was not reported.109 This result suggests that enzyme-catalyzed ligation may be a potentially useful method for the in situ triggering of gelation for cell culture applications.112 In a subsequent study, Ulijn and coworkers also reported a two-step enzyme-catalyzed method for the in situ generation of self-assembling dipeptides.111 They promoted enzymatic peptide bond formation between Fmoc-Thr and a C-terminal methyl ester derivative of Leu that resulted in the formation of a hydrogel following self-assembly of the C-terminal methyl ester Fmoc-Thr-Leu (13) dipeptide.111 When the C-terminal methyl ester, 13a, was enzymatically converted to the C-terminal acid, 13b, a gel/sol transition occurred.111 These results demonstrated that enzymes could be utilized to control both the sol/gel and gel/sol transitions corresponding to chemical changes in the assembly motif.
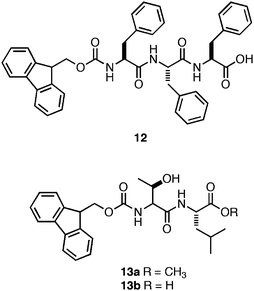 | ||
| Fig. 9 Chemical structures of Fmoc-Phe-Phe-Phetripeptide and Fmoc-Thr-Leudipeptide derivatives investigated by Ulijn et al.111 | ||
3.2 Naphthyl-protected dipeptide hydrogelators
Xu and coworkers have investigated the role of N-terminal aromatic protecting groups on the self-assembly and hydrogelation of simple peptides.113–117 They observed that in addition to N-terminal Fmoc functionalization, naphthalene functional groups also promoted the self-assembly of a Gly-β-Ala dipeptide (14, Fig. 10) into fibrils (25 nm in diameter) that formed an optically transparent hydrogel (G′ = 200 Pa, 15 mM peptide).113Fluorescence emission spectra indicated that π-π interactions between naphthyl (Nap) groups promoted assembly into fibrils. It was hypothesized that the peptide backbones then establish inter-strand hydrogen bonding networks, leading to fibril elongation.113 The authors concluded that π-π interactions between the Nap protecting groups were weaker than those formed by the Fmoc protected hydrogelators, accounting for the observed decrease in G′ of Nap-peptide hydrogels relative to Fmoc-peptide hydrogels (G′ of ∼100–200 Pa for Nap-protected dipeptides, G′ of ∼1000–10000 Pa for Fmoc-protected dipeptides).113,118 Xu et al. hypothesized that the decreased aromatic surface area of the Nap-protecting group relative to Fmoc results in less efficient π-π overlap. Alternatively, the greater conformational flexibility of the β-amino acid backbone may result in weaker hydrogen bonding interactions and rheologically weaker hydrogels.113 These results indicate that the aromatic surface area at the N-terminus can influence the rheological behavior; however, whether this is primarily a hydrophobic effect or π-π effect is unclear.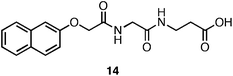 | ||
| Fig. 10 Chemical structure of the Nap-Gly-β-Aladipeptide. | ||
Xu et al. screened a series of Nap-functionalized dipeptides in order to determine the relative effects of the configuration of the N-terminal Nap group and overall hydrophobicity of the dipeptide on self-assembly and hydrogelation.119 The dipeptides (15–17, Fig. 11) were Gly-Gly, Gly-Ala, or Gly-Ser sequences with varying configurations of N-terminal Nap groups.119 A close relationship between the length of the linker joining the N-terminal Nap to the peptide backbone and self-assembly was observed. Peptides with linkers 1 or 2 atoms in length between the Nap group and the dipeptide (16 and 17) failed to self-assemble and merely precipitated from aqueous solutions; peptides with 3 carbon atom linkers (15) self-assembled to form hydrogels.119Dipeptides15a–c self-assembled into fibrils ranging in diameter from 30–50 nm and yielded optically transparent hydrogels with storage moduli of ∼4000 Pa (∼5.9 mM peptide).119 The authors investigated the cytotoxicity of 15a–c by adding 200 μM of each dipeptide to cell culture media; HeLa cell viability was then monitored via a live/dead assay. No cell death was observed following 24 h incubation times.119 Thus, conformational flexibility at the N-terminus dramatically affects the establishment of effective π-π interactions that lead to self-assembly and hydrogelation.
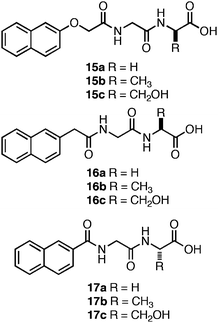 | ||
| Fig. 11 Chemical structures of Nap-protected dipeptides investigated by Xu et al.119 | ||
Adams et al. investigated the role of the C-terminus on self-assembly of N-terminal Nap-protected dipeptides (Fig. 12).120–123 They screened a library of sequences with N-terminal Nap groups bearing hydrogen, bromine, or cyano substituents at the 6-position (18–20) in order to probe the influence of varying dipeptide hydrophobicity on the C-terminal pKa.120 These Nap-Phe-Phepeptides had apparent carboxyl pKa values of ∼5–6, significantly higher than would be expected for C-terminal carboxylic acids (∼3.5).120 Consistent with the findings of Ulijn et al.,99 they observed that self-assembly did not proceed until the pH of the system was brought below the apparent pKa at which point partial protonation of the C-terminal carboxylates occurred, thereby promoting assembly into fibrils.120 The magnitude of the increase in the apparent pKa was strongly correlated to overall sequence hydrophobicity and not simply the identity of the C-terminal amino acid.120 For example, for the dipeptide series, 18e, 19e, and 20e, each possessed sequence homogeneity (Phe-Phe) and differed only in the substitution present on the N-terminal Nap-protecting group. It was observed that the apparent C-terminal pKa ranged from 6–6.8 for the series, demonstrating that the overall properties of the dipeptide must be considered when designing self-assembling hydrogels.120 In addition, sequences with an increased self-assembly propensity, such as the Phe-Phe motif in the 18e–20e series,94 possessed a higher apparent pKa (∼6 versus ∼4–5 for 18a–c, 19a–c, 20a–c). It was hypothesized that the increase is apparent pKa is due to the formation of higher order aggregates that begin to assemble and form hydrogen bonding interactions between the C-terminal carboxylates. These hydrogen bonds prevent further lowering of the pH until assembly is complete. Following completion of assembly, protonation of the exposed carboxylates on the fibril surfaces is initiated coincident with the further lowering of solution pH.120
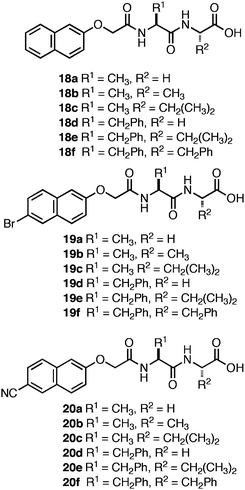 | ||
| Fig. 12 Nap-protected dipeptide derivatives investigated by Adams et al. | ||
In a separate study, Adams and coworkers observed that the 1-Nap-protected dipeptide21 (Fig. 13) initially formed an optically transparent hydrogel that was stable for ∼2 days; however, after >2 days, crystallization and precipitation was observed.122 Self-assembled, fibrillar materials are notoriously difficult to crystallize; thus, the formation of crystals from aqueous hydrogels provided a rare opportunity to gain structural insight into the packing architecture of the assembled fibrils.88,122 In addition, crystals of self-assembling compounds are typically grown under conditions where fibrilself-assembly is not observed. The relevance of these crystal structure to fibers in the hydrogel state has been questioned.86,88 Thus, crystallization under gelation conditions allowed possibly unique insight into the structure of self-assembled hydrogels.122 The resulting high resolution crystal structure indicated an antiparallel β-sheet stacking orientation similar to that proposed by Ulijn et al. for the assembly of Fmoc-Phe-Phe (Fig. 4).97,122 This orientation places the Nap groups on alternating ends of the nascent sheet; the Nap groups then form π-π interactions between sheets to form an extended two-dimensional sheet.122 Interestingly, it was observed that the X-ray scattering pattern of the crystals did not correlate with scattering patterns obtained from the solvated fibers found in the hydrogel.122 Thus, while single crystal structures provide useful insight into the noncovalent interactions that exist in these self-assembled systems caution must be exercised in extrapolating the precise structure of the hydrogel material from the crystalline material. Crystallization and hydrogelation are related processes that do not necessarily provide exactly similar structures.86,122
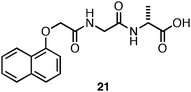 | ||
| Fig. 13 Chemical structure Nap-Gly-D-Ala. | ||
3.3 N-Functionalized amino acids as noncovalent hydrogelators
Xu and coworkers were the first to observe that N-terminal Fmoc functionalization of single amino acids (5 and 6) promotes fibrilco-assembly and hydrogelation.103 Based on this observation, Xu and coworkers developed methods for externally triggering the self-assembly and hydrogelation of Fmoc protected amino acids and peptides.124–127 They found that phosphorylated Fmoc-Tyr (22a, Fig. 14) did not self-assemble; however, enzymatic dephosphorylation of the phenolic oxygen promoted rapid self-assembly into 10 nm diameter fibrils coincident with the formation of an opaque hydrogel (G′ = 1000 Pa, ∼50 mM).124 Based on CD and fluorescence emission spectroscopic evidence, Xu et al. proposed that the Fmocgroups assemble in an antiparallel fashion, with the tyrosine side chains of neighboring residues participating in π-π and hydrogen bonding interactions (Fig. 15).124 The incorporation of an external trigger for self-assembly is highly desirable for tissue engineering applications, and Xu et al. subsequently demonstrated the generality of the enzymatic dephosphorylation trigger for assembly and hydrogelation of larger Tyr-containing peptides.127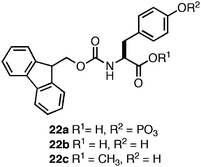 | ||
| Fig. 14 Chemical structures of Fmoc-Tyr derivatives. | ||
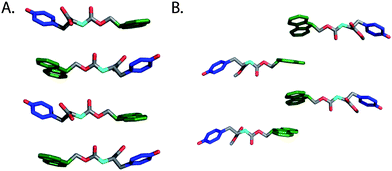 | ||
| Fig. 15 (A) A proposed structural model for fibrils derived from self-assembly of Fmoc-Tyr (Xu et al.).124 (B) Proposed structural model for fibrils derived from self-assembly of Fmoc-Tyr methyl ester (Yang et al.).128 Hydrogens omitted for clarity, Fmoc (green), phenol side chain (blue). | ||
Yang and coworkers later demonstrated that enzymatic dephosphylation could also trigger the self-assembly and hydrogelation of a C-terminal methyl ester derivative of Fmoc-Tyr (22c, Fig. 14).128,129 Following enzymatic dephosphorylation it was observed that 22c self-assembled into an opaque hydrogel (∼24 mM amino acid) that consisted of fibrils with average diameters of 1–2 μm.128 Similar CD and fluorescence emission spectra to those seen for 22b were observed and the authors proposed a distinctive packing model for self-assembly relative to that proposed for the closely related 22c (Fig. 15B).124,128 The origin of the large variation in fiber diameter between 22b and 22c may be a function of distinctive fibril lamination during the hierarchical self-assembly process. As individual monomers self-assemble into filaments, these filaments begin to laminate into fibrils.46 At sufficiently high concentrations of fibrils, further bundling into fibers can occur. The increased hydrophobicity of 22c relative to 22b may provide a sufficient driving force for further desolvation and bundling, resulting in the observation of much larger, μm diameter fibrils.
It has been previously observed that the incorporation of a photo-switch onto a self-assembling peptide hydrogelator can be used to externally trigger gel/sol and sol/gel transitions.130,131 Xu et al. designed a photoisomerizable amino acid based hydrogelator through the N-terminal functionalization of phenylalanine with a cinnamoylgroup (23, Fig. 16). It was demonstrated that the trans form of 23 was capable of self-assembling into fibrils (∼10–15 nm in diameter) which formed opaque hydrogels (2 wt %) with a G′ of ∼1000 Pa (68 mM). UV irradiation of the hydrogel initiated a trans to cisphotoisomerization of 23, coincident with a gel/sol transition of the hydrogel material.132 This result is consistent with disassembly of the fibrils due to steric constraints, which causes collapse of the hydrogel network. This process was shown to be reversible; photoisomerization of 23 from cis to trans resulted in reassembly and formation of an opaque hydrogel in timescales ranging from hours to days.132 While these studies present highly significant proof-of-concept methods to externally regulate self-assembly and hydrogelation phenomena, the resulting opaque hydrogels are not ideal materials for cell-scaffolding applications where imaging is required.31 The time for re-gelation (h/days) following photoisomerization are not amenable to encapsulation of cells within the hydrogel.31
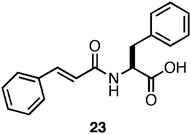 | ||
| Fig. 16 Chemical structure of a photo-switchable Phe-derived hydrogelator. | ||
Xu et al. were also interested in investigating if N-terminal functionalization of a bioactive amino acid/carbohydrate hybrid could promote self-assembly and hydrogelation while retaining bioactivity.133 They investigated the ability of Nap-functionalized phenylalanine to promote the self-assembly and hydrogelation of a scaffold that presents the wound healing agent glucosamine (24, Fig. 17).133 It was observed that 24 (4 mM in H2O) self-assembled into fibers (∼30 nm in diameter) and had a CD and fluorescence spectrum consistent with the formation of π-π interactions between the Nap groups.133 Following assembly into fibrils the formation of an optically transparent hydrogel with a G′ of 1000 Pa was observed. Xu and coworkers tested the cytotoxicity of 24 using a live/dead assay and it was found that 79% of cultured HeLa cells survived after a 24 h exposure to the surface of the hydrogel.133 In addition, the authors investigated whether the presence of the hydrogel would promote wound healing, as glucosamine had been previously observed to inhibit the formation of scar tissue at wound sites.133 They observed that mice treated with the hydrogel of 24 displayed significantly less scar tissue at the site of surgery after 6 days, indicating that the hydrogel aided wound healing.133 These results highlight the utility of N-functionalized amino acids as assembly motifs to form hydrogels that can affect multivalent display of biologically active compounds to form functional hydrogel materials.
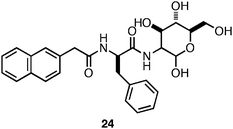 | ||
| Fig. 17 Chemical structure of Nap-Phe-D-glucosamine. | ||
In order to probe the relationship between structure of the assembly motif and hydrogelation propensity we have investigated a series of derivatives based on Fmoc-Phe and Fmoc-Tyr.97,123,124Fmoc-Phe has been shown to be capable of self-assembly and hydrogelation only under conditions where pH is carefully adjusted from basic to neutral/acidic.123 In comparison, Fmoc-Tyr undergoes spontaneous self-assembly and hydrogelation upon dilution into water from concentrated organic solutions; Fmoc-Phe does not readily self-assemble under these conditions.134 We questioned whether these differences in assembly propensity between Fmoc-Phe and Fmoc-Tyr were due to the electron withdrawing nature of the hydroxyl substitution on the phenyl side chain of Tyr promoting more efficient π-π interactions involving the side chains or whether these effects were due to differences in hydrophobicity (Tyr is significantly less hydrophobic than Phe).134,135
The comparative self-assembly and hydrogelation capacity of a series of Fmoc-Phe derivatives with varying side chain electronics and hydrophobicity was examined in order to understand these effects.134 The self-assembly and hydrogelation of Fmoc-Phe and Fmoc-Tyr was compared with that of Fmoc-pentafluorophenylalanine (25, Fig. 18A). Perfluorination of the phenyl side chain results in a significant electronic perturbation of the phenyl side chain (the side chain is much more electron deficient that that of Tyr) and an increase in hydrophobicity (25 is much more hydrophobic than Fmoc-Phe). The self-assembly propensity of 25 relative to Fmoc-Phe and Fmoc-Tyr was tested by diluting the amino acids from DMSO into water (4.2 mM amino acid in 2% DMSO/H2O by volume). It was observed that 25 rapidly self-assembled into fibrils with average diameters of 9 nm that formed an optically transparent hydrogel (3–5 min) with a G′ of 3000 Pa.134 In comparison, Fmoc-Phe precipitated from solution under these conditions and failed to assemble and Fmoc-Tyr self-assembled and formed hydrogels at much slower rates (∼30 min) and with lower rigidity (G′ = 1000 Pa). Fluorination of the Phe side chain significantly enhances the self-assembly propensity of Fmoc-Phe derivatives. Further investigation is required in order to determine if side chain π-π interactions or the enhanced hydrophobicity of 25 relative to Fmoc-Phe accounts for the enhanced hydrogel properties.91,92,134,136
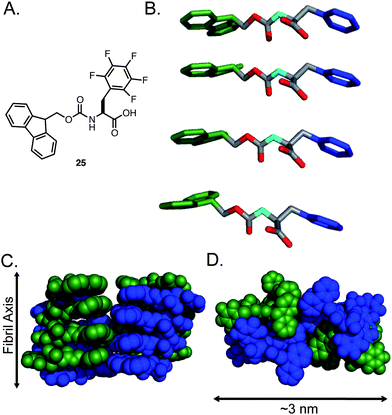 | ||
| Fig. 18 (A) Chemical structure of Fmoc-F5-Phe. (B) Proposed packing model of Fmoc-F5-Phe by Ryan et al. Hydrogens are omitted for clarity, Fmoc (green), Phe side chain (blue). (C) Proposed mode of lamination of fibrils leading to fibers, green and blue indicate individual fibrils. (D) Proposed mode of fiber formation, green and blue indicate individual fibrils. | ||
We examined the potential molecular packing mode of the fibrils using a variety of spectroscopic and imaging tools. CD, UV-vis, and fluorescence spectroscopy provided strong evidence for Fmoc–Fmoc and side chain–side chain π-π interactions, consistent with previously observed aromatic amphiphiles.134Solid-state NMR and X-ray powder diffraction measurements indicated that these compounds were assembling via an amyloid-like pathway since characteristic d-spacing at 4.7 Å and 10 Å were observed in scattering measurements.134 Based on these experimental constraints and on rudimentary molecular modeling studies we proposed a packing model that involved a parallel arrangement of the amino acids within fibrils (Fig. 19).134 It was hypothesized that hydrophobic burial of the Fmocgroups lead to the formation of slipped stacking interactions between Fmocgroups. This orientation aligns the phenyl side chains of 25 on a single face of the fibril, which aligns the carbamate backbone in a hydrogen-bonding network along the axis of the fibril. This arrangement exposes the C-terminal carboxylate groups on a single solvent accessible face of the fibril unit, allowing for increased solubility of the fibril once lamination of the basic fibril occurs to form fibers with Fmocgroups in the core (Fig. 18C–D).134 This model was based on the cumulative spectroscopic and empirical observations from of our analysis of these systems, though alternate models such as those proposed by Xu et al. and Yang et al. may also be valid (see Section 4 for a discussion of the limitations of current structural analyses).124,128
 | ||
| Fig. 19 Chemical structure of monohalogenated Fmoc-Phe derivatives. | ||
The observation that perfluorination of the Phe side chain promoted the efficient assembly and hydrogelation of 25 led us to further probe the role of side chain halogenation on self-assembly propensity.137 We investigated the effect of monohalogenation (F, Cl, or Br) at the ortho, meta, and para position of the benzyl side chain of Fmoc-Phe (26a–c, Fig. 19) on self-assembly and hydrogelation. It was observed that incorporation of even a single halogen on the benzyl ring promoted efficient self-assembly and hydrogelation of these molecules.137 There was a significant positional effect observed on self-assembly propensity and rheological properties of the resulting hydrogels. Halogenation at the meta position, 26a, promoted rapid self-assembly into fibrils (∼20 nm in diameter) that formed hydrogels with similar rates (∼3–5 min) and rigidity (G′ of ∼4000 Pa) to 25.134,137Halogenation at the para position, 26b, led to immediate (<30 sec) self-assembly into large fibril bundles (100–300 nm in diameter) that provided rheologically weak hydrogels (G′ of ∼300 Pa). Lastly, halogenation at the ortho position, 26c, resulted in slower formation of fibrils (30–40 min, ∼20 nm in diameter) that formed hydrogels with a G′ of ∼2000 Pa.137
It was hypothesized that these positional effects were the direct result of perturbation of side chain π-π interactions. For example, substitution at the ortho position, 26c, may promote the formation of complementary π-π stacking interactions between phenyl side chains of neighboring residues by placing the electronegative fluorine over the electron deficient region of the adjacent phenyl side chain.137 This interaction, while electronically favorable, would require a degree of structural reorganization due to the steric bulk of the halogen substituent, consistent with the diminished rate of assembly and hydrogel formation.137 Substitution at the meta position, 26a, a more sterically favorable position rotationally (relative to ortho) can achieve a similar stabilizing π-π interaction between adjacent side chains without a steric penalty, consistent with the observed rapid assembly and hydrogelation of the meta substituted Fmoc-Phe.137Halogenation at the para position, 26b, cannot access the same favorable π-π interactions as the ortho and meta variants. Thus, it was hypothesized that para substitution drives the rapid lamination of fibrils into the observed bundles due to exposure of the halogengroup to the exterior of the fibril structure.137 These results clearly indicate that single atom substitutions can significantly perturb side chain π-π interactions and facilitate tuning of self-assembly kinetics and hydrogelation.
We have also studied the role of the C-terminal carboxylic acid on self-assembly of halogenated Fmoc-Phe derivatives.138 Based on our structural model, the C-terminus is in direct contact with the surrounding solvent and it was hypothesized that C-terminal hydrogen bonding with solvent and with neighboring amino acids (intra- or inter-fibril) has a significant influence on self-assembly propensity.134 In our previous studies, assembly of 25 and 26 occurred in unbuffered conditions and the pH of the hydrogel was 3.5–3.7, near the pKa of the carboxylic acid.134,137 At this pH the carboxylate is partially protonated, reducing repulsive Coulombic effects and allowing hydrogen bonding between residues within and between fibrils. Attempts to assemble 25 and 26 at pH 7 promoted precipitation of the materials as amorphous aggregate due to the inability of the negatively charged carboxylate groups to self-assemble.138 It was hypothesized that substitution of the C-terminal carboxylic acid for a C-terminal amide (27a, Fig. 20) would promote self-assembly at pH 7 by removing electrostatic repulsion and maintaining the ability to hydrogen bond with the solvent.138 The C-terminal methyl ester (27b) was also investigated in order to determine whether hydrogen bonding with solvent was critical to self-assembly.138
 | ||
| Fig. 20 Chemical structure of C-terminal Fmoc-Phe derivatives. | ||
C-terminal functionalization exerted a dramatic influence on the self-assembly and hydrogelation propensity of 27 (4.9 mM in 2% DMSO/H2O).88,99,101,138 Substitution of the C-terminal carboxyl group with the C-terminal amide (27a) resulted in the formation of fibrils (21 nm in diameter) at neutral pH; however, the mechanical properties of the resulting gel were significantly diminished relative to the C-terminal acid (G′ = 100 Pa compared to 3000 Pa).138 It was hypothesized that hydrogen bonding between the C-terminal amides was not as favorable as the C-terminal acids, resulting in diminished noncovalent cross-linking of the fibrils.138 This hypothesis was further supported by the observation that substitution of the C-terminal carboxylic acid with the C-terminal methyl ester (27b) abrogated self-assembly.138 These C-terminal substitutions also increase the hydrophobicity of 27a,b relative to 26a, which also influences the self-assembly propensity of these materials by decreasing the solubility of both the monomer and fibrilassemblies.138
Based on these observations, it was hypothesized that an equimolar mixture of the C-terminal acid (26a) and C-terminal amide (27a) would facilitate the formation of hydrogen bonds and effectively screen the negatively charged carboxylates in the context of fibrils at neutral pH.138Co-assembly of compounds 26a and 27a (4.9 mM total amino acid in 2% DMSO/phosphate-buffered saline) into fibrils (13 nm in diameter) at pH 7.4 occurred much more efficiently than self-assembly of either component. These co-fibrils formed an optically transparent hydrogel with a relatively weak G′ of 160 Pa.138 This G′ was significantly reduced relative to the G′ of the same 26a/27a mixture under unbuffered conditions (G′ of 1000 Pa in H2Oversus 160 Pa in PBS). The presence of salts can influence the rheological properties of a hydrogel by perturbing the equilibrium between monomer and fibril, which results in a net decrease in fibril concentration and network entanglements.73,138NMR experiments confirmed that in phosphate buffer the fibril/monomer equilibrium of the acid/amide mixture was perturbed as evidenced by an increase of monomer that is unincorporated into the fibril network relative to assembly in unbuffered water.138 This result highlights the challenge of promoting hydrogelation of complex media solutions relative to simple aqueous solutions.
A significant hurdle to the utilization of these materials for cell scaffolding applications was the observation that these materials were not stable to shear stress.134,137,138 Following the application of sufficient stress, irreversible precipitation of the hydrogel network was typically observed. We hypothesized that the hydrophobicity of fibrils assembled from Fmoc-Phe derivatives results in a thermodynamic driving force for desolvation and precipitation of the hydrogel network (hydrogels may be kinetic products while desolvated fibrils are thermodynamic products).139 It had been previously shown that addition of polylethylene glycol (PEG) groups to self-assembling peptides increased the water solubility of the self-assembled fibrils.140–142 It was reasoned that introduction of a PEG linker at the C-terminus of Fmoc-F5-Phe (28, Fig. 21) would impart greater solubility to the monomer and the self-assembled fibrillar network, which could impart in shear recovery properties to the resulting hydrogel networks.139
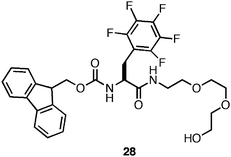 | ||
| Fig. 21 Chemical structure of Fmoc-Phe-PEG derivative. | ||
The self-assembly propensity of 28 was investigated (4.9 mM in 2% DMSO/H2O) and the formation of fibrils (15 nm in diameter) was observed.139 These fibrils formed rheologically weak (G′ = 100 Pa), optically transparent hydrogels within 3–4 hours (heating to 100 °C accelerated this process). This decrease in rigidity is likely due to reduced fibril-fibril cross-linking as a function of the non-adherent properties of PEG.139 Despite the decrease in the rheological rigidity, fibrils derived from 28 did not precipitate following application of stress; rather, the hydrogel network fully recovered following cessation of stress.139 Based on these results it was hypothesized that mixtures of 25 and 28 in varying ratios would give hydrogels with optimal rigidity and shear recoverability.139 It was observed that 9:1, 4:1, and equimolar mixtures of 25:28 formed mechanically rigid hydrogels (G′ ∼3000 Pa) with the ability to shear recover following the application of 100% strain.139 These results were consistent with the hypothesis that the statistical incorporation of a solubilizing group, such as PEG, at the fibril surface could be used to impart greater solubility and shear recovery to the fibril network.139 These methods also offer a practical method for tailoring the rheological properties of amino acid based hydrogels for potential uses in tissue engineering applications.
4. Challenges and opportunities in the development of noncovalent hydrogels derived from simple self-assembling dipeptide and amino acid motifs
While significant progress has been made in the development of noncovalent hydrogels derived from the self-assembly of minimal dipeptide and amino acids motifs, none universally meet the demanding criteria for ideal materials for three dimensional cell culture applications as outlined previously. Currently, a majority of the structure/function analyses conducted on these self-assembled systems are based on phenomenological and empirical strategies in which monomer structures are synthetically perturbed and the resulting self-assembly hydrogel behavior is characterized. These results, while successful for identifying novel hydrogelators and providing information about structure/function, do not fundamentally explain the observed hydrogelation behavior. For example, we observed that monohalogenation of the phenyl side chain of Fmoc-Phe resulted in the ability to tune the rheological strength, although the lack of detailed structural information limits understanding of the precise molecular origins of these effects.137 Further investigation into the precise atomic structure of self-assembled fibrils will become increasingly important to assist design of self-assembled, noncovalent hydrogels for sophisticated tissue culture applications.To date, the most common methods for characterizing the molecular structure of fibrils present in hydrogels are inherently low-resolution. UV-vis, IR, fluorescence, and CD spectroscopy provide information on the secondary structure and electronic structure of fibrillar materials21 that can be used to infer information about the molecular structure and orientation of monomers within fibrils.86,143Electron microscopy methods including TEM, cryo-TEM, and SEM are useful for analyzing fibril dimensions and morphology and can also be used to determine length-to-mass information for fibrilconstructs.86Diffraction methods, including small- and wide-angle X-ray, neutron, and electron diffractionprobe the molecular packing distances in the fibrils and the dimensions of the fibrils themselves in situ. Detailed structures based on these methods alone, however, rely on a high degree of inference and interpretation and can provide multiple models that can be equally supported by the data.
The molecular structure of related self-assembled systems can be highly variable. Simple changes in the molecular structure of the monomeric unit can give rise to significant changes in fibril architecture as can changes in solvent conditions, concentration, or sample handling.97,99,105,106,121 For example, IR, UV-vis, and CD spectroscopy have been used to develop a packing model for Fmoc-diphenylalanine in which the dipeptide self-assembles into antiparallel β-sheets (Fig. 4).97 For peptides there are well characterized stretches in the amide I region of the IR (∼1690 cm−1) that are commonly viewed as indicative of antiparallel β-sheet structures.90,91 In contrast, IR is less instructive concerning the self-assembly of Fmoc-Phe derivatives, since there are no amide bonds present in many of these structures (the carbamate functionality that links the Fmocgroup to the amino acid is less well characterized in this regard). In the absence of reliable FT-IR data, a parallel β-sheet model was deemed to be most consistent with all available data (Fig. 18).134 Others have also observed inconsistent spectroscopic results when studying libraries of structurally related compounds.105,107,121 Thus we find the emergence of contradictory models that may all be correct given the differences in molecular structure between the systems in question. These structural uncertainties highlight the growing need for high-resolution structural characterization of self-assembled hydrogel materials to enable understanding of the relationship between monomer structure, assembled fibril structure, and emergent hydrogel properties.
Crystallographic techniques have proven to be invaluable for characterization of atomic structure of biological molecules; however, there are some hurdles to their implementation for the characterization of self-assembled materials.86,144Amino acid based hydrogelators are inherently non-crystalline, similar to amyloid forming proteins and peptides, making them difficult to analyze by traditional crystallographic methods.144 In addition, it has been demonstrated that the scattering pattern obtained from single crystals differ from those obtained from hydrogel fibrils.122 Despite these differences, it is commonly accepted that single crystals provide good approximations of the hydrogel state and thus allow for increased insight into self-assembled architectures of self-assembled fibers at the molecular level.144,145 Despite significant technical challenges, crystallographic methods are evolving to enable detailed structure of self-assembled systems to be assessed.
Solution phase NMR is largely ineffective for investigating self-assembled materials. As monomers assemble into higher order structures, the NMR signals broaden due to anisotropic effects.146,147Solid state NMR (SS-NMR) has emerged as a useful tool in the study of amyloid materials.148 Lynn and Tycko have developed two-dimensional SS-NMR isotopic labeling methods that defined β-strand registry in several amyloid-forming proteins.149–151 These techniques are increasingly utilized in the amyloid field to characterize the structure of cross-βfibril structures that have similarities to the fibrils observed in the hydrogel networks described herein. Despite the great potential of SS-NMR, these methods have not been widely applied to structure/function studies of hydrogel-forming self-assembled materials. Judicious isotopic labeling of self-assembling amino acid and dipeptide hydrogelators would enable the acquisition of high-resolution structural constraints by SS-NMR that would facilitate the development of improved structural models for these materials. SS-NMR also enables the potential investigation of self-assembled hydrogels in the native gel state rather than as the dried xerogel.86
In order to address the critical need for further structural insight into self-assembled hydrogels it will be necessary to exploit data obtained from all the low and high-resolution techniques described above. Each of the above methods provides constraints that collectively can be incorporated into high-level computational models, leading to more accurate depictions of the molecular interactions that promote self-assembly and hydrogelation relative to isolated spectroscopic methods.86 In this regard, inspiration can be derived from recent success in developing structural models of amyloid systems.148 The added insight that high-resolution structural models of self-assembled hydrogels will enable rational, structure-based approaches to the development of next-generation noncovalent hydrogels. For example, the rational design of hydrogels with desired rheological properties, including tunable mechanical rigidity and shear recovery, through synthetic customization of the self-assembling monomer will become possible.
The mechanisms of in vivo toxicity and biodegradation of self-assembled hydrogels is also an aspect of these materials that is under-investigated.1,13,30 Much of the current data on the toxicity of dipeptide and small molecule based hydrogels has come from the results of in vitro cytotoxicity assays, however, limited insight on in vivo toxicity can be extrapolated from these studies.4,9,31,66,71,76,105,106,114 In contrast, there are several studies that comment on the immunogenicity and degradation of self-assembled peptide-based hydrogels in animal models. Stupp and coworkers investigated the in vivo immunogenic response in mice following injection of an IKVAV-displaying peptideamphiphile hydrogel to the site of a spinal cord injury and observed no inflammation or autoimmune response.5,81 Zhang and coworkers observed that injection of fibrils of the amphiphilic self-assembled (EAKA)4 and (RADA)4peptides into mice elicited no immunogenic responses after 9 days and 5 weeks and that the fibril network appeared to be degraded over time.7,51 The enzymatic degradation of self-assembled peptidefibril materials was demonstrated by Hartgerink and coworkers; noncovalent peptide based hydrogels containing an MMP-2 cleavage motif in the core sequence of the peptide were efficiently enzymatically degraded in vitro.57 These results demonstrate that self-assembled amyloid-inspired materials can be benign biomaterials that can be cleared without invoking immune responses in living organisms, although few reports of the immunogenicity and biodegradation of self-assembled amino acid and dipeptide-based materials exist. Additional studies aimed at understanding how these materials are tolerated in vivo are necessary to establish the mechanisms of biodegradation and immunogenicity.9
5. Conclusions
The work reviewed herein represents significant progress toward understanding the behavior of noncovalent hydrogels; however, some significant questions remain. Are there technical limitations to the potential use of minimal assembly motifs as noncovalent materials that match the performance of peptides and proteins? How are self-assembly kinetics and dynamics related to the emergent hydrogel viscoelastic properties of noncovalent materials? While protein-based hydrogels have the benefit of extended structures that can form numerous noncovalent hydrogen bond and side chain interactions, dipeptide and amino acids are inherently limited to a small number of hydrogen bond donors and acceptors and hydrophobic or aromatic contacts. Thus, there may be limits to the structural substitutions that can be exploited to tune self-assembly and hydrogelation of these much simpler scaffolds. Future studies aimed at elucidating the precise mechanisms of self-assembly and the molecular structure of self-assembled materials will be of critical importance in the next decade in order to understand and realize the potential of hydrogels derived from low molecular weight self-assembling molecules as scaffolds for tissue engineering applications. Structural understanding coupled with continuous structure/function analysis of these materials will facilitate advancement toward the ability to design functional, noncovalent materials from first principles, providing next-generation materials that can be tuned to meet precisely defined criteria.6. References
- W. T. Truong, S. Su, J. T. Meijer, P. Thordarson and F. Braet, Chem.–Asian J., 2011, 6, 30–42 CrossRef CAS
.
- J. P. Jung, J. Z. Gasiorowski and J. H. Collier, Biopolymers, 2010, 94, 49–59 CrossRef CAS
- O. D. Krishna and K. L. Kiick, Biopolymers, 2010, 94, 32–48 CrossRef CAS
- Y. Gao, Z. Yang, Y. Kuang, M. Ma, J. Li, F. Zhao and B. Xu, Biopolymers, 2010, 94, 19–31 CrossRef CAS
- H. Cui, M. J. Webber and I. S.S, Biopolymers, 2010, 94, 1–18 CrossRef CAS
- S. I. Stupp, Nano Lett., 2010, 10, 4783 CrossRef CAS
- X. Zhao, F. Pan, H. Xu, M. Yaseen, H. Shan, C. A. E. Hauser, S. Zhang and J. R. Lu, Chem. Soc. Rev., 2010, 39, 3480–3498 RSC
- B. Xu, Langmuir, 2009, 25, 8375–8377 CrossRef CAS
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS
- M. C. Branco and J. P. Schneider, Acta Biomater., 2009, 5, 817–831 CrossRef CAS
- S. Kyle, A. Aggeli, E. Ingham and M. J. McPherson, Trends Biotechnol., 2009, 27, 423–433 CrossRef CAS
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS
- E. K. Johnson, D. J. Adams and P. J. Cameron, J. Mater. Chem., 2011, 21, 2024–2027 RSC
- M. Zelzer and R. V. Ulijn, Chem. Soc. Rev., 2010, 39, 3351–3357 RSC
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC
- J. Kopecek and Y. Yang, Acta Biomater., 2009, 5, 805–816 CrossRef CAS
- Z. Yang and B. Xu, J. Mater. Chem., 2007, 17, 2385–2393 RSC
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822–835 RSC
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS
- S. C. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Science, 1997, 276, 1425–1428 CrossRef
- A. Birgersdotter, R. Sandberg and I. Ernberg, Semin. Cancer Biol., 2005, 15, 405–412 CrossRef
- S. Zhou, S. Matsumoto, H. Tian, H. Yamane, A. Ojida, S. Kiyonaka and I. Hamachi, Chem.–Eur. J., 2005, 11, 1130–1136 CrossRef CAS
- R. A. Hule, R. P. Nagarkar, A. Altunbas, H. R. Ramay, M. C. Branco, J. P. Schneider and D. J. Pochan, Faraday Discuss., 2007, 139, 251–264 RSC
- M. M. Stevens and J. H. George, Science, 2005, 310, 1135–1138 CrossRef CAS
- D. E. Discher, P. Janmey and Y. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS
- D. F. WIlliams, Biomaterials, 2008, 29, 2941–2953 CrossRef CAS
- D. S. Kohane and R. Langer, Chem. Sci., 2010, 1, 441–446 RSC
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS
- C. Yan and D. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC
- C. Yan, A. Altunbas, T. Yucel, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Soft Matter, 2010, 6, 5143–5156 RSC
- Z. Yang and B. Xu, Adv. Mater., 2006, 18, 3043–3046 CrossRef CAS
- Z. Yang, G. Liang and B. Xu, Soft Matter, 2007, 3, 515–520 RSC
- D. N. Woolfson, Biopolymers, 2010, 94, 118–127 CrossRef CAS
- F. W. Kotch and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3028–3033 CrossRef CAS
- J. A. Fallas, L. E. R. O'Leary and J. D. Hartgerink, Chem. Soc. Rev., 2010, 39, 3510–3527 RSC
- M. A. Cejas, W. A. Kinney, C. Chen, J. G. Vinter, H. R. Almond, K. M. Balss, C. A. Maryanoff, U. Schmidt, M. Breslav, A. Mahan, E. Lacy and B. E. Maryanoff, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8513–8518 CrossRef CAS
- M. A. Cejas, W. A. Kinney, C. Chen, G. C. Leo, B. A. Tounge, J. G. Vinter, P. P. Joshi and B. E. Maryanoff, J. Am. Chem. Soc., 2007, 129, 2202–2203 CrossRef CAS
- E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS
- C. Gribbon, K. J. Channon, W. Zhang, E. F. Banwell, E. H. C. Bromley, J. B. Chaudhuri, R. O. C. Oreffo and D. N. Woolfson, Biochemistry, 2008, 47, 10365–10371 CrossRef CAS
- A. M. Smith, S. F. A. Acquah, N. Bone, H. W. Kroto, M. G. Ryadnov, M. S. P. Stevens, D. R. M. Walton and D. N. Woolfson, Angew. Chem., Int. Ed., 2005, 44, 325–328 CrossRef CAS
- M. G. Ryadnov and D. N. Woolfson, Nat. Mater., 2003, 2, 329–332 CrossRef CAS
- I. Cherny and E. Gazit, Angew. Chem., Int. Ed., 2008, 47, 4062–4069 CrossRef CAS
- I. W. Hamley, Angew. Chem., Int. Ed., 2007, 46, 8128–8147 CrossRef CAS
- A. M. Smith and T. Schiebel, Macromol. Chem. Phys., 2010, 211, 127–135 CrossRef CAS
- S. Cavalli, F. Albericio and A. Kros, Chem. Soc. Rev., 2010, 39, 241–263 RSC
- J. Kisiday, M. Jin, B. Kurz, H. Hung, C. Semino, S. Zhang and A. J. Grodzinsky, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9996–10001 CrossRef CAS
- D. M. Marini, W. Hwang, D. A. Lauffenburger, S. Zhang and R. D. Kamm, Nano Lett., 2002, 2, 295–299 CrossRef CAS
- T. C. Holmes, S. Lacalle, X. Su, G. Liu, A. Rich and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6728–6733 CrossRef CAS
- M. R. Caplan, P. N. Moore, S. Zhang, R. D. Kamm and D. A. Lauffenburger, Biomacromolecules, 2000, 1, 627–631 CrossRef CAS
- C. A. E. Hauser and S. Zhang, Macromol. Symp., 2010, 295, 30–48 CrossRef CAS
- C. A. E. Hauser and S. Zhang, Chem. Soc. Rev., 2010, 39, 2780–2790 RSC
- R. G. Ellis-Behnke, Y. Liang, D. K. C. Tay, P. W. F. Kau, G. E. Schneider, S. Zhang, W. Wu and K. So, Nanomed.: Nanotechnol., Biol. Med., 2006, 2, 207–215 CrossRef CAS
- L. Aulisa, H. Dong and J. D. Hartgerink, Biomacromolecules, 2009, 10, 2694–2698 CrossRef CAS
- K. M. Galler, L. Aulisa, K. R. Regan, R. N. D'Souza and J. D. Hartgerink, J. Am. Chem. Soc., 2010, 132, 3217–3223 CrossRef CAS
- C. J. Bowerman and B. L. Nilsson, J. Am. Chem. Soc., 2010, 132, 9526–9527 CrossRef CAS
- J. Guilbaud, E. Vey, S. Boothroyd, A. M. Smith, R. V. Ulijn, A. Saiani and A. F. Miller, Langmuir, 2010, 26, 11297–11303 CrossRef CAS
- H. Rapaport, H. Grisaru and T. Silberstein, Adv. Funct. Mater., 2008, 18, 2889–2896 CrossRef CAS
- S. Ramachandran, J. Trewhella, Y. Tseng and Y. B. Yu, Chem. Mater., 2006, 18, 6157–6162 CrossRef CAS
- S. Ramachandran, P. Flynn, Y. Tseng and Y. B. Yu, Chem. Mater., 2005, 17, 6583–6588 CrossRef CAS
- A. Aggeli, M. Bell, L. M. Carrick, C. W. G. Fishwick, R. Harding, P. J. Mawer, S. E. Radford, A. E. Strong and N. Boden, J. Am. Chem. Soc., 2003, 125, 9619–9628 CrossRef CAS
- J. H. Collier, B. Hu, J. W. Ruberti, J. Zhang, P. Shum, D. H. Thompson and P. B. Messersmith, J. Am. Chem. Soc., 2001, 123, 9463–9464 CrossRef CAS
- J. H. Collier, J. S. Rudra, J. Z. Gasiorowski and J. P. Jung, Chem. Soc. Rev., 2010, 39, 3413–3424 RSC
- A. Altunbas, S. J. Lee, S. A. Rajasekaran, J. P. Schneider and D. J. Pochan, Biomaterials, 2011, 32, 5906–5914 CrossRef CAS
- C. M. Micklitsch, P. J. Knerr, M. C. Branco, R. Nagarkar, D. Pochan and J. P. Schneider, Angew. Chem., Int. Ed., 2011, 50, 1577–1579 CrossRef CAS
- R. V. Rughani, M. C. Branco, D. J. Pochan and J. P. Schneider, Macromolecules, 2010, 43, 7924–7930 CrossRef CAS
- C. Veerman, K. Rajagopal, C. S. Palla, D. Pochan, J. P. Schneider and E. M. Furst, Macromolecules, 2006, 39, 6608–6614 CrossRef CAS
- M. S. Lamm, K. Rajagopal, J. P. Schneider and D. Pochan, J. Am. Chem. Soc., 2005, 127, 16692–16700 CrossRef CAS
- J. K. Kretsinger, L. A. Haines, B. Ozbas, D. J. Pochan and J. P. Schneider, Biomaterials, 2005, 26, 5177–5186 CrossRef CAS
- L. A. Haines, K. Rajagopal, B. Ozbas, D. A. Salick, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2005, 127, 17025–17029 CrossRef CAS
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Macromolecules, 2004, 37, 7331–7337 CrossRef CAS
- D. Pochan, J. P. Schneider, J. Kretsinger, B. Ozbas, K. Rajagopal and L. Haines, J. Am. Chem. Soc., 2003, 125, 11802–11803 CrossRef CAS
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS
- M. Gungormus, M. Branco, H. Fong, J. P. Schneider, C. Tamerler and M. Sarikaya, Biomaterials, 2010, 31, 7266–7274 CrossRef CAS
- M. A. Greenfield, J. R. Hoffman, M. Olvera de la Cruz and S. I. Stupp, Langmuir, 2009, 26, 3641–3647 CrossRef
- N. L. Angeloni, C. W. Bond, Y. Tang, D. A. Harrington, S. Zhang, S. I. Stupp, K. E. McKenna and C. A. Podlasek, Biomaterials, 2011, 32, 1091–1101 CrossRef CAS
- J. E. Goldberger, E. J. Berns, R. Bitton, C. J. Newcomb and S. I. Stupp, Angew. Chem., Int. Ed., 2011, 50, 6292–6295 CrossRef CAS
- R. N. Shah, N. A. Shah, M. M. del Rosario Lim, C. Hsieh, G. Nuber and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3293–3298 CrossRef CAS
- V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, J. Neurosci., 2008, 28, 3814–3823 CrossRef CAS
- E. T. Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef CAS
- K. L. Niece, J. D. Hartgerink, J. J. J. M. Donners and S. I. Stupp, J. Am. Chem. Soc., 2003, 125, 7146–7147 CrossRef CAS
- M. J. Krysmann, V. Castelletto, A. Kelarakis, I. W. Hamley, R. A. Hule and D. J. Pochan, Biochemistry, 2008, 47, 4597–4605 CrossRef CAS
- Y. Hu, H. Wang, J. Wang, S. Wang, W. Liao, Y. Yang, Y. Zhang, D. Kong and Z. Yang, Org. Biomol. Chem., 2010, 8, 3267–3271 CAS
- J. H. van Esch, Langmuir, 2009, 25, 8392–8394 CrossRef CAS
- R. A. Gortner and W. F. Hoffman, J. Am. Chem. Soc., 1921, 43, 2199–2202 CrossRef CAS
- F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679–11691 CrossRef CAS
- F. M. Menger, Y. Yamasaki, K. K. Catlin and T. Nishimi, Angew. Chem., Int. Ed. Engl., 1995, 34, 585–586 CrossRef CAS
- F. T. Senguen, T. M. Doran, E. A. Anderson and B. L. Nilsson, Mol. BioSyst., 2011, 7, 497–510 RSC
- F. T. Senguen, N. R. Lee, X. Gu, D. M. Ryan, T. M. Doran, E. A. Anderson and B. L. Nilsson, Mol. BioSyst., 2010, 7, 486–496 RSC
- C. J. Bowerman, D. M. Ryan, D. A. Nissan and B. L. Nilsson, Mol. BioSyst., 2009, 5, 1058–1069 RSC
- E. Gazit, FASEB J., 2002, 16, 77–83 CrossRef CAS
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS
- A. M. Smith, R. J. WIlliams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS
- W. Helen, P. Leonardis, R. V. Ulijn, J. Gough and N. Tirelli, Soft Matter, 2011, 7, 1732–1740 RSC
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS
- Z. Yang, G. Liang, M. Ma, Y. Gao and B. Xu, Small, 2007, 3, 558–562 CrossRef CAS
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC
- D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Soft Matter, 2010, 6, 1971–1980 RSC
- Z. Yang, H. Gu, Y. Zhang, L. Wang and B. Xu, Chem. Commun., 2004, 208–209 RSC
- Z. Yang, L. Wang, J. Wang, P. Gao and B. Xu, J. Mater. Chem., 2010, 20, 2128–2132 RSC
- V. Jayawarna, S. M. Richardson, A. R. Hirst, N. W. Hodson, A. Saiani, J. E. Gough and R. V. Ulijn, Acta Biomater., 2009, 5, 934–943 CrossRef CAS
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS
- G. Cheng, V. Castelletto, R. R. Jones, C. J. Connon and I. W. Hamley, Soft Matter, 2011, 7, 1326–1333 RSC
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS
- S. Toledano, R. J. WIlliams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. H. van Esch, S. Santabarbara, N. T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2, 1089–1094 CrossRef CAS
- A. K. Das, R. Collins and R. V. Ulijn, Small, 2008, 4, 279–287 CrossRef CAS
- R. J. WIlliams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19–24 CrossRef CAS
- Z. Yang, G. Liang and B. Xu, Chem. Commun., 2006, 738–740 RSC
- Y. Zhang, Y. Kuang, Y. Gao and B. Xu, Langmuir, 2011, 27, 529–537 CrossRef CAS
- M. Ma, Y. Kuang, Y. Gao, Y. Zhang, P. Gao and B. Xu, J. Am. Chem. Soc., 2010, 132, 2719–2728 CrossRef CAS
- F. Zhao, Y. Gao, J. Shi, H. M. Browdy and B. Xu, Langmuir, 2011, 27, 1510–1512 CrossRef CAS
- G. Liang, Z. Yang, R. Zhang, L. Li, Y. Fan, Y. Kuang, Y. Gao, T. Wang, W. W. Lu and B. Xu, Langmuir, 2009, 25, 8419–8422 CrossRef CAS
- Y. Zhang, H. Gu, Z. Yang and B. Xu, J. Am. Chem. Soc., 2003, 125, 13680–13681 CrossRef CAS
- Z. Yang, M. Ma, Y. Gao and B. Xu, J. Mater. Chem., 2007, 17, 850–854 RSC
- L. Chen, S. Revel, K. Morris, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS
- D. J. Adams, K. Morris, L. Chen, L. Serpell, J. Bacsa and G. M. Day, Soft Matter, 2010, 6, 4144–4156 RSC
- S. Sutton, N. L. Campbell, A. I. Cooper, M. Kirkland, W. J. Frith and D. J. Adams, Langmuir, 2009, 25, 10285–10291 CrossRef CAS
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315–226 CrossRef CAS
- Z. Yang and B. Xu, Chem. Commun., 2004, 2424–2425 RSC
- Z. Yang, G. Liang, L. Wang and B. Xu, J. Am. Chem. Soc., 2006, 128, 3038–3043 CrossRef CAS
- J. Gao, H. Wang, L. Wang, J. Wang, D. Kong and Z. Yang, J. Am. Chem. Soc., 2009, 131, 11286–11287 CrossRef CAS
- H. Wang, C. Yang, M. Tan, L. Wang, D. Kong and Z. Yang, Soft Matter, 2011, 7, 3897–3905 RSC
- Y. Huang, Z. Qiu, Y. Xu, J. Shi, H. Lin and Y. Zhang, Org. Biomol. Chem., 2011, 9, 2149–2155 CAS
- Y. Lin, Y. Qiao, P. Tang, Z. Li and J. Huang, Soft Matter, 2011, 7, 2762–2769 RSC
- J. Shi, Y. Gao, Z. Yang and B. Xu, Beilstein J. Org. Chem., 2011, 7, 167–172 CrossRef CAS
- Z. Yang, G. Liang, M. Ma, A. S. Abbah, W. W. Lu and B. Xu, Chem. Commun., 2007, 843–845 RSC
- D. M. Ryan, S. B. Anderson, F. T. Senguen, R. E. Youngman and B. L. Nilsson, Soft Matter, 2010, 6, 475–479 RSC
- J. L. Fauchere, M. Charton, L. B. Kier, A. Verloop and V. Pliska, Int. J. Pept. Protein Res., 1988, 32, 269–278 CrossRef CAS
- C. J. Bowerman, W. Liyanage, A. J. Federation and B. L. Nilsson, Biomacromolecules, 2011, 12, 2735–2745 CrossRef CAS
- D. M. Ryan, S. B. Anderson and B. L. Nilsson, Soft Matter, 2010, 6, 3220–3231 RSC
- D. M. Ryan, T. M. Doran, S. B. Anderson and B. L. Nilsson, Langmuir, 2011, 27, 4029–4039 CrossRef CAS
- D. M. Ryan, T. M. Doran and B. L. Nilsson, Chem. Commun., 2011, 47, 475–477 RSC
- N. Tzokova, C. M. Fernyhough, P. D. Topham, N. Sandon, D. J. Adams, M. F. Butler, S. P. Armes and A. J. Ryan, Langmuir, 2009, 25, 2479–2485 CrossRef CAS
- N. Tzokova, C. M. Fernyhough, M. F. Butler, S. P. Armes, A. J. Ryan, P. D. Topham and D. J. Adams, Langmuir, 2009, 25, 11082–11089 CrossRef CAS
- V. Castelletto, G. E. Newby, Z. Zhu and I. W. Hamley, Langmuir, 2010, 26, 9986–9996 CrossRef CAS
- H. Friedrich, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2010, 49, 7850–7858 CrossRef CAS
- K. Morris and L. Serpell, Chem. Soc. Rev., 2010, 39, 3445–3453 RSC
- L. Goldschmidt, P. K. Teng, R. Riek and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3487–3492 CrossRef CAS
- A. R. Hirst, I. A. Coates, T. R. Boucheteau, J. F. Miravet, B. Escuder, V. Castelletto, I. W. Hamley and D. K. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS
- B. Escuder, M. L. Lusar and J. F. Miravet, J. Org. Chem., 2006, 71, 7747–7752 CrossRef CAS
- R. Tycko, Q. Rev. Biophys., 2006, 39, 1–55 CrossRef CAS
- A. K. Paravastu, R. D. Leapman, W. Yau and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18349–18354 CrossRef CAS
- A. K. Mehta, K. Lu, W. S. Childers, Y. Liang, S. N. Dublin, J. Dong, J. P. Snyder, S. V. Pingali, P. Thiyagarajan and D. G. Lynn, J. Am. Chem. Soc., 2008, 130, 9829–9835 CrossRef CAS
- Y. Liang, S. V. Pingali, A. S. Jogalekar, J. P. Snyder, P. Thiyagarajan and D. G. Lynn, Biochemistry, 2008, 47, 10018–10026 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2012 |