Synthesis of multi-block copolymer stars using a simple iterative Cu(0)-mediated radical polymerization technique†
Cyrille
Boyer
,
Aurelia
Derveaux
,
Per B.
Zetterlund
and
Michael R.
Whittaker
*
Centre for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: mikey.whittaker@unsw.edu.au
First published on 30th September 2011
Abstract
In this communication, we describe a simple and highly efficient route to well-defined multi-block star copolymers based on copper(0)-mediated living radical polymerization. The technique involves a core first approach using a multi-functional initiator in connection with iterative copper(0)-mediated radical polymerization steps. Importantly, purification is not required between the successive chain extension steps as complete monomer conversion is reached before the addition of each consecutive monomer type.
Introduction
Star polymers can be regarded as singular well-defined and structurally complex nanoparticles, and have found applications in such diverse areas as nanomedicine,1–4 catalysis5 and photonics.10,11 These uniquely structured materials have attracted considerable interest due to their dendrimer-like properties such as high level of functionality and unique solution properties, as dictated by the relationship between solvent viscosity, number of arms and arm molecular weight.6–9 Importantly, while the synthesis of star polymers offers significant advantages over the synthesis of dendrimers (ease of purification, reduced synthesis time, etc), there are still problems associated with the routine introduction of high degrees of structural complexity (e.g. blocks) into star polymers.In general, well-defined star polymers have been prepared via two different routes; the arm first approach or the core first approach.10–14 In the arm first approach, pre-synthesized arms are either attached to a suitable multifunctional core or linked iteratively together to form a star (usually through cross-linking chemistry to give a cross-linked core). However, in both these cases, steric effects generally come into play at high arm molecular weights and/or high numbers of arms, resulting in less than quantitative yields.10 The use of orthogonal ‘click chemistry’ strategies, e.g. Huisgen cycloaddition, thiol-ene and others, has allowed the routine preparation of MIKTO-arm stars in high yields.15–23 Multifunctional microgels or nanogels have also proved to be suitable cores for polymer stars synthesis, where residual vinyl groups are used for subsequent arm attachment. Controlled/living radical polymerization (CLRP)2,24–28 has been applied to the iterative linking together of polymer arms via cross-linking chemistry. This approach was pioneered and optimized for ATRP by Matyjaszewski,11 and later expanded on by Qiao and co-workers, both reporting star polymers with very low polydispersities (PDIs).29–31
The core first approach entails the synthesis of a suitable macro-initiator bearing a defined number of initiating groups, each of which is subsequently transformed into a polymer chain viaCLRP. The approach was first demonstrated using ionic polymerization techniques, which are ideally suited to this star synthesis procedure as bimolecular termination (especially coupling) does not occur, and therefore star-star coupling by-products are not present.32–34 On the contrary, the CLRP techniques of ATRP/transition metal-mediated radical polymerization,11,13,35–40 RAFT/MADIX12,41–51 and NMP24,27,52 all suffer from bimolecular termination reactions to various extents, thus limiting their applicability to multi-arm star polymer synthesis. Tedious optimization of the polymerization conditions is required to minimize these side reactions and ensure low polydispersities.1,53–56
Cu(0)-mediated radical polymerization (SET-LRP) has been exploited to prepare star polymervia the core first approach. For example, Whittaker et al.57 and Ding et al.58 have reported the synthesis of 4-arm poly(MA) stars with a PDI of approximately 1.4. The high PDI was attributed to star-star coupling. In the present work, we have optimized the synthesis of multi-block copolymer star polymers using a core first approach with a very high control of the polymer structure. To achieve this, we have used our recent strategy to prepare linear multi-block copolymers with a high level of structural complexity.59 The approach, inspired by work of Haddleton,60,61 Matyjaszewski35,62–65 and Percec and co-workers,66–69 entails iterative Cu(0)-mediated radical polymerization steps in the presence of initially added Cu(II). The experimental conditions have been optimized to minimize any star-star coupling reactions by adjusting the deactivator (Cu(II)) concentration. The effect of adding CuBr2 to copper mediated polymerization is still not perfectly understood. However, as a deactivator it aids in the preservation of high end-group functionality (CH–Br), as recently demonstrated by our team for the polymerization of methyl acrylate70 and other groups.60,61,66–69 The ratio [Cu(II)]/[CH–Br] is critical with regards to minimization of star-star coupling, which is usually observed at very high monomer conversion. To illustrate the versatility of our approach, we have prepared five different star polymers with predesigned multi-block arms comprising short blocks (250–1 000 g mol−1) of a variety of monomers (hydrophobic, hydrophilic and functional monomers).
Experimental
Materials
Copper(II) bromide (Sigma-Aldrich, 99%), tetrahydrofuran (THF, Sigma, 9) and dimethyl sulphoxide (DMSO, UNIVAR, AR) were all used as received. Copper wire (diameter = 1.25 mm) was activated by washing in sulfuric acid for 10 min. Tris(2-(dimethylamino)ethyl)amine (Me6TREN) was synthesized according to literature procedures71 and stored under nitrogen prior to use.Monomers methyl acrylate (MA, Aldrich, 99%), n-butyl acrylate (nBA, Sigma-Aldrich, 99%), ethyl acrylate (EA, Sigma-Aldrich, 99%), tert-butyl acrylate (tBA, Aldrich, 99%), 2-ethyl hexylacrylate (2-EHA, Sigma-Aldrich, 99%) were de-inhibited by percolating over a column of basic alumina (Ajax, AR).
5 arm star glucose core synthesis
Synthesis of a pentafunctional ATRP initiator, 1,2,3,4,6-penta-O-isobutyryl bromide-α-D-glucose, was implemented according to a method described in the literature.72,73A typical Cu(0)-meditated iterative polymerization with [Cu(II)]0: [CH–Br]0 = 0.04: 1
5 arm star core (initiator) synthesized above (0.103g, 0.112 mmol, 5 eq. of CHBr), MA (0.2 mL, 2.22 mmol, 20 eq), DMSO (1.8 mL), Me6TREN (0.034 mL, 0.15 mmol, 0.30 eq), CuBr2 (5 mg, 0.022 mmol) and a magnetic stir bar were charged to a polymerization flask fitted with a rubber septum and the mixture was degassed vianitrogen for 10 min. A slight positive pressure of nitrogen was then applied and the pre-activated copper wire (0.5 cm) was carefully added under a nitrogen blanket. The polymerization flask was then resealed and polymerization was allowed to proceed at room temperature for 24 h. A sample of the reaction mixture was carefully removed for 1H NMR, GPC and mass spectroscopy analysis. The samples for 1H NMR were simply diluted with CDCl3, while the sample for GPC analysis was first diluted with THF and then passed over an aluminum oxide column to remove metal salts.For the subsequent chain extension (i.e. the next iterative polymerization), a further 0.4 mL of a degassed monomer was then carefully added via gas tight syringe and again the solution was allowed to polymerize at room temperature for another 24 h with stirring.
The above iterative polymerization-sampling-extension procedure was repeated as required.
Other [Cu(II)]0: [CH–Br]0 ratios (0.08![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 0.16: 1) were tested using the similar recipe.
:1 and 0.16: 1) were tested using the similar recipe.
Characterizations
DMAc GPC analysis of the polymers was performed in N,N-dimethylacetamide [DMAc; 0.03% w/v LiBr, 0.05% 2, 6-di-butyl-4-methylphenol (BHT)] at 50 °C (flow rate = 1 mL min−1) using a Shimadzu modular system comprised of an SIL-10AD auto-injector, a PL 5.0-mm bead-size guard column (50 × 7.8 mm) followed by four linear PL (Styragel) columns (105, 104, 103, and 500 Å) and an RID-10A differential refractive-index detector. Calibration was achieved with commercial polystyrene standards ranging from 500 to 106 g/mol.
Results and discussion
Various five arm multi-block star copolymers have been prepared by iterative Cu(0)-mediated radical polymerization in the presence of initially added Cu(II) using a pentafunctional initiator.59 This approach entails polymerization to (close to) full conversion during formation of each block, prior to the addition of new monomer in one continuous iterative polymerization process. To achieve this, two major difficulties have to be overcome: firstly, excessive loss of end-group functionality (livingness) at ultra-high monomer conversion, generally observed for CLRP such as ATRP, NMP, RAFT polymerization and others,25,26,74–76 must be minimized. Secondly, the formation of star-star coupling by-products due to bimolecular termination (coupling reaction), commonly observed during star synthesis by CLRP,10 must also be avoided. In our previous works,44,59 we have demonstrated that the addition of Cu(II) limits termination events and serves to maintain high end-group fidelity in Cu(0)-mediated radical polymerization during synthesis of linear multi-block copolymers. We demonstrated that high end group fidelity can be maintained even under post-polymerization conditions (i.e. essentially no unreacted monomer remaining) for as long as three days.44Scheme 1 depicts the process used in this study. Typically, the polymerizations reached close to full monomer conversion (>95%) in less than 3 h. However, to demonstrate the facile nature of this approach, the polymerizations were continued for 24 h for the synthesis of each block before addition of “the next” monomer. This iterative process was repeated several times to yield multi-block star copolymer.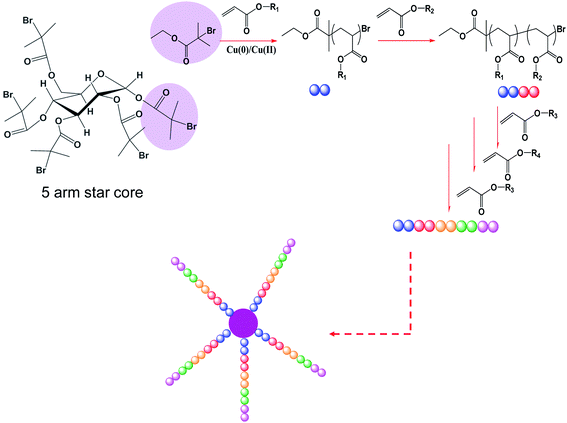 | ||
| Scheme 1 Synthesis of 5-arm star copolymersvia iterative Cu(0)-mediated radical polymerization. | ||
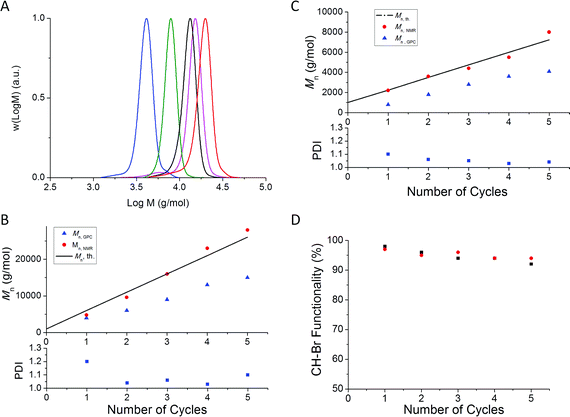
In preliminary experiments, we followed our previous work on the synthesis of linear multi-block copolymers,44,59 using a similar [Cu(II)]: [CH–Br] ratio of 0.04: 1, and the same amount of Cu(0) in the presence of the penta-functional initiator. To simplify the characterization, methyl acrylate was initially used as a model acrylate monomer, with a targeted block length of Mn = 500 g/mol. GPC analysis of the polymer formed after the first iteration (Fig. 1A) reveals a highly multimodal molecular weight distribution (MWD) with a prominent high molecular weight peak due to star-star coupling. Further successive chain extensions (by addition of degassed MA/DMSO mixture (50/50 V-%) without polymerpurification) resulted in the MWDs gradually shifting to higher molecular weights as anticipated (Fig. 1A), but the broadness of the MWDs increased (PDI > 1.5 and Mn > Mn, th; Fig. 2) indicating the formation of star-star coupled products.
![Molecular weight distributions obtained in cycles no. 1–5 during synthesis of multi-block 5 arm stars by Cu(0)-mediated radical polymerization of methyl acrylate in the presence of different initial amounts of Cu(ii): (A) [Cu(ii)]: [CH–Br] = 0.04: 1, (B) [Cu(ii)]: [CH–Br] = 0.08: 1 and (C) [Cu(ii)]: [CH–Br] = 0.16: 1.](/image/article/2012/PY/c1py00384d/c1py00384d-f1.gif) | ||
| Fig. 1 Molecular weight distributions obtained in cycles no. 1–5 during synthesis of multi-block 5 arm stars by Cu(0)-mediated radical polymerization of methyl acrylate in the presence of different initial amounts of Cu(II): (A) [Cu(II)]: [CH–Br] = 0.04: 1, (B) [Cu(II)]: [CH–Br] = 0.08: 1 and (C) [Cu(II)]: [CH–Br] = 0.16: 1. | ||
![(A) Evolution of molecular weight (Mn, GPC) and PDI versus number of cycles during synthesis of multi-block 5 arm stars by Cu(0)-mediated radical polymerization of methyl acrylate in the presence of different initial amounts of Cu(ii). Mn, th corresponds to the theoretical values calculated by the following equation: Mn, th. = (5 × [M]/[Initiator]) × MWMonomer + MWInitiator, with [M], [Initiator], MWMonomer and MWInitiator corresponding to monomer and initiator concentrations, and molar masses of monomer and initiator, respectively; (B) Evolution of molecular weight determined by NMR analysis (Mn, NMR) using the following equation: Mn, NMR = (∫CH3O3.6 ppm/3 × ∫CH6.3 ppm) × MWMA + MWInitiatorversus the number of cycles.](/image/article/2012/PY/c1py00384d/c1py00384d-f2.gif) | ||
| Fig. 2 (A) Evolution of molecular weight (Mn, GPC) and PDI versus number of cycles during synthesis of multi-block 5 arm stars by Cu(0)-mediated radical polymerization of methyl acrylate in the presence of different initial amounts of Cu(II). Mn, th corresponds to the theoretical values calculated by the following equation: Mn, th. = (5 × [M]/[Initiator]) × MWMonomer + MWInitiator, with [M], [Initiator], MWMonomer and MWInitiator corresponding to monomer and initiator concentrations, and molar masses of monomer and initiator, respectively; (B) Evolution of molecular weight determined by NMR analysis (Mn, NMR) using the following equation: Mn, NMR = (∫CH3O3.6 ppm/3 × ∫CH6.3 ppm) × MWMA + MWInitiatorversus the number of cycles. | ||
Increasing the amount of initially added Cu(II) by a factor of two ([Cu(II)]: [CH–Br] ratio = 0.08: 1) resulted in a significant improvement of the control over the MWD with the PDI remaining lower than 1.3 (Fig. 2A). However the occurrence of star-star coupling during the first and second cycle is still evidenced by high molecular weights shoulders in the MWDs (Fig. 1B). A further increase in the amount of Cu(II) by a factor of two led to excellent control over the MWDs throughout all five polymerization cycles with PDI < 1.10 (Fig. 1C) and Mn, GPC ≈ Mn, th (Fig. 2), confirming the formation of a well-defined 5 arm star polymer after 5 successive chain extensions (Fig. 1C). 1H NMR analysis of each iteration, (without purification) reveals monomer conversions >95%, indicating that Cu(II) does not cause any marked reduction in the rate of the polymerization in agreement with results of Haddleton and co-workers.60,61
After purification of the star polymers, 1H NMR analysis was performed to determine the end group fidelity by use of the characteristic signal at 4.3 ppm (i.e., –CH–Br). The presence of the initiator was confirmed by the signals at 6.3, 5.7, 5.0–5.5 and 4.1 ppm attributed to the carbohydrate ring (Fig. 3A). Poly(MA) signals at 3.6 and 1.0–2.5 ppm are attributed to –OCH3, –CH– and –CH2–, respectively.
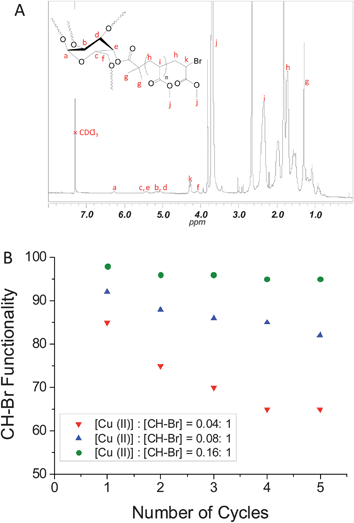 | ||
| Fig. 3 (A) 1H NMR spectrum of poly(MA)5 star polymer obtained by Cu(0)-mediated radical polymerization of methyl acrylate in five successive iterations in the presence of different initial amounts of Cu(II); (B) Chain end functionality versus the number of cycles for different initial amounts of Cu(II). | ||
The CH–Br functionality (i.e. livingness) was calculated based on the equation: %CH–Br = (∫CH–Br4.3 ppm/(5 × ∫CH 6.3 ppm)) × 100, with the CH intensity corresponding to the CH of the initiator at 6.3 ppm (Fig. 3A and Figure S1 in the ESI†). Fig. 3B shows the thus obtained CH–Br functionalities versus number of cycles. As already inferred from the GPC data above, it is immediately apparent that an increase in the amount of initially added Cu(II) greatly increases the CH–Br functionality. At the highest Cu(II) content, i.e. [Cu(II)]: [CH–Br] = 0.16: 1, the CH–Br functionality after the first cycle is as high as 98%. As pointed out in our previous work,59 this is a remarkable feature of this technique considering the high level of monomer conversion. It appears that the amount of Cu(II) plays a crucial role in preserving the CH–Br functionality. At lower concentrations of Cu(II), the functionality decreases markedly with increasing number of cycles, reaching as low as 65% after 5 cycles using the lowest Cu(II) concentration. At the highest Cu(II) concentration, the CH–Br functionality is maintained above 95% after 5 cycles. This is consistent with the corresponding MWDs (Fig. 1C), which are narrow and do not feature any prominent low or high molecular weight shoulders, indicative of minimal presence of nonfunctional arms and limited, if any, star-star coupling.
The molecular weights determined by GPC for the first two blocks (for all [Cu(II)]: [CH–Br] ratios) are lower than the theoretical molecular weights (Mn, th) calculated by the equation: Mn, th. = (5 × [M]/[Initiator]) × Monomerconversion × MWMonomer + MWInitiator, with [M], [Initiator], MWMonomer and MWInitiator corresponding to monomer and initiator concentrations, and molar masses of monomer and initiator, respectively (Fig. 2A). This discrepancy is attributed to the lower hydrodynamic volume of a star polymer than a linear polymer (i.e. the GPC standards) of the same molar mass. To obtain more accurate Mn values, 1H NMR analysis was used. The molecular weight (Mn, NMR) was calculated using the equation: Mn, NMR = (∫CH3O3.6 ppm/3 × ∫CH6.5 ppm) × MWMA + MWInitiator, with ∫CH3O3.6 ppm and ∫CH6.3 ppm corresponding to the intensities from methyl acrylate (methyl ester) and initiator (CH in adjacent position of CH2O), respectively. This approach is based on the assumption that all chains contain one initiator moiety, i.e. the occurrence of bimolecular termination reactions by coupling (star-star coupling) would cause the thus obtained value of Mn to be underestimated. A better agreement between Mn, NMR and Mn, th. was obtained (Fig. 2B).
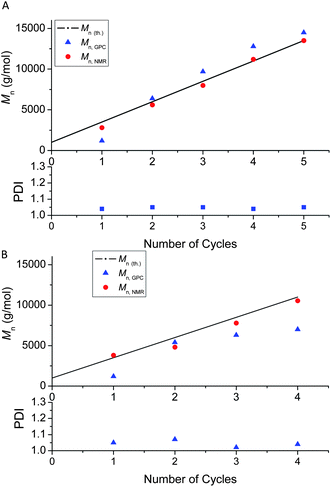
The versatility of this synthetic route to multi-block star polymers was further demonstrated by preparing two 5 arm star polymers (P(MA)5) using the same initiator as above and [Cu(II)]: [CH–Br] = 0.16:1, comprising blocks of targeted Mn of 250 and 1000 g mol−1, respectively. In both cases, well-defined star polymers were obtained after 5 cycles as evidenced by good agreement between Mn, NMR and Mn, th. and narrow MWDs (Fig. 4A–C). The CH–Br functionality decreases with number of cycles, but remains greater than 90% after 5 cycles for both target Mns (Fig. 4D). The functionality is slightly higher for the shorter block lengths as anticipated; the shorter the chains generated, the shorter is the cumulative time that each chain end spends in the active state, thus minimizing side reactions (e.g. bimolecular termination). We are currently exploring the preparation of material comprised of higher molecular weight blocks (Mn > 10 000 g mol−1) using this copper mediated polymerization technique. Theoretically, this technique can easily be extrapolated to higher molecular weight block materials, however, the ratios of the components need to be optimized.77
 | ||
| Fig. 4 Molecular weight and end group functionality data for multi-block star poly(MA) obtained via iterative Cu(0)-mediated radical polymerization: (A) MWDs vs. the number of cycles (block length 1000 g mol−1); (B) Mn and PDI vs. the number of cycles (block length 1000 g mol−1); (C) Mn and PDI vs. number of cycles (block length 250 g mol−1); D- End group functionality (% CH–Br chain ends at α-end) vs. number of cycles: (red dot) block length 250 g mol−1, (black square) block length 1000 g/mol. | ||
Finally, the synthetic methodology was applied to short multi-block star copolymers comprising a range of different monomers. We decided to use various hydrophobic and hydrophilic acrylates: hydrophobic: ethyl acrylate (EtA), tert-butyl acrylate (tBA), 2-ethylhexyl acrylate (HEA) and n-butyl acrylate (nBA), dodecyl acrylate (DA); hydrophilic: 2-dimethylaminoethyl acrylate (DMAEA) and hydroxyl acrylate (HEA). Two different star copolymers were synthesized, namely poly(PMA-b-PHEA-b-PDMEA-b-PDA) and poly(PMA-b-PnBA-b-PEA-b-PEHA-b-PtBA) (Table S1 in the ESI†). In both cases, the targeted block length was a molecular weight of approximately 500 g/mol. In the case of poly(PMA-b-PnBA-b-PEA-b-PEHA-b-PtBA), GPC analysis reveals increasing molecular weights with increasing number of cycles (Figure S4 in the ESI†), with exceptionally good control of the MWD (PDI well below 1.10) (Fig. 5A–B and Figure S2 in the ESI†). Excellent control over the polymer structure was also obtained for poly(PMA-b-PHEA-b-PDMEA-b-PDA) (Table S1 and Figure S3 in the ESI†). The Mn,th values are significantly lower than the molecular weights obtained by GPC in both cases, attributed to GPC error associated with linear poly(styrene) standards as discussed above. Therefore, the molecular weights were again estimated by 1H NMR, although the task is more complex in the case of copolymers. For example, Mn of poly(MA-b-nBA) was calculated via the equation: Mn (NMR) = (∫CH3O3.6 ppm/3 × ∫CH6.3 ppm) × MWMA + (∫CH24.1 ppm/2 × ∫CH6.3 ppm) × MWnBA + MWInitiator, where ∫CH3O3.6 ppm, ∫CH31.3 ppm, ∫CH6.3 ppm, MWMA, MWnBA and MWInitiator correspond to intensities from MA (methyl ester), CH2–O (ester) and CH of the initiator (at 6.3 ppm), molar mass of MA, nBA and initiator, respectively. For both multi-block star copolymers, the Mn values calculated by 1H NMR are in good accord with the theoretical values (Table 1 in the ESI, and Figure S2 and S3 in the ESI†). These results clearly illustrate the versatility of the present approach, which provides a simple and efficient route to multi-block star copolymers with excellent control over the polymer microstructure for a wide range of hydrophobic and hydrophilic acrylates.
 | ||
| Fig. 5 M n and PDI values (A) for poly(PMA-b-PnBA-b-PEA-b-PEHA-b-PtBA) and (B) poly(PMA-b-PBA-b-PEA-b-PEHA-b-PtBA) multi-block star copolymers obtained via iterative Cu(0)-mediated polymerization (approximate molecular weight of each block = 500 g mol−1, degree of polymerization ≈ 4–5). | ||
Conclusions
A recently developed, highly efficient and convenient synthetic route to multi-block copolymers involving iterative Cu(0)-mediated radical polymerization in the presence of initially added Cu(II)59 has been applied to the synthesis of multi-block five arm stars. It has been shown that the amount of initially added Cu(II) plays a crucial role in minimizing side reactions such as star-star coupling, allowing synthesis of multi-block star polymers with excellent control over the MWD (PDI < 1.1) and high end group functionality (livingness). The versatility of the method has been illustrated by the efficient synthesis of multi-block star copolymers comprising various hydrophobic, hydrophilic and functional monomers. Importantly, the above syntheses were all conducted in one pot via successive monomer additions, without polymerpurification between successive iterative polymerization steps.Acknowledgements
The authors thank the Australian Research Council and UNSW for funding. CB acknowledges an APD-ARC Fellowship.References
- M. E. Fox, F. C. Szoka and J. M. J. Fréchet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. B. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- J. T. Wiltshire and G. G. Qiao, Aust. J. Chem., 2007, 60, 699–705 CrossRef CAS.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS.
- V. Rodionov, H. Gao, S. Scroggins, D. A. Unruh, A.-J. Avestro and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 2570–2572 CrossRef CAS.
- B. M. Erwin, M. Cloitre, M. Gauthier and D. Vlassopoulos, Soft Matter, 2010, 6, 2825–2833 RSC.
- S. Coppola, N. Grizzuti, G. Floudas and D. Vlassopoulos, J. Rheol., 2007, 51, 1007–1025 CrossRef CAS.
- T. K. Goh, K. D. Coventry, A. Blencowe and G. G. Qiao, Polymer, 2008, 49, 5095–5104 CrossRef CAS.
- M. Sugimoto, T. Koizumi, T. Taniguchi, K. Koyama, K. Saito, D. Nonokawa and T. Morita, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 2226–2237 CrossRef CAS.
- C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Aust. J. Chem., 2006, 59, 719–727 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551–595 CrossRef CAS.
- A. Blencowe, J. F. Tan, T. K. Goh and G. G. Qiao, Polymer, 2009, 50, 5–32 CrossRef CAS.
- B. Boutevin, G. David and C. Boyer, Adv. Polym. Sci., 2007, 206, 31–135 CrossRef CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959–4961 RSC.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Polymer, 2009, 50, 3158–3168 CrossRef CAS.
- J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromol. Rapid Commun., 2008, 29, 1421–1421 CrossRef CAS.
- W. Lin, R. Jing, G. Wang and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2802–2810 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- Z. Zhang, G. Wang and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2811–2817 CrossRef CAS.
- X. Tang, X. Liang, Q. Yang, X. Fan, Z. Shen and Q. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4420–4427 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- G. David, C. Boyer, J. Tonnar, B. Ameduri, P. Lacroix-Desmazes and B. Boutevin, Chem. Rev., 2006, 106, 3936–3962 CrossRef CAS.
- P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747–3794 CrossRef CAS.
- L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034–5068 CrossRef CAS.
- J. Ferreira, J. Syrett, M. Whittaker, D. Haddleton, T. P. Davis and C. Boyer, Polym. Chem., 2011, 2, 1671–1677 RSC.
- J. F. Tan, A. Blencowe, T. K. Goh, I. T. M. Dela Cruz and G. G. Qiao, Macromolecules, 2009, 42, 4622–4631 CrossRef CAS.
- M. Spiniello, A. Blencowe and G. G. Qiao, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2422–2432 CrossRef CAS.
- T. K. Goh, A. P. Sulistio, A. Blencowe, J. W. Johnson and G. G. Qiao, Macromolecules, 2007, 40, 7819–7826 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS.
- K. Khanna, S. Varshney and A. Kakkar, Polym. Chem., 2010, 1, 1171–1185 RSC.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS.
- W. Li and K. Matyjaszewski, Macromol. Rapid Commun., 2011, 32, 74–81 CrossRef CAS.
- G. Deng, M. Cao, J. Huang, L. He and Y. Chen, Polymer, 2005, 46, 5698–5701 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Acc. Chem. Res., 2008, 41, 1120–1132 CrossRef CAS.
- S. Hou, E. L. Chaikof, D. Taton and Y. Gnanou, Macromolecules, 2003, 36, 3874–3881 CrossRef CAS.
- O. G. Schramm, M. A. R. Meier, R. Hoogenboom, H. P. van Erp, J.-F. Gohy and U. S. Schubert, Soft Matter, 2009, 5, 1662–1667 RSC.
- G. Moad, Aust. J. Chem., 2006, 59, 661–662 CrossRef CAS.
- H. Chaffey-Millar, M. H. Stenzel, T. P. Davis, M. L. Coote and C. Barner-Kowollik, Macromolecules, 2006, 39, 6406–6419 CrossRef CAS.
- M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4498–4512 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- K. Ranganathan, R. Deng, R. K. Kainthan, C. Wu, D. E. Brooks and J. N. Kizhakkedathu, Macromolecules, 2008, 41, 4226–4234 CrossRef CAS.
- D. Taton, J.-F. Baussard, L. Dupayage, Y. Gnanou, M. Destarac, C. Mignaud and C. Pitois, in Controlled/Living Radical Polymerization, American Chemical Society, 2006, vol. 944, pp. 578–594 Search PubMed.
- A. Duréault, D. Taton, M. Destarac, F. Leising and Y. Gnanou, Macromolecules, 2004, 37, 5513–5519 CrossRef.
- E. Setijadi, L. Tao, J. Liu, Z. Jia, C. Boyer and T. P. Davis, Biomacromolecules, 2009, 10, 2699–2707 CrossRef CAS.
- J. A. Syrett, D. M. Haddleton, M. R. Whittaker, T. P. Davis and C. Boyer, Chem. Commun., 2011, 47, 1449–1451 RSC.
- J. Liu, L. Tao, J. Xu, Z. Jia, C. Boyer and T. P. Davis, Polymer, 2009, 50, 4455–4463 CrossRef CAS.
- J. Moraes, T. Maschmeyer and S. Perrier, Aust. J. Chem., 2011, 64, 1047–1053 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- S. A. Bencherif, H. Gao, A. Srinivasan, D. J. Siegwart, J. O. Hollinger, N. R. Washburn and K. Matyjaszewski, Biomacromolecules, 2009, 10, 1795–1803 CrossRef CAS.
- H. Y. Cho, H. Gao, A. Srinivasan, J. Hong, S. A. Bencherif, D. J. Siegwart, H.-J. Paik, J. O. Hollinger and K. Matyjaszewski, Biomacromolecules, 2010, 11, 2199–2203 CrossRef CAS.
- S. Abraham, J. H. Choi, C.-S. Ha and I. Kim, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5559–5572 CrossRef CAS.
- C.-H. Lu, J.-H. Wang, F.-C. Chang and S.-W. Kuo, Macromol. Chem. Phys., 2010, 211, 1339–1347 CrossRef CAS.
- M. R. Whittaker, C. N. Urbani and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6346–6357 CrossRef CAS.
- W. Ding, C. Lv, Y. Sun, H. Luan, T. Yu and G. Qu, Polym. Bull., 2011 DOI:10.1007/s00289-011-0468-1.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS.
- M. E. Levere, I. Willoughby, S. O'Donohue, A. de Cuendias, A. J. Grice, C. Fidge, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1086–1094 RSC.
- M. E. Levere, I. Willoughby, S. O'Donohue, P. M. Wright, A. J. Grice, C. Fidge, C. R. Becer and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1753–1763 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482–4486 CrossRef CAS.
- W. Jakubowski, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 39–45 CrossRef CAS.
- K. Matyjaszewski, N. V. Tsarevsky, W. A. Braunecker, H. Dong, J. Huang, W. Jakubowski, Y. Kwak, R. Nicolay, W. Tang and J. A. Yoon, Macromolecules, 2007, 40, 7795–7806 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4684–4695 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2745–2754 CrossRef CAS.
- G. Lligadas, B. M. Rosen, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8360–8364 CrossRef CAS.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5109–5119 CrossRef CAS.
- F. Nyström, A. H. Soeriyadi, C. Boyer, P. B. Zetterlund and M. Whittaker, J. Polym. Sci. Part A: Polym. Chem., 2011 DOI:10.1002/pola.2510.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1966, 5, 41–44 CrossRef CAS.
- S. D. Angus and T. P. Davis, Langmuir, 2002, 18, 9547–9553 CrossRef CAS.
- D. Nyström, E. Malmström, A. Hult, I. Blakey, C. Boyer, T. P. Davis and M. R. Whittaker, Langmuir, 2010, 26, 12748–12754 CrossRef.
- C. Boyer, J. Liu, V. Bulmus, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 5641–5650 CrossRef CAS.
- J. F. Lutz and K. Matyjaszewski, Macromol. Chem. Phys., 2002, 203, 1385–1395 CrossRef CAS.
- W. Jakubowski, B. Kirci-Denizli, R. R. Gil and K. Matyjaszewski, Macromol. Chem. Phys., 2008, 209, 32–39 CrossRef CAS.
- C. Boyer, A. H. Soeriyadi, P. B. Zetterlund and M. Whittaker, Macromolecules, 2011 DOI:10.1021/ma201529j.
Footnote |
| † Electronic supplementary information (ESI) available: Additional molecular weights and NMR of poly(MA) homopolymer star and block-copolymer. See DOI: 10.1039/c1py00384d |
| This journal is © The Royal Society of Chemistry 2012 |