Solvent-assisted thermal annealing of disulfonated poly(arylene ether sulfone) random copolymers for low humidity polymer electrolyte membrane fuel cells
Chang Hyun
Lee
a,
Kwan-Soo
Lee
a,
Ozma
Lane
a,
James E.
McGrath
*a,
Ying
Chen
b,
Sungsool
Wi
b,
So Young
Lee
cd and
Young Moo
Lee
cd
aMacromolecules and Interfaces Institute, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA. E-mail: jmcgrath@vt.edu; Fax: +1 540-231-8517; Tel: +1 540-231-5976
bDepartment of Chemistry, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA
cWCU Department of Energy Engineering, College of Engineering, Hanyang University, Seoul 133-701, Republic of Korea
dSchool of Chemical Engineering, College of Engineering, Hanyang University, Seoul 133-791, Republic of Korea
First published on 30th November 2011
Abstract
Fundamental studies of a new thermal annealing strategy for maximizing proton conductivity and single cell performance of glassy sulfonated hydrocarbon fuel cell membrane materials under both fully and partially hydrated conditions are reported. Directly copolymerized disulfonated poly(arylene ether sulfone) random copolymers (BisA-XX, XX is the mole percent of hydrophilic moieties, XX = 20 to 40) with 2,2′-isopropylidene diphenol swivel ((CH3)2–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) units in their polymer backbone were cast from N,N-dimethylacetamide (DMAc) and dried under two different protocols; one set was dried at 60 °C (BisA-XX_60 °C), and the other at 150 °C under vacuum immediately after the initial drying (BisA-XX_150 °C). Small amounts of DMAc solvent remained during the second drying step which depressed the glass transition temperatures (Tg) of the BisA copolymers lower than 150 °C. BisA-XX_150 °C samples were essentially thermally annealed when dried at 150 °C. This increased the density of BisA copolymer chains; their Tg values were increased. Moreover, T1 values of the protons in BisA-XX aromatic phenylene rings measured via solid-state NMR technique were lower as a result of the improved 1H–1H dipolar interaction. Interestingly, proton conductivity was improved after thermal annealing, possibly because the sulfonic acid density in the fully hydrated state (i.e., IECv(wet)) was enhanced. The synergistic effect of reduced water uptake and more developed hydrophilic–hydrophobic nanochannel formation may be important. The thermal annealing significantly influenced proton conductivity particularly at a low humidity (30 to 80% relative humidity (RH)). As a result, BisA-XX_150 °C samples derived from identical materials exhibited electrochemical polymer electrolyte membrane fuel cell (PEMFC) performances similar and superior to Nafion® 112 and BisA-XX_60 °C samples measured at 65% RH at 80 °C, respectively.
) units in their polymer backbone were cast from N,N-dimethylacetamide (DMAc) and dried under two different protocols; one set was dried at 60 °C (BisA-XX_60 °C), and the other at 150 °C under vacuum immediately after the initial drying (BisA-XX_150 °C). Small amounts of DMAc solvent remained during the second drying step which depressed the glass transition temperatures (Tg) of the BisA copolymers lower than 150 °C. BisA-XX_150 °C samples were essentially thermally annealed when dried at 150 °C. This increased the density of BisA copolymer chains; their Tg values were increased. Moreover, T1 values of the protons in BisA-XX aromatic phenylene rings measured via solid-state NMR technique were lower as a result of the improved 1H–1H dipolar interaction. Interestingly, proton conductivity was improved after thermal annealing, possibly because the sulfonic acid density in the fully hydrated state (i.e., IECv(wet)) was enhanced. The synergistic effect of reduced water uptake and more developed hydrophilic–hydrophobic nanochannel formation may be important. The thermal annealing significantly influenced proton conductivity particularly at a low humidity (30 to 80% relative humidity (RH)). As a result, BisA-XX_150 °C samples derived from identical materials exhibited electrochemical polymer electrolyte membrane fuel cell (PEMFC) performances similar and superior to Nafion® 112 and BisA-XX_60 °C samples measured at 65% RH at 80 °C, respectively.
Introduction
Polymer electrolyte membrane fuel cells (PEMFCs) are eco-friendly alternative power sources with high energy efficiency, fuel flexibility (e.g., hydrogen, and methanol) and zero pollutant emission.1 In particular, PEMFCs operated effectively at a relatively low humidity, exhibit additional advantages such as easy water management and simple system design due to no need for humidifiers.2 However, proton transport is severely restricted in polymer electrolyte membranes (PEMs) as well as electrodes,3 since protons are transported as complexes with water molecules (i.e., H3O+, H5O2+, and H9O4+), either through hydrophilic channels formed by fixed charged anions (e.g., –SO3− and –COO−) in polymer main chains, and/or hopping via hydrogen bonding with proton acceptable water media.4–7 Several approaches compensate for this reduced proton conductivity in the dehydrated PEMs. The approaches include the fabrication of inorganic acid-doped polymer membranes,8–10 multiblock copolymer membranes,10–14 and organic–inorganic composite membranes15–19 as well as membrane surface treatments.20,21 These successful low-humidity membrane studies were accomplished by incorporating proton-conducting inorganic acids8–10 and/or inorganic fillers,15–19 or by improving the water retention level within polymer matrices via well-defined hydrophilic–hydrophobic phase-separated morphology formation10–14 and strengthened hydrogen bonding with water molecules.10–14,18,20,21Appropriately controlling membrane preparation conditions may also enhance the physicochemical properties (e.g., proton conductivity) of polymer electrolyte membranes, particularly those made up of sulfonated hydrocarbon materials. Most of sulfonated hydrocarbon materials used for PEMs are amorphous glassy polymers in the non-equilibrium state. Thus, the thermal history during membrane preparation may influence the packing density of their polymer chains and may change the hydrophilic and hydrophobic domain characteristics, such as size and phase separated morphologies.22
Thermal annealing is a simple route for stabilizing glassy polymers via the densification of their polymer chains and is accomplished through thermal treatment of the glassy thermoplastics. This has been widely used in applications such as aerospace,23 gas separation,24,25 solar cells,26,27 and light emitting diodes28 to improve resistance to vapor sorption/gas permeation23–25 or to develop better nanoscale phase separation.26–28 In fuel cell membrane applications, some positive contributions of thermal annealing have been reported.29–31 For example, thermal annealing of post-sulfonated random poly(arylene ether sulfone) copolymer (Fig. 1(a)) membranes, which were prepared via direct sulfonation of copolymers, resulted in reduced methanol permeability as well as improved dimensional stability, when the membranes were used as PEMs for direct methanol fuel cell applications.29 On the other hand, their proton conductivities were reduced after the thermal treatment. Also, undesirable side-reactions (e.g., crosslinking, heterogeneous reaction, and chain-scission) occurred during the post-sulfonation,32 which made it difficult to fabricate PEM materials with high sulfonation degrees. In addition to the random copolymer study, thermal treatments around the glass transition temperature (Tg) of hydrophobic blocks of multiblock copolymers led to more developed hydrophilic–hydrophobic nanophase-separated morphologies and improved proton conductivity both in liquid water and at a relatively low humidity.30,31 In those multiblock studies, the thermal treatments were conducted at temperatures higher than about 200 °C in the solvent-free membrane state where polymer chain motion is less free than that under residual solvents. However, the importance of thermal history on the electrochemical performances of glassy hydrocarbon PEMs is not clearly established.
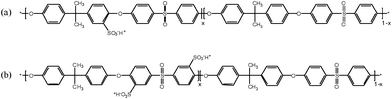 | ||
| Fig. 1 Chemical structures of (a) post-sulfonated poly(arylene ether sulfone) (PAES) and (b) directly copolymerized poly(arylene ether sulfone) random (BisAH-XX) copolymers in proton form (–SO3−H+). Here, x means the molar ratio of hydrophilic moieties. | ||
This paper reports a new strategy to maximize the proton conductivity of sulfonated polymer electrolyte membranes and thus to improve electrochemical single cell performance via a simple thermal treatment at relatively low temperatures. The objective is to investigate the changes of fundamental film characteristics, including morphological transformation, after a thermal treatment of sulfonated copolymers, particularly with directly copolymerized poly(arylene ether sulfone) random copolymers. We employed solid-state NMR, which is sensitive enough to monitor infinitesimal changes in the polymer system environment. The goal of this study was also to verify the electrochemical efficacy of the thermal annealing approach, particularly under partially hydrated PEMFC conditions.
Experimental
Materials
A series of directly copolymerized disulfonated poly(arylene ether sulfone) copolymers containing 2,2′-isopropylidene diphenol units (BisA-XX, XX mean mole percent of hydrophilic moieties) (Fig. 1(b)), were initially synthesized in potassium salt form (–SO3−K+) via a step or polycondensation reaction of 4,4′-dichlorodiphenyl-sulfone (DCDPS, Solvay Advanced Polymers), 3,3′-disulfonate-4,4-dichlorodiphenylsulfone (SDCDPS, Akron Polymer Systems) and 2,2′-isopropylidene diphenol (Solvay Advanced Polymers) monomers following published procedures.33–36 The SDCDPS was prepared as earlier reported.36,37 The resulting BisA-XX copolymers were converted into proton form BisAH-XX copolymers in the membrane state following Method II, which will be described in Membrane fabrication. The sulfonation degrees (DS) of BisA-XX copolymers were controlled from 20 to 40 mol% (x = 0.2 to 0.4) by varying the ratio of SDCDPS and DCDPS. Basic information on BisA-XX copolymers is summarized in Table 1. N,N-Dimethyl acetamide (DMAc) (Aldrich Chemical Co.) was used as a casting solvent without purification.| Polymer materials | Target DS | Measured DSa | Intrinsic viscosityb | Number average molecular weight (Mn)c |
|---|---|---|---|---|
| [mol%] | [mol%] | [—] | [Da] | |
| a Calculated from the relative 1H NMR integrals of protons between the aromatic resonances. b Measured at 30 °C in NMP with 0.05 M LiBr. c Determined by GPC using NMP with 0.05 M LiBr.38 | ||||
| BisA-20 | 20.0 | 20.0 | 1.10 | 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
| BisA-25 | 25.0 | 25.0 | 0.81 | 34000 |
| BisA-30 | 30.0 | 29.0 | 1.06 | 46000 |
| BisA-35 | 35.0 | 34.0 | 0.83 | 41000 |
| BisA-40 | 40.0 | 39.9 | 1.06 | 38700 |
Membrane fabrication
BisA-20 (2 g) was completely dissolved in 18 g of DMAc at 30 °C (10 wt% concentration). The solution was filtered using 0.45 μm of PTFE syringe filter, degassed under vacuum at 25 °C for at least 1 day and cast on a clean glass plate. Then, the cast solution was dried under two different thermal protocols; one set was dried at 60 °C in an oven without vacuum for 2 days (BisA-XX_60 °C), while the other set was thermally treated under vacuum at 150 °C for an extra 2 days after initially being dried at 60 °C for 2 days (BisA-XX_150 °C). The nominal thickness of all membranes was approximately 30–40 μm. After the thermal treatments, each set was immersed in deionized water for 1 week to remove the residual solvent. The deionized water was replaced with clean water on a daily basis. After the water treatment, each salt form membrane was converted into a proton form (BisAH-XX) membrane by treating in boiling 0.5 M H2SO4 for 2 h and, then, in boiling deionized water for 2 h (Method II).10,13,33–35Characterization
The amount of residual solvents in the BisA copolymer films fabricated following the thermal drying protocols was investigated using a thermogravimetric analyzer (TGA) (TA instruments Q500 TGA) operated at a heating rate of 10 °C min−1 from 50 to 400 °C in a 60 mL min−1 nitrogen (N2) sweep gas. Prior to the thermal decomposition measurement, all samples were kept in a N2 purged TGA furnace at 110 °C for 15 min to minimize the content of water molecules interacting with –SO3−K+ groups.A TA Instrument DSC Q-1000 was used to determine the glass transition temperature (Tg) of BisA copolymers both in the presence of residual DMAc and under the solvent-free condition. For DMAc-depressed Tg measurement, the membrane samples were placed in thermally sealed high volume DSC pans (TA Instrument) which can withstand pressure up to 600 psi. Then, the samples were heated at a rate of 5 °C min−1 under a N2 atmosphere. The Tg of each sample was obtained from the first scan. For Tg measurement of solvent-free BisA copolymer membranes, all samples were placed in normal DSC pans (TA Instrument). Prior to DSC measurement, those samples were preheated to 350 °C for 5 min under a N2 atmosphere and cooled down to 50 °C. Tg values were obtained from the second and the third scan, whose differences were not higher than 2 °C.
Water uptake (%) of BisAH-XX was calculated from the ratio of the mass of absorbed water molecules (Ww - Wd) to the measured mass of the dry film samples (Wd). Here, Ww is the measured mass of fully hydrated film samples. Each sample (dimension = ∼5 × 5 cm2) was soaked in deionized water at 25 °C for 1 day before measuring Ww. Then, the sample was dried in a vacuum oven at 110 °C for 1 day before measuring Wd.
Atomic force microscopy (AFM) images in the tapping mode were obtained by using a Digital Instruments MultiMode scanning probe microscope with a NanoScope Iva controller. A silicon probe (Vecco, end radius < 10 nm and force constant = 5 Nm−1) was used for morphological images of the film surface. The set-point ratio was 0.82. All samples were kept in an oven set at 30 °C and 40% relative humidity (RH) for at least 12 h.
Cross-polarization magic-angle spinning (CPMAS) 13C NMR spectra were performed with a Bruker Avance II 300 MHz wide-bore spectrometer operating at Larmor frequencies of 75.47 MHz for 13C and 300.13 MHz for 1H nuclei. Samples (50–60 mg) in thin film forms were cut into small pieces for packing into 4 mm MAS rotors for MAS experiments. Spin-lock pulses of 1 ms duration were applied along both 1H and 13C channels for CP, employing a 50 kHz rf-field at the 13C channel and a ramped rf pulse at the 1H channel whose rf-field strength changes linearly over a 25% range centered at 38 kHz. A pulse technique known as total suppression of spinning side bands (TOSS)39 was combined with a CP sequence to obtain sideband-free 13C MAS spectra at 6 kHz MAS spinning speed. NMR signal averaging was achieved by co-adding 2048 transients with a 4 s acquisition delay time. 1H and 13C π/2 pulse lengths were 4 μs and 5 μs, respectively. The small phase incremental alternation with 64 steps (SPINAL-64)40 decoupling sequence at 63 kHz power was used for proton decoupling during 13C signal detection.
Proton conductivity (σ, Scm−1) of each sample (rectangular size = 1 × 4 cm2) was obtained from an equation of σ = l/(R × S), where l and S represent the distance between reference electrodes and the cross-sectional area of the sample, respectively.41 Ohmic resistance (R, Ω) in liquid water at 30 °C, at 95% RH and 60 °C, and under different RH conditions at 80 °C was measured via four-point probe alternating current impedance spectroscopy using a combination of Solartron 1287 and 1260. The sample dimension in a certain hydrated state was measured immediately after the resistance measurement. For the RH conductivity test, all samples were kept in deionized water for at least 1 day and, then installed in the test cell setup within a temperature-humidity controlled oven. The ohmic resistance was measured from a high RH to a low RH (95, 80, 70, 60, 50, 40, and 30% RH) with an equilibrium interval of at least 45 min per each RH. Each measurement was repeated at least five times to ensure good reproducibility.
Single cell tests were conducted at 80 °C with pure H2 and O2 gases under ambient pressure. Their gas flow rate was 100 ml min−1. The relative humidity applied for test cells was controlled in both 85% and 65% RH by bubbling heated water in reservoirs. Current–voltage polarization behavior was monitored by managing an electronic loader (Smart wload, Wonatech, Korea). All membrane-electrode assemblies (MEAs) were fabricated by transferring catalyst slurry (mixture of 0.72 g of 20 wt% Pt/C and 4 g of 5 wt% Nafion® solution in 200 ml of isopropyl alcohol) onto membranes via a screen-printing method.42
Results and discussion
Fig. 2(a) shows dynamic TGA thermograms of BisA-40 films measured in the temperature range of 50 to 400 °C. Considering that potassium salt form BisA-XX films are thermally stable up to about 400 °C, characteristic weight losses observed between 150 and 400 °C were ascribed to thermal evaporation of DMAc (boiling point at 760 mmHg = 163–165 °C43). The residual DMAc content measured right after drying at 60 °C was high, but the residual content was reduced after the additional drying process under vacuum at 150 °C. Though 150 °C is higher than the boiling point of DMAc under vacuum (e.g., ∼96 °C at 80 mmHg43), it was difficult to remove the residual solvent thoroughly from BisA-40_150 °C because DMAc strongly interacts with –SO3−K+ groups. Conversely, the water treatment remarkably weakened the interaction via solvent exchange from DMAc to water and strong hydrogen bond formation with polar water molecules allowing extraction of residual DMAc. The weight losses observed in both BisA-40_60 °C and BisA-40_150 °C after water treatment were negligible and supposed to be derived from water molecules tightly bound to –SO3−K+ groups. The same trend was observed in all BisA-XX films, irrespective of DS.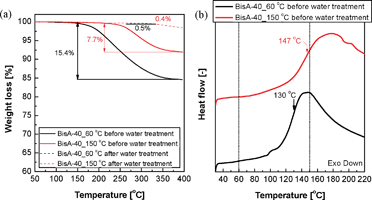 | ||
| Fig. 2 (a) Residual solvent contents in BisA-40 films obtained as a result of thermal treatments at different temperatures and subsequent water treatment, and (b) Tg of BisA-40 in the presence of residual DMAc. | ||
The Tg of the sulfonated copolymers usually increases as DS increases, since –SO3−K+ groups in the polymers become bulky and their ionic natures are enhanced.44 BisA-40 has the highest Tg value among BisA-XX with the same fabrication history. The Tg of BisA-40_60 °C in the presence of residual DMAc was lowered by 90 °C (Fig. 2(b)), when compared with that of solvent-free BisA-40_60 °C (238 °C, measured by DSC after water treatment). The Tg depression is attributed to the plasticizing effect of DMAc which enhances polymer chain mobility. Interestingly, the Tg of BisA-40_60 °C under residual DMAc was lower than 150 °C which was selected as the second drying temperature in this study. Meanwhile, the Tg of BisA-40_150 °C measured immediately after drying at 150 °C was still between 60 and 150 °C. Other BisA-XX_60 °C films with low DS also have Tg lower than 150 °C under residual DMAc. It means that BisA-XX_60 °C was thermally annealed to BisA-XX_150 °C during the thermal treatment at 150 °C higher than their depressed Tg values (“solvent-assisted thermal annealing”). The resulting BisA-XX_150 °C was expected to show thermally induced film characteristics different from non-annealed BisA-XX_60 °C.
Fig. 3 shows the thermal annealing effect on the Tg of solvent-free BisA-XX copolymers. Here, all film samples for the DSC measurements were used in –SO3−K+ form to avoid the degradation of thermally weak sulfonic acid (–SO3−H+) groups. Similar to other sulfonated polymer systems, an increase of disulfonation level in BisA-XX led to an increase of Tg. Moreover, the Tg of BisA-XX was raised by 5 to 8 °C after the heat treatment at 150 °C. Note that the Tg difference slightly increased with DS and become most distinct for BisA-40. The high Tg in BisA-XX_150 °C is associated with the formation of compact, dense BisA microstructures. A similar thermal annealing effect on Tg was also reported in post-sulfonated PAES with the same polymer backbone (Fig. 1(a)).29
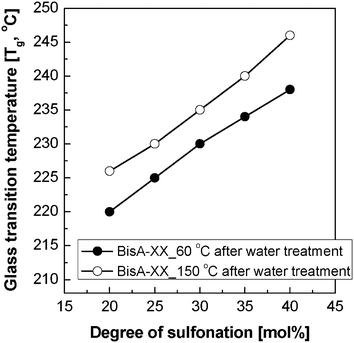 | ||
| Fig. 3 The Tg change in BisA-XX measured by DSC after thermal annealing at 150 °C and subsequent water treatment. | ||
Fig. 4 shows (a) motional dynamics and (b) molecular ordering information of sodium ions of BisA-XX, depending on DS and thermal annealing history, studied by solid-state NMR. 1H T1 data (detected via directly attached 13C signal by employing a short cross-polarization (∼150 μs)) in Fig. 4(a) provides information on the segmental vibrations of protons in BisA-XX aromatic phenylene rings whose frequency range is near the proton's Larmor frequency (ω0 = 3 × 108 Hz under 7.05 T). Here, the data were obtained in the fully hydrated state at an ambient temperature. 1H T1 was shortened as the DS value of BisA-XX copolymers increased, because highly absorbed water molecules in high DS BisA-XX systems act as plasticizers softening the polymer chains. The T1 was much reduced after the thermal treatment at 150 °C. The T1 difference between BisA-XX_60 °C and BisA-XX_150 °C becomes larger at a high DS. This behavior can be explained by a basic 1H T1 relaxation mechanism governed by 1H–1H homonuclear dipolar interactions (Eqn 145):
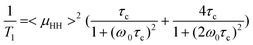 | (1) |
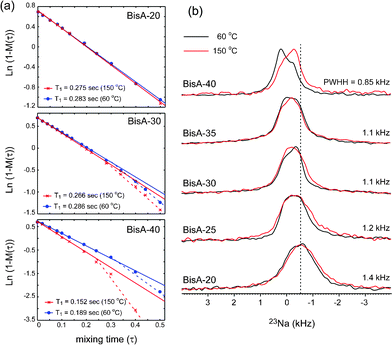 | ||
| Fig. 4 (a) 1H–T1 plot data and (b) 23Na NMR spectra of BisA-XX_60 °C and BisA-XX_150 °C measured by solid-state NMR techniques. | ||
23Na solid-state NMR spectroscopy (Fig. 4(b)) was employed to investigate the thermally induced molecular order/disorder in the hydrophilic domains in BisA-XX. For this, all BisA-XX samples were successively treated in boiling 0.5 M H2SO4 for 2 h, boiling deionized water for 2 h,10,13,33–35 0.5 M NaCl for 3 days, and deionized water for 1 week to convert counter ions to –SO3− from K+ to Na+ thoroughly. Here, Na+ ions were used as probes to observe the molecular ordering environment in hydrophilic domains, as Na+ ions make a complex (–SO3−Na+) with –SO3− groups in BisA-XX and their quadrupolar interaction (spin 3/2) would be sensitive over the structural and dynamic environments. The characteristic 23Na peak was sharpened as the DS increased, indicating that the packing order or distribution of sodium ions became more uniform. Furthermore, the 23Na peaks of non-annealed BisA-XX_60 °C shifted slightly with increasing DS, but their shifted peaks, particularly in BisA-40, returned to positions similar to others, after the thermal treatment. This behavior may be related to the changes of sulfonic acid density in the BisAH-XX matrix after the thermal annealing, which is discussed in volume-based ion exchange capacity.
Fig. 5 exhibits the water uptake of BisAH-XX as a function of ion exchange capacity based on dry polymer mass (IECw) and fully hydrated polymer volume (IECv(wet)). Water uptake of BisAH-XX in Fig. 5(a) was compared with that of previously reported post-sulfonated PAES29 with an identical polymer backbone as a reference sample (Fig. 1(a)). Water uptake of both BisAH-XX and post-sulfonated PAES continuously increased with IECw whose value is proportional to the employed sulfonic acid concentrations (DS). At a similar IECw, post-sulfonated PAES has a water uptake higher than that of BisAH-XX. It may be due to an irregular distribution of bulky sulfonic acid groups derived from the synthetic route (e.g., electrophilic substitution) and chemistry different from those of BisAH-XX. IECw values of BisAH-XX and post-sulfonated PAES were independent of the thermal treatment at 150 °C, since IECw was obtained through a simple calculation of atomic weight per sulfonic acid group. However, the water uptake of thermally annealed post-sulfonated PAES and BisAH-XX was reduced, irrespective of their sequential difference. This reduced water swelling is expected to contribute to improved dimensional stability in the fully hydrated state.
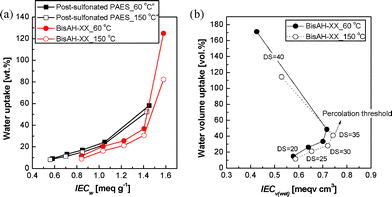 | ||
| Fig. 5 Comparison of (a) water uptake of post-sulfonated PAES and BisAH-XX as a function of IECw and (b) water volume uptake of BisAH-XX as a function of IECv(wet) according to thermal annealing. a Obtained from Ref. 29. | ||
It is interesting to discuss the swelling behavior of BisAH-XX with volume-related parameters such as water volume uptake (WU(vol)) and IECv(wet) defined in the following equations:46
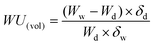 | (2) |
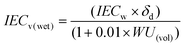 | (3) |
Fig. 6(a) shows the proton conductivity of post-sulfonated PAES and BisAH-XX as a function of IECw. Here, the proton conductivity measurement of BisAH-XX was conducted under almost identical measurement conditions (temperature, humidity, and test apparatuses and protocols) to those of post-sulfonated PAES used for comparison. Proton conductivity of both materials increased as IECw increased, irrespective of their synthetic routes. Under the same film drying protocols, BisAH-XX exhibited proton transport behavior faster than post-sulfonated PAES with a similar IECw. This difference in proton transport originates from the sequences and positions of –SO3H groups in their polymer backbones (see Fig. 1). In post-sulfonated PAES, –SO3H groups are randomly introduced into ortho-position of electron-rich diphenyl ether (–O–) units, since the post-sulfonation is an electrophilic substitution reaction. On the other hand, the sulfonation of DCDPS monomer leads to the formation of meta-positioned sulfonated diphenyl sulfone monomer (SDCDPS), where electron-withdrawing sulfone (SO) units in the phenyl rings makes adjacent –SO3H groups more acidic. In addition, DS in BisAH-XX is controlled by varying the molar ratio of SDCDPS to DCDPS. It enables –SO3H groups in SDCDPS to be arranged with certain sequences during polymer synthesis, which affords relatively well-defined hydrophilic moieties in BisAH-XX. Accordingly, protons from –SO3H groups in BisAH-XX are dissociated more easily than those in the corresponding PAES.
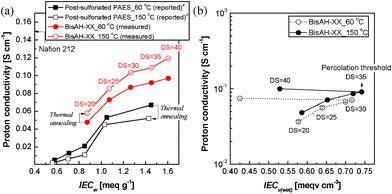 | ||
| Fig. 6 (a) Proton conductivity comparison of post-sulfonated PAES and BisAH-XX as a function of IECw (measurement condition: 60 °C and 95% RH) and (b) proton conductivity of BisAH-XX as a function of IECv(wet) (measurement condition: liquid water at 30 °C) according to thermal annealing. a Reported in Ref. 29. | ||
Surprisingly, the thermal annealing effects on proton conductivity were fairly different for post-sulfonated PAES and BisAH-XX. Post-sulfonated PAES_150 °C lost its proton conduction capability after the thermal annealing. However, this proton conductivity loss has not been clearly understood yet. It was suspected from either thermal decomposition or thermally-induced crosslinking. Unlike in salt form, sulfonated polymers in proton form are susceptible to thermal degradation. Their –SO3H groups begin to decompose at around 200 °C and the initial decomposition temperatures may be somewhat changed depending on polymer architectures and characteristics, residual solvents including water vapor, and ionic pairs.47 A portion of –SO3H groups can also be utilized as crosslinkable sites where sulfonated polymers undergo inter- or intra-molecular covalent bond formation via thermal conversion from –SO3H to sulfone bridge groups in a temperature range between 120 to 170 °C.48,49 The sacrifice of some –SO3H groups resulted in slightly reduced IECw and unavoidable proton conductivity loss. In contrast, proton conductivity of BisAH-XX was improved after the thermal annealing. This positive electrochemical contribution of thermal treatment was remarkably observed in BisAH-XX_150 °C with a high DS. These changes in proton conductivity may be attributed to the density of sulfonic acid groups which were improved even under a fully hydrated state. Note that IECv(wet) in Fig. 6(b) was enhanced after the thermal treatment and the biggest IECv(wet) change was observed in BisAH-40 samples. The percolation threshold to proton conductivity was 35 mol%.
Fig. 7 presents the changes of surface morphologies of BisAH-40 after the thermal annealing. AFM images generally provide information on membrane surface morphology. However, the surface morphology of disulfonated poly(arylene ether sulfone) random copolymers are not largely different from their corresponding bulk morphology. Both images contain soft hydrophilic phases (dark regions) and hard hydrophobic phases (bright region). Unlike the other random copolymers with low DS values where hydrophilic domains are randomly distributed within hydrophobic polymer matrices, hydrophilic domains in BisAH-40 are interconnected with each other, which is associated with its hydrophilic mole fraction being higher than percolation threshold of BisAH-XX (i.e., 35 mol%). The BisAH-40_60 °C sample has relatively large hydrophilic domains in the average sizes of 25–30 nm, which may be advantageous in absorbing a large amount of water molecules. After the thermal treatment, the average diameters of interconnected hydrophilic domains became smaller by 9–11 nm. Note that the hydrophilic domains are homogeneously distributed with a relatively high concentration in the same area, which may be correlated with improved sulfonic acid density after the thermal treatment, since the hydrophilic domains are obtained as a result of self-assembly of sulfonic acid groups in BisAH-XX. This peculiar morphology was expected to be helpful in maintaining a relatively high water retention level even at a low humidity.
 | ||
| Fig. 7 AFM surface topology of (a) BisAH-40_60 °C and (b) BisAH-40_150 °C in tapping mode. The dimensions of the images are 500 × 500 nm2. The phase scale is 0–20°. The measurement was carried out at 35% RH. | ||
Fig. 8 shows proton conduction behavior of sulfonated copolymers, including Nafion® 212 for comparison, in a partially hydrated state. Their proton conductivities decreased, as applied humidity was reduced from 95% RH to 30% RH. This conventional conductivity drop at a low humidity was expected to be retarded by increasing their sulfonation levels,50,51 since highly sulfonated copolymers can form strong hydrogen bonds with water molecules. Different from our expectation, though BisAH-40_60 °C showed proton conductivity superior to BisAH-35_60 °C, their RH conductivity trends were analogous. In contrast, thermal annealing of the same copolymers at 150 °C led to much improved proton conductivity in the tested RH ranges. For example, proton conductivity of BisAH-35_150 °C was one order of magnitude higher than that of BisAH-35_60 °C at 30% RH. Furthermore, BisAH-XX_150 °C showed relatively stable proton conduction behavior even at a low RH when compared with BisA-XX_60 °C. High proton conductivity under the partially hydrated conditions may result from an increase in water retention capability via synergistic effects of enhanced sulfonic acid density and strong complexation of water molecules with –SO3H groups in compactly packed and hydrophilic–hydrophobic nanophase-separated BisAH-XX chains after the thermal treatment at 150 °C. Note that annealed BisAH-35 and BisAH-40 have proton conductivities comparable to Nafion® 212 at a low humidity (< 80% RH).
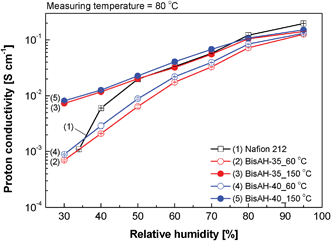 | ||
| Fig. 8 Proton conductivity measured in a humidity range from 30 to 95% RH at 80 °C. | ||
Fig. 9 shows how the thermal history of glassy BisAH-XX materials significantly influences their single cell performances under partially hydrated conditions. Here, all MEAs including Nafion® 212 MEA were obtained with the same fabrication procedures and electrode compositions. Unlike in Nafion® 212 and BisAH-35_150 °C MEAs, BisAH-35_60 °C MEA showed a fast electrochemical loss in the ohmic polarization region beginning from ∼40 mA cm−2. It may be due to the combination of its relatively low proton conductivity at a low humidity and the poor interface formation with Nafion® electrodes as a result of incompatibility with the hydrocarbon membrane and higher water swelling than BisAH-35_150 °C at the same RH. Interestingly, after the thermal annealing, enhanced current density and maximum power density were exhibited by BisAH-35 by 67% and 95%, respectively. The current–voltage polarization curve of BisAH-35_150 °C at 65% RH is comparable with that of corresponding Nafion® 212. Note that their proton conductivities at 65% RH are almost the same (see Fig. 8). Their electrochemical difference in a high current density region over 1060 mA cm−2 may be derived from different fuel (H2/O2) accessibility. Another peculiar feature was observed in the case of dehydration from 85% RH to 65% RH. The electrochemical performance of BisAH-35_150 °C was not largely reduced, which is fairly different from that of Nafion® 212. It strongly demonstrated that thermal annealing is a promising strategy for improving PEMFC performances and preserving a relatively high water retention level within a glassy PEM in a partially dehydrated state.
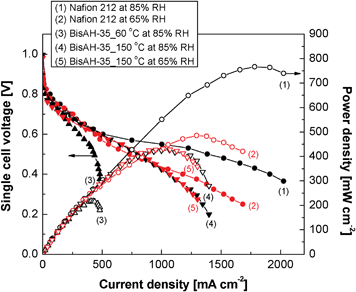 | ||
| Fig. 9 PEMFC single cell performance measured under partially hydrated conditions at 80 °C. H2/O2 flow rates = 100 ml min−1/100 ml min−1. Active area and Pt content of each MEA were 5 cm2 and 0.3 mg cm−2, respectively. | ||
Conclusions
The thermally annealed membranes dried at 150 °C showed Tg values higher than those of non-annealed membranes treated only at 60 °C. It suggested that the annealing resulted in densification under higher temperature conditions. The thermal annealing also changed the molecular dynamics of BisA-XX copolymers as judged by solid state NMR. Segmental vibrations of protons in BisA-XX aromatic phenyl rings (or 1H-1H dipolar interaction) were enhanced and, thus, their T1 relaxation values became shorter. The characteristic peak of 23Na solid-state NMR, where Na+ ions were used as probes to monitor the molecular ordering environment of BisA-XX hydrophilic domains, was also sharpened after the thermal annealing. This implies that the packing order or distribution of sodium ions (in other words, sulfonic acid groups) in BisA-XX became more uniform after annealing. This behavior was more clearly observed in BisA-XX that had a high disulfonation content.Membranes obtained after acidification of thermally annealed salt form copolymers had reduced weight-percent water uptake and improved volume-based sulfonic acid density (i.e., IECv(wet)) when compared with corresponding materials dried at 60 °C. Tapping mode AFM measurement on an annealed membrane exhibited a more developed hydrophilic–hydrophobic nanophase separation and higher sulfonic acid density than on a non-annealed membrane. Consequently, the proton conductivity of thermally annealed BisAH-XX was improved in liquid water and even under partially hydrated states. Importantly, the electrochemical contribution of the thermal annealing was significant at low humidities (e.g. 30 to 80% RH). The proton conductivity of annealed BisAH-XX at the partially hydrated state was comparable to that of Nafion® 212. This may be due to the synergistic effect of the improved sulfonic acid density and the strong complexation of water with sulfonic acid groups. Finally, it was demonstrated that the simple solvent-assisted thermal annealing afforded improved lower humidity PEMFC performances similar to that of the state-of-the-art fuel cell membrane, Nafion®.
Our current and future studies will focus on the systematic thermal annealing of glassy multiblock copolymers which have demonstrated proton conductivities higher than those of the corresponding random copolymers at a low humidity owing to their well-defined hydrophilic–hydrophobic nanophase separation.31
Acknowledgements
We thank the US Department of Energy (DE-FG36-06G016038) for its support for this research. This work was also supported by the Korean Foundation for International Cooperation of Science & Technology (KICOS) through a grant provided by the Korean Ministry of Education, Science & Technology (K20701010356-07A0100-10610). YML also acknowledges the support from the project by the WCU program, National Research Foundation (NRF) of the Korean Ministry of Science and Technology (No. R31-2008-000-10092-0).References
- C. E. Health and A. G. Revfsz, Science, 1973, 11, 542–544 Search PubMed.
- Q. Li, J. O. Jensen, R. F. Savinell and N. J. Bjerrum, Prog. Polym. Sci., 2009, 34, 449–477 CrossRef CAS.
- C. Song, C. Jensenchua, Y. Tang, J. Zhang, J. Li, K. Wang, S. McDermid and P. Kozak, Int. J. Hydrogen Energy, 2008, 33, 2802–2807 CrossRef CAS.
- K.-D. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- A. A. Kornyshev, A. M. Kuznetsov, E. Spohr and J. Ulstrup, J. Phys. Chem. B, 2003, 107, 3351–3366 CrossRef CAS.
- M. Eikerling, A. A. Kornyshev, A. M. Kuznetsov, J. Ulstrup and S. Walbran, J. Phys. Chem. B, 2001, 105, 3646–3662 CrossRef CAS.
- T. J. F. Day, U. W. Schmitt and G. A. Voth, J. Am. Chem. Soc., 2000, 122, 12027–12028 CrossRef CAS.
- J. S. Wainright, J. T. Wang, D. Weng, R. F. Savinell and M. Litt, J. Electrochem. Soc., 1995, 142, L121–L123 CrossRef CAS.
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587–4612 CrossRef CAS.
- H. S. Lee, A. Roy, O. Lane and J. McGrath, Polymer, 2008, 49, 5387–5396 CrossRef CAS.
- A. S. Badami, A. Roy, H.-S. Lee, Y. Li and J. E. McGrath, J. Membr. Sci., 2009, 328, 156–164 CrossRef CAS.
- H. Ghassemi, J. McGrath and T. Zawodzinskijr, Polymer, 2006, 47, 4132–4139 CrossRef CAS.
- Y. Li, A. Roy, A. Badami, M. Hill, J. Yang, S. Dunn and J. McGrath, J. Power Sources, 2007, 172, 30–38 CrossRef CAS.
- A. Roy, H. Lee and J. McGrath, Polymer, 2008, 49, 5037–5044 CrossRef CAS.
- Y. S. Kim, F. Wang, M. A. Hickner, T. A. Zawodzinski and J. E. McGrath, J. Membr. Sci., 2002, 212, 263–282 CrossRef.
- J. D. Halla, M. Mamak, D. E. Williams and G. A. Ozin, Adv. Funct. Mater., 2003, 13, 133–138 CrossRef CAS.
- A. B. Bourlinos, K. Raman, R. Herrera, Q. Zhang, L. A. Archer and E. P. Giannelis, J. Am. Chem. Soc., 2004, 126, 15358–15359 CrossRef CAS.
- K. Kanamura, T. Mitsui and H. Munakata, Chem. Mater., 2005, 17, 4845–4851 CrossRef CAS.
- M. L. Hill, Y. S. Kim, B. R. Einsla and J. E. McGrath, J. Membr. Sci., 2006, 283, 102–108 CrossRef CAS.
- J. Roeder, V. Zucolotto, S. Shishatskiy, J. R. Bertolino, S. P. Nunes and A. T. N. Pires, J. Membr. Sci., 2006, 279, 70–75 CrossRef CAS.
- C. H. Lee, S. Y. Lee, Y. M. Lee, S. Y. Lee, J. W. Rhim, O. Lane and J. E. McGrath, ACS Appl. Mater. Interfaces, 2009, 1, 1113–1121 CAS.
- C. H. Park, C. H. Lee, M. D. Guiver and Y. M. Lee, Prog. Polym. Sci., 2011, 36, 1443–1498 CrossRef CAS.
- J. J. Singh and T. L. St.clair, NASA Technical Reports Server (NTRS), 1986 Search PubMed , NASA-TM-8773419860018657.
- M. B. Moe, W. J. Koros and D. R. Paul, J. Polym. Sci., Part B: Polym. Phys., 1988, 26, 1931–1945 CrossRef CAS.
- B. W. Rowe, B. D. Freeman and D. R. Paul, Polymer, 2010, 51, 3784–3792 CrossRef CAS.
- J. Jo, S.-S. Kim, S.-I. Na, B.-K. Yu and D.-Y. Kim, Adv. Funct. Mater., 2009, 19, 866–874 CrossRef CAS.
- F. Padinger, R. S. Rittberger and N. S. Sariciftci, Adv. Funct. Mater., 2003, 13, 85–88 CrossRef CAS.
- J. Liu, T.-F. Guo and Y. Yang, J. Appl. Phys., 2002, 91, 1595 CrossRef CAS.
- H. B. Park, H. S. Shin, Y. M. Lee and J. W. Rhim, J. Membr. Sci., 2005, 247, 103–110 CrossRef CAS.
- R. Guo, O. Lane, D. VanHouten and J. E. McGrath, Ind. Eng. Chem. Res., 2010, 49, 12125–12134 CrossRef CAS.
- Y. Chen, R. Guo, C. H. Lee, M. Lee and J. E. McGrath, International Journal of Hydrogen Energy, 2011DOI: 10.1016/j.ijhydene.2011.06.139 Search PubMed.
- B. C. Johnson, İ. Yilgör, C. Tran, M. Iqbal, J. P. Wightman, D. R. Lloyd and J. E. McGrath, J. Polym. Sci., Polym. Chem. Ed., 1984, 22, 721–737 CrossRef CAS.
- F. Wang, M. A. Hickner, Y. S. Kim, T. A. Zawodzinski and J. E. McGrath, J. Membr. Sci., 2002, 197, 231–242 CrossRef CAS.
- M. J. Sumner, W. L. Harrison, R. M. Weyers, Y. S. Kim, J. E. McGrath, J. S. Riffle, A. Brink and M. H. Brink, J. Membr. Sci., 2004, 239, 199–211 CrossRef CAS.
- Y. Li, F. Wang, J. Yang, D. Liu, A. Roy, S. Case, J. Lesko and J. McGrath, Polymer, 2006, 47, 4210–4217 CrossRef CAS.
- Y. Li, R. A. VanHouten, A. E. Brink and J. E. McGrath, Polymer, 2008, 49, 3014–3019 CrossRef CAS.
- M. Sankir, V. A. Bhanu, W. L. Harrison, H. Ghassemi, K. B. Wiles, T. E. Glass, A. E. Brink, M. H. Brink and J. E. McGrath, J. Appl. Polym. Sci., 2006, 100, 4595–4602 CrossRef CAS.
- J. Yang, Y. Li, A. Roy and J. McGrath, Polymer, 2008, 49, 5300–5306 CrossRef CAS.
- W. T. Dixon, J. Chem. Phys., 1982, 77, 1800–1809 CrossRef CAS.
- B. M. Feng, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef.
- C. H. Lee, H. B. Park, Y. M. Lee and R. D. Lee, Ind. Eng. Chem. Res., 2005, 44, 7617–7626 CrossRef CAS.
- D. S. Hwang, C. H. Park, S. C. Yi and Y. M. Lee, International Journal of Hydrogen Energy, 2011 Search PubMed.
- The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, Merck Research Laboratories, Division of Merck & Co. , Inc., Whitehouse station, NJ, 1996 Search PubMed.
- K. Xu, K. Li, P. Khanchaitit and Q. Wang, Chem. Mater., 2007, 19, 5937–5945 CrossRef CAS.
- R. Kimmich, NMR-Tomography, Diffusometry, Relaxometry: Part II. Molecular Motion, Springer, 1997 Search PubMed.
- Y. S. Kim and B. S. Pivovar, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 123–148 CrossRef CAS.
- R. W. Kopitzke, C. A. Linkous and G. L. Nelson, Polym. Degrad. Stab., 2000, 67, 335–344 CrossRef CAS.
- M. L. Di Vona, E. Sgreccia, S. Licoccia, G. Alberti, L. Tortet and P. Knauth, J. Phys. Chem. B, 2009, 113, 7505–7512 CrossRef CAS.
- M. L. Di Vona, E. Sgreccia, M. Tamilvanan, M. Khadhraoui, C. Chassigneux and P. Knauth, J. Membr. Sci., 2010, 354, 134–141 CrossRef CAS.
- K. Miyatake, Y. Chikashige and M. Watanabe, Macromolecules, 2003, 36, 9691–9693 CrossRef CAS.
- B. Bae, K. Miyatake and M. Watanabe, Macromolecules, 2010, 43, 2684–2691 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |