Hydrophilic porous carbon with tailored nanostructure and its sensitive hydrogen peroxide biosensor
Shu Juan
Bao†
abc,
Chun Xian
Guo†
ab and
Chang Ming
Li
*ab
aInstitute for Clean Energy & Advanced Materials, Southwest University, Chongqing 400715, P. R. China
bSchool of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore. E-mail: ecmli@ntu.edu.sg; Fax: +65 67911761; Tel: +65 67904485
cInstitute of Applied Chemistry, Xinjiang University, Urumqi, 830046, P. R. China
First published on 30th November 2011
Abstract
A microwave irradiation approach is used to prepare a novel hydrophilic porous carbon with a large specific surface area of 1040 m2 g−1 and uniform pore size distribution. In particular, the nanostructure can be tailored by changing microwave treatment time. As an application demonstration, the hydrophilic porous carbon is immobilized with protein haemoglobin for a hydrogen peroxide biosensor, showing higher direct electron transfer rate of haemoglobin for much higher sensitivity (0.15 mA mM−1) and lower detection limit (0.22 μM) over reported works.
1. Introduction
Construction of materials with tailored nanostructures has brought a great momentum to energy conversion/storage systems, environmental monitoring devices and biomedical applications.1–5 In particular, the non-siliceous porous-structured materials have been used to prepare functional electrodes for high energy conversion/storage devices and sensitive biosensors because of their high specific surface area and desired pore distribution.6,7 The porous structure may shorten the diffuse distance for ions/analytes to access the electrode and to promote the storage capacity/bioelectrocatalytic activity.8 Among those porous materials, porous carbon is a very important versatile material due to its high surface area, low cost, chemical inertness and conductive properties.9,10 Nevertheless, a good electrode material should possess not only a large surface area and good conductivity, but also good hydrophilicity and tailored nanostructure. These still remain great scientific challenges in research and development.During the last decade, significant efforts have been devoted to the synthesis of nanostructured porous carbons utilizing the “nanocasting” technique, which involves impregnation of a porous silica template with an appropriate carbon source, carbonization of the carbon precursor, and subsequent removal of the template.11 A highly ordered porous carbon was synthesized by carbonization of the impregnated sucrose inside the pore channels of a mesoporous silica template at high temperature (>800 °C) followed by removal of the silica framework.12 To date, various porous silica templates and carbon precursors including furfuryl alcohol, phenol, and aromatic hydrocarbons have been developed to fabricate porous carbons.13–15 However, the high carbonization temperature usually leads to products containing limited oxygen-based functional groups and then results in hydrophobicity from loss of oxygen-based functionality (e.g., alcohols). The increased hydrophobicity prohibits the ions/analytes to access the electrode surface, resulting in poor device performance, in particular in aqueous solution based systems.9 Recently, in order to enhance the hydrophilicity of the carbon materials, a number of methods have been explored by modifying the carbon surface, such as oxidation of carbons with nitric acids or ozone to produce some oxygenated functionalities. However, these methods could decrease the surface reactivity and damage the nanostructure of the carbons during the oxidation treatment.16,17 It is a great challenge to develop an efficient approach to fabricate porous carbon with tailored nanostructure and good hydrophilicity.
We report here a microwave irradiation approach to prepare a novel hydrophilic porous carbon with tailored nanostructure. Compared with conventional heating methods, the microwave heating can deliver energy to the materials through molecular interactions with an electromagnetic field, resulting in uniform, rapid and volumetric heating without sacrificing the quality of the product.18,19 The structure and hydrophilicity of the novel porous carbon are characterized by electron microscopy, Raman, X-ray diffraction, nitrogen adsorption–desorption isotherms and contact angle measurement. The as-prepared carbon is also used as an electrode material to construct an enzymatic biosensor for hydrogen peroxide detection.
2. Experimental procedures
2.1 Synthesis of novel porous carbon
The new mesoporous carbon was synthesized by using polyethylene glycol (PEG 400, liquid form), with a large amount of hydrophilic groups, as the carbon precursor and porous silica SBA-15 as the template under microwave irradiation. The process could be described as follows: 1 g SBA-15 silica was first dispersed into 10 mL PEG under ultrasonic condition for 1 h, and then PEG was infiltrated into the channels of SBA-15 silica adequately in a vacuum of 0.1 MPa for 2 h. The obtained mixture was stored under an air atmosphere overnight for silica depositing. After removal of the redundant PEG, a viscous gel was obtained. 1 mL concentrated H2SO4 (98%) was dropped into the viscous gel under vigorous stirring. The obtained precursor was placed in a drying oven for 12 h at 150 °C. The carbonization was completed in a microwave oven (LG, MS-2744B) using graphite as the heat-conducting layer for different times. The carbonization was carried out in a procedure of 5 min on and 1 min off and 5 min on again. After 15 and 30 min microwave treatment, a black and a grey sample was obtained, respectively. When the time further increased to 45 min and longer, the product became white, completely indicating that carbon had totally disappeared and only silica template was left. Finally the carbon–silica composite was washed with 5 wt% hydrofluoric acid at room temperature to remove the silica template. For comparison, conventional mesoporous carbon was prepared using the same chemicals and preparation procedures as those for the hydrophilic porous carbon, but was pyrolized using a furnace at 900 °C under N2 protection for 3 h.2.2 Material characterizations
The crystal structure of the product was characterized by X-ray diffraction (XRD, Bruker). Morphology and microstructure of the synthesized materials were investigated by field emission scanning electron microscopy (FESEM, JSM-6700F, Japan) and high-resolution transmission electron microscopy (HRTEM, JEM-2100F, Japan). Nitrogen adsorption–desorption experiments were carried out at 77.3 K by means of an Autosorb-1 (Quantachrome Instruments) analyzer. The surface area was calculated by using the Brunauer–Emmett–Teller (BET) equation. Pore-size distributions were calculated by the Barrett–Joyner–Halenda (BJH) method. The Raman study of the samples was carried out by a setup with an integrated confocal Raman microscope system (CRM200, WITec, Germany). The excitation wavelength is 632.8 nm, and the highest grating was adopted to reach a resolution of less than 2 cm−1.2.3 Biosensor preparation and testing
H2O2, a reactive oxygen species produced in various physiological processes is considered as a biochemical mediator in cellular pathology and can be used as an early indicator for cytotoxic events and physiological disorders.5 Therefore, a sensitive and selective H2O2 sensor is essential to understand its role in cellular physiology and also to provide reliable diagnosis of pathological conditions. Enzymatic biosensors possess high selectivity.20,21Hb, a heme-protein has specific catalytic activity towards H2O2 and is often used to construct H2O2 biosensors for high signal to noise ratio.31 To prepare the Hb based biosensor, glassy carbon electrodes of 3 mm in diameter (GCE, CH Instruments, USA) were polished with 0.3 and 0.05 μm alumina powder, followed by thorough rinsing with deionized water. After sonicating in 1 M nitric acid, acetone and deionized water, respectively, the electrodes were dried at room temperature. 10 mg new mesoporous carbon was dispersed into 2 mL double-distilled water under ultrasonic conditions for 2 h. Then, 1 mL upper solution after depositing for 1 h was transferred into a fresh tube, followed by adding 10 mg haemoglobin (Hb) under ambient conditions and shaken 30 min. The mixture obtained was finally stored at 4 °C for protein adsorption. Before each experiment, the bioconjugates obtained were re-shaken, and then 2 μL of the suspension was deposited onto the centre of the pre-treated GCE. 3 μL of 0.5% Nafion was subsequently placed on the whole electrode surface to form a Nafion membrane, which was used to fix Hb impregnated materials on to the electrode. Then, the modified electrode was left to dry at room temperature. The amount of Hb adsorption was determined by UV absorption at 407 nm using a Hitachi U2800 spectrophotometer. About 31% protein was adsorbed on the mesoporous carbon.The electrochemical measurements were carried out using a three-electrode system, employing a platinum wire as the counter electrode, a saturated calomel electrode (SCE) as the reference, and the new mesoporous carbon modified glassy carbon electrode (GCE) as the working electrode. Three trials were performed to obtain a calibration curve of the sensor.
3. Results and discussion
3.1 Materials characterization
The structural ordering of the as-synthesized carbon materials obtained under different carbonization conditions was first evaluated by XRD. The XRD pattern of the material prepared under microwave treatment for 15 min (Fig. 1A) displays a weak peak of (100) and a broad peak at 2θ ranging from 1.7 to 1.98°, which is due to the overlap of two additional peaks of (110) and (200), indicating a ordered 2-d hexagonal structure.22 With microwave treatment for 30 min, the intensity of the three well-defined peaks in the low-angle region increases significantly, which is very similar to that of the carbon material synthesized by conventional synthesis. This suggests that a well ordered mesoporous structure of the sample similar to the carbon material fabricated by conventional synthesis was obtained. The high-angle XRD patterns of the samples exhibit two peaks, possibly associated with the graphite-like structure, i.e., (002) and (101) planes. The (002) peak of the carbons synthesized by different carbonization conditions should correspond to the carbon interlayer distance.23 The d002 value for the carbon obtained by microwave treatment 30 min is very close to that of the carbon prepared by conventional synthesis. This result indicates that the microwave method used in this work can reduce the reaction time significantly without sacrificing the quality of the products.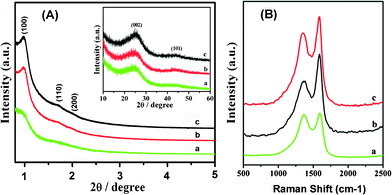 | ||
| Fig. 1 (A) XRD patterns for as-synthesized carbon materials. The inset shows the large-angle XRD patterns of the samples. (B) Raman spectra of as-synthesized carbon materials. The excitation wavelength is 632.8 nm (a, Carbon obtained by microwave treatment for 15 min; b, Carbon obtained by microwave treatment for 30 min; c, Carbon obtained by conventional synthesis). | ||
Representative Raman spectra (Fig. 1B) of the carbons prepared with different carbonization conditions exhibit two bands at ∼1357 cm−1 (D band) and ∼1587 cm−1 (G band). The peak at 1587 cm−1 corresponds to an E2g mode of the hexagonal graphite and is related to the vibration of sp2-bonded carbon atoms in a 2-dimensional lattice, such as in a graphite layer. The D-band at about 1357 cm−1 is associated with vibrations of carbon atoms with dangling bonds in plane terminations of disordered graphite or glassy carbons.24 According to the Raman spectra results, the carbons are not fully graphitized under the three different carbonization conditions. Some disordered carbon or defects in the samples are illustrated by the result. The ID/IG ratios of the carbons prepared by conventional synthesis, microwave treatment for 30 min and 15 min are 0.81, 0.65, and 0.98, respectively. As reported in literature, the ratio of the amplitude of the D- and G-bands is related to the disorder degree of the carbon materials.19 This result shows that more disordered carbon is present in the bulk sample synthesized with microwave treatment for 15 min, which is in accordance with the XRD results. The sample prepared by the microwave treatment for 30 min is more ordered than other two samples, further demonstrating that the microwave treatment is a good carbonization method for the synthesis of a porous carbon.
The effect of carbonization condition on the microstructure and morphology of the synthesized material was studied by FESEM. SBA-15 silica, the template used in the experiments, illustrates rope-like morphologies with a relatively uniform size (Fig. 2A). Its high-resolution SEM image reveals that the external surface of the SBA-15 silica is composed by a uniform array of parallel channels (Fig. 2B). The SEM images of the carbon prepared by conventional synthesis with SBA-15 silica template exhibit very similar morphologies to those of the template (Fig. 2C and 2D). However, the sample obtained by the microwave treatment for 15 min displays a very different morphology that consisted of amorphous flakes, most likely due to a lower growth rate (Fig. 2E and 2F). Interestingly, when the microwave treatment time increases to 30 min, the sample tends to form loose and porous bundles (Fig. 2G) composed of very thin paralleled nanowires (Fig. 2H).
 | ||
| Fig. 2 Low and high magnification FESEM of the samples. (A and B, SBA-15; C and D, Carbon prepared by conventional synthesis; E and F, Carbon obtained by microwave treatment for 15 min; G and H, Carbon obtained by microwave treatment for 30 min.) | ||
The detailed structure of the mesoporous carbon fabricated under different carbonization conditions was investigated by TEM and high magnification FESEM. The representative TEM images of the carbon materials synthesized under different heating methods are shown in Fig. 3. As shown in Fig. 3B and 3E, both the samples have well-ordered mesoporous structures. The relatively bright lines are the images of nanopores of the carbon, while the black lines are carbon wires filled in the pores of SBA-15 (Fig. 3B, 3E and 3F). However, the samples fabricated by different heating methods also present significantly different microstructures (Fig. 3A and 3C); the surface of the carbon produced by the microwave treatment for 30 min is more slack and porous than the material prepared by the conventional synthesis. The high-magnification TEM further illustrates that the slack surface of the carbon formed by the microwave treatment for 30 min is composed of uniformly distributed mesostructured cells with a diameter of ∼10 nm (Fig. 3D).
 | ||
| Fig. 3 TEM (A and B) images of carbon obtained by conventional synthesis; TEM (C, D and E) and FESEM (F) images of carbon obtained by microwave treatment for 30 min. | ||
In Fig. 4, the nitrogen adsorption–desorption isotherms and pore-size distribution measured from different mesoporous carbons are compared with those of SBA-15 silica. The ordered SBA-15 silica shows type IV with type H1 hysteresis loops for a typical mesoporous and well-organized hexagonal structure with amorphous pore walls and narrow pore size distributions centered at 6.3 nm. The specific surface area for silica SBA-15 is 821 m2 g−1, and little micropores are present in its pore structure. The structure of the silica template can determine the primary structure of the resulting mesoporous carbon because the space occupied by the silica framework forms the mesopores after the removal of the silica template. The nitrogen adsorption–desorption isotherm for the mesoporous carbon prepared by the conventional synthesis in Fig. 4A(II) displays a reverse replica of the SBA-15 ordered silica with hexagonal structure, which is analogous to the ordered mesoporous carbon reported.22 A comparison of two carbon materials prepared under different microwave treatment times shows that the adsorption capacity increases and the position of the capillary condensation step shifts to relatively higher pressures as the microwave treatment time increases. The BET surface area of the carbon prepared by the microwave treatment for 30 min is up to 1040 m2 g−1, which is much larger than that of the carbon synthesized under a shorter time of 15 min (775 m2 g−1), and also the carbon synthesized by the conventional method (884 m2 g−1), demonstrating that this simple microwave approach can maintain the microstructure, high surface area and uniform pore size distribution in the as-prepared carbons and even much smaller pores (< 3 nm) in the carbon when using microwave treatment for 30 min (Fig. 4B(III)).
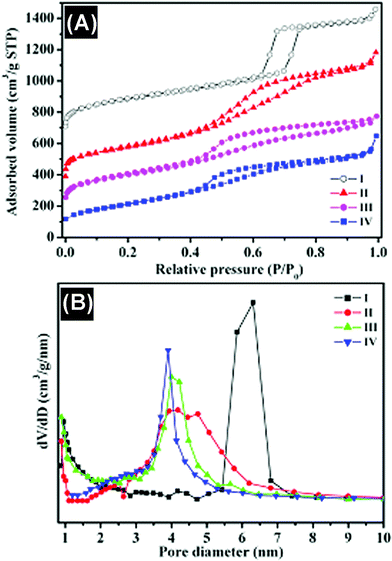 | ||
| Fig. 4 (A) Nitrogen adsorption–desorption isotherms and (B) BJH pore-size distributions of different samples. I, SBA-15 silica; II, carbon prepared by conventional synthesis; III, carbon obtained by microwave treatment for 15 min; IV, carbon obtained by microwave treatment for 30 min. | ||
The hydrophilicity of the carbons fabricated was evaluated by water contact angle measurements. The water contact angles of the carbons obtained by the microwave treatments for 15, 30 min and conventional approach were 9.3°, 23° and 38.6°, respectively, which were much smaller than 79.1° observed for the mesoporous carbon material reported.9 This result clearly demonstrates that the hydrophilicity can be tailored by the microwave treatment time. This could be explained by FTIR spectra of the samples shown in Fig. 5. The broad absorption bands centered at 3450 and 1620 cm−1 correspond to the stretching and bending modes of the OH group, and the absorption bands at 1234 and 1385 cm−1 are assigned to the stretching and bending vibrations of C–OH. The results indicate that the hydroxyl group remains in the prepared carbons when using polyethylene glycol as the precursor, thus leading to improved hydrophilicity. In contrast, the high carbonization temperature in the conventional synthesis usually causes significant loss of oxygen-based functional groups for poor hydrophobicity. In addition, FTIR spectrum for carbon obtained by microwave treatment for more than 45 min shows no carbon-related characteristic peaks (Inset of Fig. 5), further confirming that only silica is left.
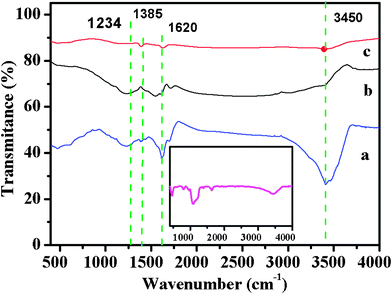 | ||
| Fig. 5 FTIR spectra of the carbons obtained by different carbonization conditions (a, Carbon obtained by microwave treatment for 15 min; b, carbon obtained by microwave treatment for 30 min; c, carbon prepared by conventional synthesis). The inset is FTIR spectrum of material obtained by microwave treatment for more than 45 min. | ||
3.2 Biosensing
It was recently reported that the nanopore could be a stoichiometric electron acceptor and host for a variety of electron-donating guest species, and thus is favorable to a wide variety of reactions involving electron transfer to a non-siliceous mesoporous host lattice.25–29 Hence, the unique structure could be used to immobilize proteins for direct electrochemistry. The cyclic voltammogram (CV) in Fig. 6a shows that Hb has direct electrochemistry behavior on mesoporous carbons; however, the microwave derived mesoporous carbon-impregnated Hb exhibits a greater reversible redox reaction with higher peak current than that of the conventional mesoporous carbon, indicating that the former has a much stronger direct electrochemistry of Hb. The cyclic voltammograms in Fig. 6b show no obvious redox peaks for both Hb and mesoporous carbon alone modified electrodes (curves 1 and 2), but a pair of well-defined redox peaks for the Hb-mesoporous carbon modified glass carbon (GC) electrode (curve 3), demonstrating the direct electron transfer ability. The anodic peak potential (Epa) and the cathodic peak potential (Epc) are located at −0.323 V and −0.371 V, respectively, corresponding to a peak-to-peak separation (ΔEp) of 48 mV. The small value of ΔEp indicates a fast electron-transfer rate for Hb-mesoporous carbon GCE.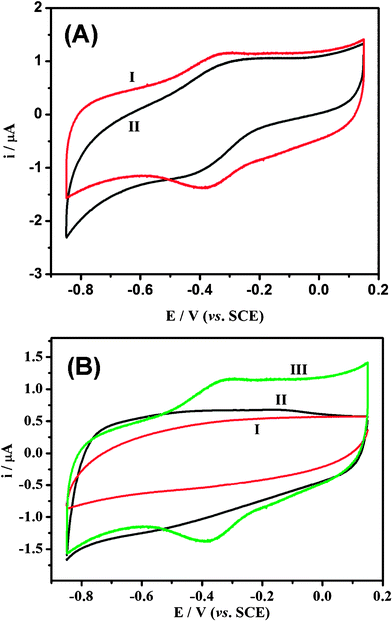 | ||
| Fig. 6 Cyclic voltammograms of different materials: modified GCEs in 0.01 M pH 7.4 nitrogen-saturated phosphate buffered saline (PBS) solution at 100 mV s−1. (GCE: glass carbon electrode; SCE: saturated calomel electrode.) ((A) Hb-mesoporous carbon, 1: mesoporous carbon obtained by microwave; 2: mesoporous carbon prepared by conventional synthesis. (B) 1, Hb/Nafion; 2, mesoporous carbon/Nafion; 3, mesoporous carbon/Hb/Nafion.) | ||
The effect of the scan rate on the electrochemical response of the Hb-mesoporous carbon modified electrode is shown in Fig. 7. Within the scan rate range 100–800 mV s−1, the reduction and oxidation peak currents exhibit a linear relationship against the scan rate (shown in Fig. 8b with r = 0.996). The charge consumed in coulombs, Q (obtained from integrating the anodic or cathodic peak area in CVs), with the background correction, is invariable at different scan rates. These results confirm that the electrochemical reaction of Hb on the mesoporous carbon exhibits a surface-controlled behavior, in which all electroactive ferric proteins (protein-FeIII) produced by the oxidation of ferrous proteins (protein-FeII) on the anodic scan could be reduced to protein-FeII on the cathodic scan. The surface concentration of the electroactive Hb (Γ*) in the mesoporous carbon could be calculated by integrating CV reduction peak based on the formula of Q = nFAΓ*, where A is the effective area of GC electrode (0.07 cm2) and other symbols have their known usual meanings. The average Γ* value over the scan rate range is 7 × 10−10 mol cm−2. The measured percentage Hb adsorption of the mesoporous carbon is 31 wt%. Thus, about 21.5% Hb immobilized on the mesoporous carbon remains its electrochemical activity, indicating that the direct electrochemistry only occurs on a part of immobilized proteins.
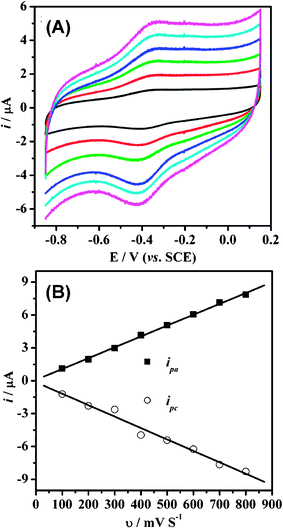 | ||
| Fig. 7 (A) Cyclic voltammograms of Hb-mesoporous carbon modified GC electrode in a 0.01 M PBS buffer (pH 7.4) at different scan rates (from 100 to 600 mV s−1, inside to outside). (B) The plots of the reduction and oxidation peak currents i against the scan rate v. | ||
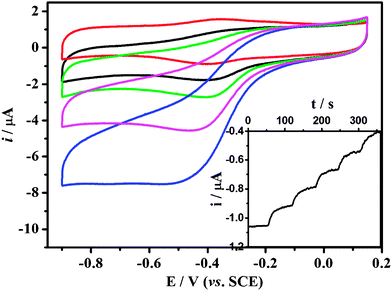 | ||
| Fig. 8 Cyclic voltammograms of Hb-mesoporous carbon modified GC electrode in a 0.01 M PBS buffer (pH 7.4) containing different H2O2 concentrations at 100 mV s−1. (0 μM, 6.7 μM, 13 μM, 27 μM, 53 μM; from top to bottom.) The inset is a typical steady state response of the biosensor on successive injection of Hb solution into 0.01 M PBS while stirring, with an applied potential of −0.4 V. | ||
The apparent heterogeneous electron transfer rate constant ks of Hb in the mesoporous carbon modified electrode could be estimated by using the equation derived by Laviron for diffusionless CV.30 The calculated value of ks, 5.3 s−1 indicates that the Hb at the mesoporous carbon modified electrode has a faster electron transfer rate than that observed from Hb immobilized on black carbon (1.02 s−1), CNTs (0.062 s−1), PANI/mesoporous carbon (2.07 s−1), and also our previous work.31–35
Based on its direct electrochemistry behavior, the Hb-mesoporous carbon modified electrode was employed to electrocatalyze H2O2 reduction for a sensitive biosensor. The catalytic reduction peak of H2O2 at Hb-mesoporous carbon modified electrode is shown in Fig. 8, in which the half-wave potential of H2O2 reduction is very close to the redox potential of the reversible electrochemical reaction for the direct electron transfer between Hb and electrode. After addition of H2O2 in the test solution, the reduction peak current of Hb increases, accompanied by the decrease and even disappearance of the oxidation peak, indicating the reduction of H2O2 is catalyzed by the Hb immobilized in the mesoporous carbon. The amperometric response of the Hb-mesoporous carbon based sensor with successive additions of H2O2 solution at an applied potential of −0.40 V is also investigated (inset of Fig. 9). Upon addition of an aliquot of H2O2 to the electrochemical cell, the reduction current increases rapidly to reach a stable value, showing a very fast electrocatalytic response. Two control experiments using Hb modified GCE and mesoporous carbon modified GCE to sense H2O2 were also carried out. The results show that mesoporous carbon modified GCE cannot catalyze H2O2 reduction in the same potential window as the Hb-modified electrode and Hb modified GCE has much lower current response compared with that of Hb-mesoporous carbon modified GCE, clearly indicating that the mesoporous carbon can significantly increase the sensitivity of a Hb-based H2O2 biosensor. It is noted that the response is dependent on electrode preparation time. The best response is achieved by immersing mesoporous carbon in Hb solution for 3 days and decreases with further increased time.
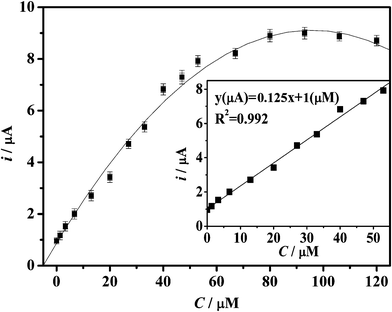 | ||
| Fig. 9 Calibration curve of the reduction peak current against the concentration of H2O2. The inset is the linear region plot. | ||
The relationship between the reduction peak current and the H2O2 concentration is displayed in Fig. 9. The respective peak current changes linearly with the H2O2 concentration (inset) ranging from 1.3 × 10−6 to 4 × 10−5 M (R2 = 0.992), with a reactive sensitivity of 0.15 mA mM−1, which is much superior to most reported works, making it possible to fabricate an extremely sensitive H2O2 biosensor. The detection limit is 0.22 μM. The performance is much superior than that of reported works.31–35 In addition, the linear detection range (1.3 × 10−6 to 4 × 10−5 M) of our sensor is important because it covers the H2O2 concentrations in most cellular physiological and pathological conditions.5 When the concentration of H2O2 is higher than 7.7 × 10−6 M, the calibration curve tends to plateau and then drops down with further addition H2O2, implying a progressive enzyme inactivation in the presence of higher concentrations of H2O2 that coincides with a Michaelis–Menten kinetics model.35 During 2 weeks of storage and measurements, no desorbed Hb was detected by UV-Vis, which should be attributed to the suitable porosity from novel carbon and/or a good barrier from the Nafion membrane. The stability is also critical for biosensors.36,37 The stability of the Hb-mesoporous carbon based biosensor was studied. To investigate the stability, the sensor was measured every three days and stored at 4 °C. The current response for H2O2 reduction decreased by only 8.5% of the original value after two weeks. In addition, there was no response change after 3 h of continuous operation in 0.3 mM H2O2, showing good stability. The results of 5 successive measurements (the plot with error bars not shown) show a relative standard deviation of 5.1% and the relative standard deviation of the current responses over 4 sensors prepared with the same procedure is less than 8.0%.
4. Conclusions
In conclusion, we report a microwave irradiation approach to prepare a novel porous carbon material with a large specific surface area up to 1040 m2 g−1, uniform pore size distribution and good hydrophilicity. The nanostructure can be tailored by simply changing the microwave treatment time. The porous carbon is further immobilized with the protein Hb for a hydrogen peroxide biosensor. The immobilized Hb exhibits much higher direct electron transfer rate of 5.3 s−1, resulting in much higher sensitivity (0.15 mA mM−1) and lower detection limit (0.22 μM) for a biosensor towards hydrogen peroxide detection than the reported works. This study not only demonstrates a suitable platform for sensing applications, but also provides an efficient method to fabricate carbon materials with good hydrophilicity and tailored nanostructure.Acknowledgements
We acknowledge the financial support from Institute for Clean Energy & Advanced Materials, Southwest University, Chongqing, P.R. China, the Center for Advanced Bionanosystems, Nanyang Technological University, and National Natural Science Foundation of China (No. 20963011 and 21063014).References
- S. J. Bao, C. M. Li, C. X. Guo and Y. Qiao, J. Power Sources, 2008, 180, 676 CrossRef CAS.
- C. X. Guo, Y. Lei and C. M. Li, Electroanalysis, 2011, 23, 885 CrossRef CAS.
- C. X. Guo, H. B. Yang, Z. M. Sheng, Z. S. Lu, Q. L. Song and C. M. Li, Angew. Chem., Int. Ed., 2010, 49, 3014 CrossRef CAS.
- C. X. Guo, X. T. Zheng, S. R. Ng, Y. C. Lai, Y. Lei and C. M. Li, Chem. Commun., 2011, 47, 2652 RSC.
- C. X. Guo, X. T. Zheng, Z. S. Lu, X. W. Lou and C. M. Li, Adv. Mater., 2010, 22, 5164 CrossRef CAS.
- S. J. Bao, C. M. Li, J. F. Zang, X. Q. Cui, Y. Qiao and J. Guo, Adv. Funct. Mater., 2008, 18, 591 CrossRef CAS.
- C. X. Guo, F. P. Hu, X. W. Lou and C. M. Li, J. Power Sources, 2010, 195, 4090 CrossRef CAS.
- W. Sun, C. X. Guo, Z. H. Zhu and C. M. Li, Electrochem. Commun., 2009, 11, 2105 CrossRef CAS.
- S. Wu, H. X. Ju and Y. Liu, Adv. Funct. Mater., 2007, 17, 585 CrossRef CAS.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073 CrossRef CAS.
- Y. Wan and D. Y. Zhao, Chem. Rev., 2007, 107, 2821 CrossRef CAS.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743 CrossRef CAS.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169 CrossRef CAS.
- R. Ryoo, I. S. Park, S. Jun, C. W. Lee, M. Kruk and M. Jaroniec, J. Am. Chem. Soc., 2001, 123, 1650 CrossRef CAS.
- J. Lee, K. Sohn and T. Hyeon, J. Am. Chem. Soc., 2001, 123, 5146 CrossRef CAS.
- Z. J. Li, W. F. Yan and S. Dai, Langmuir, 2005, 21, 11999 CrossRef CAS.
- J. L. Bahr and J. M. Tour, J. Mater. Chem., 2002, 12, 1952 RSC.
- E. H. Hong, K. H. Lee, S. H. Oh and C. G. Park, Adv. Funct. Mater., 2003, 13, 961 CrossRef CAS.
- L. Dennany, P. sherrell, J. Chen, P. C. Innis, G. G. Wallace and A. I. Minett, Phys. Chem. Chem. Phys., 2010, 12, 4135 RSC.
- D. Mackey, A. J. Killard, A. Ambrosi and M. R. Smyth, Sens. Actuators, B, 2007, 122, 395 CrossRef.
- X. L. Luo, A. Morrin, A. J. Killard and M. R. Smyth, Electroanalysis, 2006, 18, 319 CrossRef CAS.
- Z. L. Li, J. Zhang, Y. Li, Y. J. Guan, Z. C. Feng and C. Li, J. Mater. Chem., 2006, 16, 1350 RSC.
- T. W. Kim, R. Ryoo, K. P. Gierszal, M. Jaroniec, L. A. Solovyov, Y. Sakamoto and O. Terasaki, J. Mater. Chem., 2005, 15, 1560 RSC.
- Z. Y. Sun, Z. M. Liu, J. M. Du, Y. Wang, B. X. Han and T. C. Mu, J. Phys. Chem. B, 2004, 108, 9811 CrossRef CAS.
- S. Sotiropoulou, V. Vamvakaki and N. A. Chaniotakis, Biosens. Bioelectron., 2005, 20, 1674 CrossRef CAS.
- L. M. Bonanno, T. C. Kwong and L. A. DeLouise, Anal. Chem., 2010, 82, 9711 CrossRef CAS.
- N. Massad-Ivanir, G. Shtenberg, A. Tzur, M. A. Krepker and E. Segal, Anal. Chem., 2011, 83, 3282 CAS.
- N. Massad-Ivanir, G. Shtenberg, T. Zeidman and E. Segal, Adv. Funct. Mater., 2010, 20, 2269 CrossRef CAS.
- B. Sciacca, E. Secret, S. Pace, P. Gonzalez, F. Geobaldo, F. Quignard and F. Cunin, J. Mater. Chem., 2011, 21, 2294 RSC.
- C. X. Guo and C. M. Li, Phys. Chem. Chem. Phys., 2010, 12, 12153 RSC.
- C. X. Guo, F. P. Hu, C. M. Li and P. K. Shen, Biosens. Bioelectron., 2008, 24, 819 CrossRef CAS.
- Q. Lu and C. M. Li, Biosens. Bioelectron., 2008, 24, 767 CrossRef CAS.
- G. X. Ma, T. H. Lu and Y. Y. Xia, Bioelectrochemistry, 2007, 71, 180 CrossRef CAS.
- Y. D. Zhao, Y. H. Bi, W. D. Zhang and Q. M. Luo, Talanta, 2005, 65, 489 CrossRef CAS.
- G. X. Ma, Y. G. Wang, C. X. Wang, T. H. Lu and Y. Y. Xia, Electrochim. Acta, 2008, 53, 4748 CrossRef CAS.
- C. M. Li, C. Q. Sun, S. Song, V. E. Choong, G. Maracas and X. J. Zhang, Front. Biosci., 2005, 10, 180 CrossRef CAS.
- C. M. Li and C. S. Cha, Front. Biosci., 2004, 9, 3324 CrossRef CAS.
Footnote |
| † Equal contribution to this work |
| This journal is © The Royal Society of Chemistry 2012 |