Microwave assisted radical organic syntheses
Roy T.
McBurney
*,
Fernando
Portela-Cubillo
and
John C.
Walton
*
School of Chemistry, EaStChem, University of St. Andrews, St.Andrews, Fife, KY16 9ST, UK. E-mail: jcw@st-andrews.ac.uk; Fax: +44 (0)1334 463808; Tel: +44 (0)1334 463864.
First published on 20th December 2011
Abstract
Applications of microwave (MW)-assistance to radical-mediated organic preparations and procedures are reviewed. Radical additions and cyclisations onto a wide range of acceptors benefited from the technique, as did organic halide reductions by tin hydrides and numerous cascade and sequential processes. Reaction times of chain processes initiated by AIBN were, in several cases, reduced to a few minutes. Regioselectivity in radical additions and ring closures was normally maintained under MW conditions, although stereoselectivities were usually poorer. MW methods have been developed for generation of C-, N-, O-, S- and P-centred radicals. As a result of this a notable number and diversity of heterocycles have been accessed. MW-methods have opened up several new and innocuous alternatives to toxic organotin reagents. Alkoxyamines smoothly release C-centred radicals and oxime ethers provide a variety of iminyl radicals on MW heating. In addition, several types of organo-element and organometallic precursors have yielded novel C-centred and hetero-radicals. To date applications of MW-assistance to homolytic processes are comparatively limited, but it is clear the alliance of the two holds a lot of promise.
 Roy T. McBurney | Roy T. McBurney was born and raised in Carnoustie, Scotland. He received a MChem in Chemistry at the University of Edinburgh (2000–2005) and later obtained his PhD under the supervision of Prof. David Leigh, investigating metal template strategies to interlocked architectures. In 2008 he took up a postdoctoral position at Durham University with Prof. David Parker studying responsive MRI probes. In 2010 he joined the group of Prof. John Walton at the University of St Andrews. His research interests include semiconductors for use as photocatalysts in organic synthesis and tin-free methods for the direct generation of organic radicals. |
 Fernando Portela Cubillo | Fernando Portela-Cubillo was born in Aranda de Duero (Burgos, Spain) in 1975. He obtained his first degree from the University of Valladolid in 2002. He subsequently studied under the supervision of Prof. John Walton and graduated with a PhD degree from the University of St Andrews in 2009. He continued his scientific education as an Ecole Polytechnique Postdoctoral Fellow at the Ecole Polytechnique (Palaiseau, France) with Prof. Sam Zard. Currently, he holds an Alexander von Humboldt Fellowship and is a postdoctoral fellow in Prof. Armido Studer’s laboratory at the University of Münster (Münster, Germany). |
 John C. Walton | John C. Walton was born in St. Albans, England, and was educated at Stanborough School and Watford Grammar School. He studied for his PhD under the supervision of Professor Lord Tedder at Sheffield and St. Andrews Universities. He joined Dundee University in 1967 and moved to St. Andrews University in 1970 becoming Research Professor of Chemistry in 2007. His research interests include: (i) clean homolytic synthetic methods based on oxime derivatives, (ii) semiconductor photocatalysts, (iii) applications of EPR spectroscopy to organic reactions. He has published over 275 articles in learned journals as well as 3 books. |
1. Introduction
The growth of organic syntheses mediated by free radicals has been spectacular over the last two decades.1 Many of the most successful preparations utilised an organotin hydride, or a less toxic mimic such as tristrimethylsilylsilane,2 in conjunction with thermal initiation by an azo-compound or a diacyl peroxide. In parallel with this, the exploitation of microwave (MW) heating has also grown exponentially and a host of MW assisted organic syntheses (MAOS) have been described.3 There appear to be good grounds to expect a synergistic alliance of homolytic and MW methods.The energy of a MW photon (∼0.15 kJ mol−1) is far too low to break chemical bonds, thus MW irradiation itself does not achieve chemical transformations. Instead MW assisted heating occurs via dipoles or ions within a sample attempting to realign themselves with the applied electromagnetic field. As the field is constantly oscillating, usually at 2.45 GHz, MW energy is rapidly converted to heat through molecular friction and dielectric loss. Details of how MW or dielectric heating works can be found in pertinent literature.4 MW irradiation offers the ability to rapidly heat a reaction mixture beyond its boiling point (usually by 20–30 °C). MAOS advantages, namely reduced reaction times, improved yields and more efficient reactions are believed to stem from this overheating.5 Excellent solvents for MW reactions include water, ethylene glycol, ethanol and DMSO which have high loss factors.6 Radical reactions can be conducted in t-butanol or acetonitrile, but they are usually carried out in non-polar solvents such as benzene, toluene or cyclohexane which have low loss factors. However, non-polar solvents can still be used for MW reactions in conjunction with polar additives to increase the MW absorbance of the reaction medium. Ionic liquids (IL), e.g.1-ethyl-3-methylimidazolium hexafluoro-phosphate (emimPF6), fill this role admirably and are easily removed during work-up by filtration.
Thermally initiated radical processes often occur viachain reactions containing both propagation and termination steps. The higher temperatures associated with MW heating are expected to increase the rates of propagation steps. Termination steps consist of radical combinations and/or disproportionations which are fast but low activation energy process for all except very sterically crowded radicals. Thus, under MW conditions, the rates of terminations will not increase to the same extent as propagation steps and consequently longer chain reactions and better yields can be expected. MW heating will also significantly increase the rate of thermal dissociation of the azo, peroxide or other initiators and hence faster initiation will occur. Depletion of the initiator could outrun reactant conversion unless the propagation steps are matchingly fast.
In the search for benign alternatives to organostannanes,7 and in order to expand the scope of free-radical based syntheses, molecules that homolytically dissociate at higher temperatures have come under scrutiny. One option for such thermolyses is heating in sealed tubes or metal autoclaves; however these techniques are inconvenient and prevent continuous monitoring of reaction progress. Another alternative is flash vacuum pyrolysis (FVP). This is a comparatively “soft” technique but, because of the much higher temperatures (typically 600–800 K) and low pressures, homolytic dissociations are often accompanied by unwanted eliminations and rearrangements.8 Thus MW assistance appears a very attractive option.
Modern MW instruments irradiate a single reaction space in what is known as a monomode (also referred to as single-mode) reactor. High throughput of samples can easily be achieved by using an auto-sampler arm and carousel, readily available for most commercial MW reactors. Most modern instrumentation can provide a detailed report of how the reaction proceeds in terms of temperature and pressure. A wide range of synthesis scales are catered for, from small sealed-tube reactions (0.2 to 50 mL, up to 250 °C and ∼20 bar) to larger open vessels (∼150 mL) under reflux conditions.
In spite of the evident “fit” between radical-mediated preparations and MW assistance the uptake of MAOS for radical processes has been relatively slow. The aim of this review is to highlight the early pioneers who used MAOS for radical syntheses, and to document the recent advances in the field. Attention will be drawn to the advantages of MW heating over conventional heating. More importantly, we hope to encourage many organic radical chemists to discover for themselves the benefits of MW assisted syntheses.
2. Applications of microwaves to radical-mediated syntheses
2.1. Early pioneers
Surprisingly, despite the advantages offered by MAOS, growth in applications of MW within the free radical community has been rather gradual. The first report appeared from the Bose group in 1991.9 Amongst other organic transformations, including Diels–Alder reactions and catalytic hydrogenations, the stereoselective free radical dehalogenation of 6,6-dibromopenicillanic acid, 1, with tributyltin hydride in the presence of 2,2-azobisisobutyronitrile (AIBN) in anhydrous THF was studied (Scheme 1). After 3 min irradiation at low power in a conventional MW oven, dibromo-acid 1 was converted into the potent β-lactamase inhibitorcis-6β-bromopenicillanic acid with minor amounts of the corresponding trans-compound (2). Compared to an almost identical radical dehalogenation of 1, in the absence of MW irradiation,10 the much improved yield and shortened reaction time signalled the impact that MW's could have on radical organic chemistry.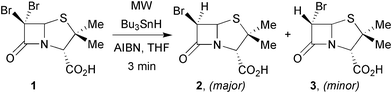 | ||
| Scheme 1 Bose's pioneering use of MW in dehalogenation reactions.9 | ||
Remarkably, it was another 8 years before the second radical mediated example was reported. Curran, Hallberg and co-workers11 described the use of highly fluorous F-21 organo-tin reagents for Stille couplings and small series of radical-mediated reactions. A radical mediated dehalogenation of adamantyl bromide with fluorous-tin reagent HSn(CH2CH2C10F21)3 in the presence of AIBN was achieved in 81% yield after 5 min at 35 W. It was notable that under conventional non-MW heating, this reaction proved irreproducible mostly giving just starting material. Radical cyclisations were also tested under these highly fluorous conditions. Aryl iodide 4 cyclised to indoline derivative 5 in excellent yield (Scheme 2). An alkyl bromide was also able to undergo a similar 5-exo-cyclisation, albeit in slightly lower yield, even at a higher wattage (50 W). The fluorous tin reagent was able to turn over catalytically (10 mol% loading) in the radical addition of adamantyl bromide to acrylonitrile yielding 77% of an addition product after 10 min at 60 W. The authors attribute the success of these reactions to: “rapid heating, superheating of the solvent, smaller temperature gradients in the reaction mixture and the absence of wall effects.” Furthermore, the F-21 tin reagent, coupled with the advantages of MW irradiation, caused biphasic reaction mixtures to coalesce upon heating to create homogenous solutions with high organic/fluorous interface areas, possibly increasing the efficiency of the reactions.
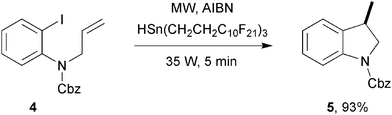 | ||
| Scheme 2 Radical cyclisation mediated by a fluorous F-21 tin reagent.11 | ||
2.2. Carbon centred radicals
 | ||
| Scheme 3 MW-assisted atom transfer radical additions.12 | ||
Horikoshi, Serpone and co-workers13 investigated the MW-enhanced radical synthesis of 3-cyclohexyl-1-phenyl-1-butanone 6 from crotonophenone 7 and iodocyclohexane 8 in a triethylborane/THF solution in the presence of t-butylhydroperoxide (Scheme 4). After 60 min of MW irradiation at ambient temperature the yield of product was almost quantitative (93%, increasing to 95% at 120 min). MW conditions at 78 °C yielded only 56% product (decreasing to 53% at 120 min). Oxygen is probably formed in several steps so that initiation takes place from its interaction with Et3B to release ethyl radicals which abstract iodine from iodocyclohexane. The cyclohexyl radical then adds to the 3-position of crotonophenone, followed by H-abstraction to yield product 6. The MW reactions proceeded much better than conventional conditions at room temperature which only yielded 40% product after 60 min. The yields under conventional conditions mirrored the MW yields in decreasing at higher temperatures. This suggested product degradation and/or by-product formation set in as temperature was raised.
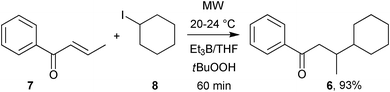 | ||
| Scheme 4 MW-assisted addition to crotonophenone.13 | ||
Studer and co-workers examined intermolecular additions of malonyl radicals onto unactivated alkenes during their extensive researches with alkoxyamines.14 Reversible thermal dissociation of alkoxyamine 9 released the persistent 2,2,6,6-tetramethylpiperidinyl-1-oxyl radical 10 (TEMPO) and stabilised malonyl radical 11 (Scheme 5). Rapid addition of 11 onto an olefin delivered adduct radical 12 that reacted selectively and irreversibly with TEMPO, due to the Persistent Radical Effect (PRE),15 to yield the carboaminoxylation product 13. However, despite the simplicity of the reactions, their usefulness was limited by protracted heating (3 days at reflux) in various high boiling solvents. Switching to MW irradiations in DMF at 180 °C for 10 min led, in some cases, to a doubling of the yield (Scheme 5a).16 Even comparing the results where the yields remained constant, a 430-fold acceleration was attained upon employing MW conditions. 1,3-Dithiane alkoxylamines, 14, were also investigated as carbon-centred radical precursors (Scheme 5b).17 Mixtures of alkylidenedithiane, 15, and TEMPO-adduct, 16, were obtained. Interestingly, reaction of 14 with n-butyl acrylate in DMF in the presence of triethylamine gave only the dithiane 15.
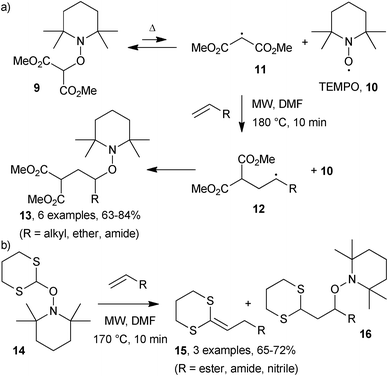 | ||
| Scheme 5 Alkoxyamine radical additions.16,17 | ||
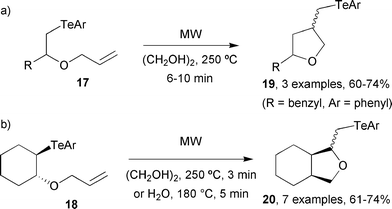 | ||
| Scheme 6 Group transfer cyclisations of (a) primary and (b) secondary organo-tellurium compounds.18 | ||
Copper-catalysed atom-transfer radical cyclisation (ATRC) reactions are well known,19 but thus far there has only been one report on the use of MW irradiation. Struggling with poor yields and long reaction times Quayle and co-workers turned to MW irradiation for their benzannulation reactions.20 They were delighted to observe that cyclisation of trichloroacetate 21 (R = H) in 1,2-dichloroethane in the presence of a catalytic amount of CuCl and ligand 22, afforded 1-chloronaphthalene, 23, in 84% yield with complete regiospecificity (Scheme 7). The observation that the progress of the reaction was dependent on the presence of copper catalyst 22-Cu(I), led Quayle's group to postulate that after initial 8-endocyclisation of 21 to lactone 24, radical dehalogenation occurred, generating radical 25, which underwent a 4-exo cyclisation to produce spirocyclic lactone 26. Rapid loss of CO2, followed by double dehydrochlorination yielded aromatic product 23. Various substituents were tolerated enabling a series of 1,6- and 1,8-substituted naphthalenes to be generated. Multiple benzannulations were also viable as shown by the synthesis 4,10-dichlorochrysene in 34% yield. The synthetic utility of this reaction was also exemplified in the construction of some novel steroids starting from estrone.
 | ||
| Scheme 7 Copper-catalysed benzannulations.20 | ||
While investigating the effect of MW on the thiol-induced cyclisations of alkenyl isocyanides, Kilburn's group found distinct advantages over conventional heating.21 In some cases the yields of pyrrolines were doubled and in every case the reaction time was reduced from hours to minutes (Scheme 8a). Thermolysis of thioethanol or thiophenol generated a thiyl radical that added to the carbon of the isocyanide group present in 27. The thioimidoyl radical, 28, underwent a 5-exo cyclisation and subsequent hydrogen abstraction to generate both cis and trans-pyrrolines 29. In most cases the cis-geometry was slightly favoured (∼1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) but the cis/trans ratio was insensitive to the mode of heating. These thioimidoyl radicals also cyclised onto alkynyl radical traps in similar yields (Scheme 8b).
:1) but the cis/trans ratio was insensitive to the mode of heating. These thioimidoyl radicals also cyclised onto alkynyl radical traps in similar yields (Scheme 8b).
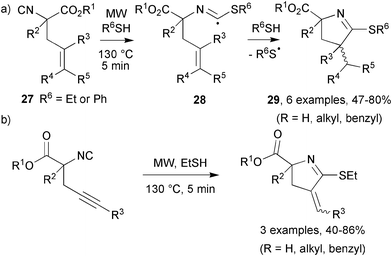 | ||
| Scheme 8 Thioimidoyl radical cyclisation on to (a) alkenyl and (b) alkynyl acceptors.21 | ||
Studer's group reported a remarkably efficient one-pot sequence linking a homolytic aromatic substitution with a subsequent ionic Horner–Wadsworth–Emmons (HWE) olefination.22 MW heating of alkoxyamines 30 in DMF for 2 min at 180 °C generated oxindoles 31via intramolecular homolytic aromatic substitution (Scheme 9). Olefination via the HWE reaction was achieved by addition of an aryl aldehyde and potassium t-butoxide and subjection to more MW irradiation (6 min at 180 °C) yielding α,β-unsaturated oxindoles 32. These sequential reactions were tolerant of substituents on the aniline ring: o-, m- and p-methoxy groups all gave similar yields of unsaturated oxindoles. The N-methyl group was also successfully replaced with a benzyl group, to facilitate later removal; possibly by hydrogenation.
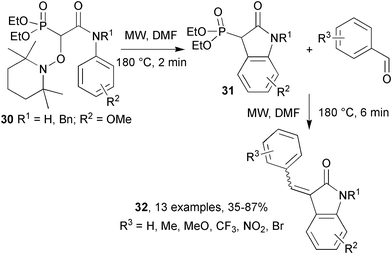 | ||
| Scheme 9 Sequential homolytic aromatic substitution and Horner–Wadsworth–Emmons olefination.22 | ||
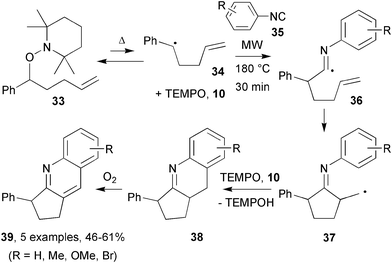
Thermolyses of alkoxyamines were again used by Studer's group for tandem radical reactions, first trapping with isonitriles then subsequent cyclisations and aromatisations.23 As before, the sequence started with thermal reversible homolysis of alkoxyamine 33 to generate carbon-centred radical 34 and TEMPO 10 (Scheme 10). Carbon centred radical 34 reacted with either TEMPO to reform starting material 33 or phenyl isonitrile 35 to afford imidoyl radical 36. 5-Exo cyclisation of imidoyl radical 36 produced primary alkyl radical 37 which underwent homolytic aromatic substitution with the aryl group to yield imine 38. Studer's group had previously demonstrated that homolytic aromatic substitution is possible using TEMPO-alkoxyamines, in which TEMPO oxidises the intermediate cyclohexadienyl radical.22Imines 38 oxidised during work-up to yield quinolines 39. Under conventional heating (DMF, 3 days, 140 °C) the yields of quinolines 39 were moderate to good. Switching to MW irradiation the reaction time was reduced to 30 min but the yield remained about the same. After optimisation, the team studied a series of substituted isonitriles; similar yields were obtained (Scheme 10). Importantly, bromo-quinolines were accessible via this method, whereas when using tin hydride conditions, aryl-bromides would be dehalogenated. The radical stabilising group present on the aryloxyamine was also varied, with production of similar amounts of quinolines. The 5-exo-cyclisations also occurred onto alkynyl derivatives, albeit in lower yields.
 | ||
| Scheme 10 TEMPO mediated preparation of quinolines.23 | ||
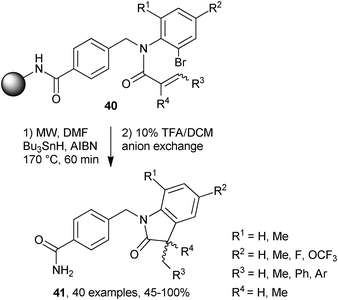
Fukase and co-workers prepared a library containing 40 different N-(2-bromophenyl)acrylamides 40 by reacting α,β-unsaturated acid chlorides with various anilines followed by attachment to Rink amide resins.24 MW irradiations of the polymer supported acrylamides with Bu3SnH and AIBN were carried out in DMF at 170 °C for 60 min. 5-Exo cyclisations followed by cleavage from the resin (treatment with a 10% solution of trifluoroacetic acid in dichloromethane) afforded indol-2-ones 41 in good to excellent yields (Scheme 11). This combinatorial approach is ideal for generating lead candidates for drug discovery because the indol-2-one sub-unit is known to be a pharmacophore of various drugs.
 | ||
| Scheme 11 Library of indol-2-ones via solid-supported acrylamides.24 | ||
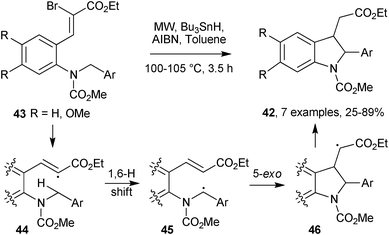
Reiser and co-workers discovered a novel synthetic route to 2-arylinoles 42 from substituted 2-(2-aminophenyl)acrylates 43.25 MW heating of 43, in the presence of Bu3SnH and AIBN in toluene at 100 °C (Scheme 12), brought about a significant increase in yields and decrease in reaction times compared to conventional heating. Bromine abstraction released vinyl radical 44, which underwent a 1,6-hydrogen shift generating benzylic radical 45. 5-Exo cyclisations and H-abstractions from the tin hydride furnished indolines 46 in good to high yields (64-89%). The exceptionally low yield of indoline from the naphthyl analogue (25%) can probably be attributed to the extensive resonance stabilisation of the naphthyl radical 45 (Ar = Naph). This directed the reaction away from ring closure into the fast H-atom abstraction channel. MW irradiation also led to improved yields in the 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) mediated aromatisations of indolines 42 to the corresponding indoles, as compared to conventional heating.
 | ||
| Scheme 12 Preparations of 2-arylindoles from aminophenylacrylates.25 | ||
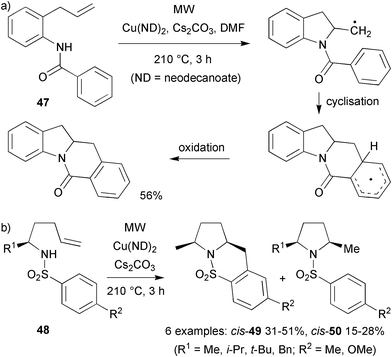
Whilst investigating various copper(II)-catalysed diamination cyclisations of N-arylsulfonyl-2-allylanilines, Chemler and co-workers observed that carbon centred radicals played a key role in the final cyclisation step.26 MW irradiation was found to greatly speed up the cyclisations of both arylsulfonamides and N-benzoyl-2-allylaniline 47 (Scheme 13a) compared to conventional heating (3 h vs. 72 h or 24 h respectively). However, the yields were comparable. A range of sulphonamides 48 was studied in the diastereoselective formation of pyrrolidines (Scheme 13b). Using conventional heating (190 °C, 72 h) or MW irradiation (210 °C, 3 h) 2,5-cis-pyrrolidines were formed with a very high diastereoselectivities (>20:1) in moderate yields (31–51%). Regardless of the R group, the products were observed as ∼2:1 mixtures of carboamination 49 and hydroamination adducts 50. The authors discussed several pathways for the first cyclisation, the formation of the C–N bond, settling on a syn aminocupration pathway. The second cyclisation was thought to involve a C-centred radical that either underwent intramolecular addition onto an aryl acceptor or hydrogen abstraction from the reaction mixture. Evidence for the radical pathway was provided by deuterium labelling studies that indicated the presence of a sp2-hybridised intermediate, most likely a primary radical, and also by trapping the radical with TEMPO.
 | ||
| Scheme 13 Copper(II)-catalysed diamination cyclisations.26 | ||
2.3. Nitrogen centred radicals
Oxime esters, R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–OC(O)R′, are easier to prepare, handle and characterise than peroxides or azo compounds. They contain weak N–O bonds and are a category of precursors for C- and N-centred radicals with promise of considerable generality. Photolytic initiation has been applied with considerable success to several varieties including benzophenone oxime esters,30oxime benzoates,31aldoxime esters,32 and dioxime oxalates.33 Zard used Ni powder and acetic acid as an alternative initiation method.34 However, all types of oxime esters have resisted thermal methods for dissociation including conventional heating and MW assistance.35 Even subjecting oxime esters to FVP led to eliminations and electro-cyclisations rather than clean N–O bond scission.36
N–OC(O)R′, are easier to prepare, handle and characterise than peroxides or azo compounds. They contain weak N–O bonds and are a category of precursors for C- and N-centred radicals with promise of considerable generality. Photolytic initiation has been applied with considerable success to several varieties including benzophenone oxime esters,30oxime benzoates,31aldoxime esters,32 and dioxime oxalates.33 Zard used Ni powder and acetic acid as an alternative initiation method.34 However, all types of oxime esters have resisted thermal methods for dissociation including conventional heating and MW assistance.35 Even subjecting oxime esters to FVP led to eliminations and electro-cyclisations rather than clean N–O bond scission.36
Oxime
ethers, R2CN–OR′, on the other hand, displayed a remarkable contrast in behaviour. O-Alkyl arylaldoxime ethers R2CN–OCHR′2 underwent radical-induced homolysis by a range of radicals by way of H-atom abstraction followed by β-scission.37 Conventional thermolyses of O-benzyl ketoximes R1R2CN–OCH2Ph in H-donor solvents at about 150 °C led to formation of products from both O–C and N–O bond scission.38 Careful evaluation of the product distributions indicated that when using 9,10-dihydrophenanthrene and 9,10-dihydroanthracene as solvents, a reverse radical disproportionation reaction, in which a hydrogen atom was transferred from the solvent to the oxime ether, followed by β-scission of the resultant aminoalkyl radical, accompanied the direct homolysis.
O-Phenyl oxime
ethers, RR′CN–OPh, can be made in good yields from a wide range of aldehydes and ketones simply by stirring them overnight with the commercially available O-phenyl hydroxylamine hydrochloride. It was expected that the resonance stabilisation of the released phenoxyl radical would encourage selective scission of the N–O bond of these precursors. Clean N–O bond homolyses were indeed observed at moderate temperatures for diaryl, arylalkyl and dialkyl O-phenyl ketoxime ethers yielding iminyl and phenoxyl radicals.39 Attempts to develop synthetic methodology around conventional heating were frustrated by the long reaction times needed and complications from side-processes. Dramatically improved thermolyses leading to superior preparations of several types of heterocycles were, however, achieved on application of MAOS techniques.
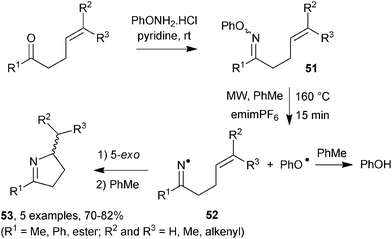
Dihydropyrrole rings are present in numerous biologically active alkaloids40 and so represent an attractive target for synthetic methodologies. When alkenone O-phenyl oxime ethers 51, in toluene solution containing emimPF6 ionic liquid, were subjected to MW irradiation in a Biotage Initiator reactor (nominally 300 MHz) at 160 °C for 15 min, clean dissociation to iminyl 52 and phenoxyl radicals took place (Scheme 15). 5-Exo-ring closure of the iminyls, and subsequent H-abstraction from the solvent, afforded dihydropyrroles 53 in very good yields.41 The phenoxyl radicals also abstracted hydrogen from the solvent forming phenol which was easily separated.
 | ||
| Scheme 15 MW assisted preparation of dihydropyrroles from alkenone O-phenyl oxime ethers.41 | ||
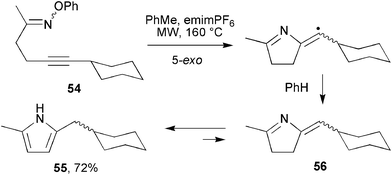
MW irradiation of an analogous O-phenyl precursor with an alkynyl acceptor, 54, gave pyrrole 55 rather than the expected alkylidene-dihydropyrrole 56 (Scheme 16). It appears that the MW conditions prompted two 1,3-proton migrations resulting in the rapid and complete formation of the aromatic pyrrole 55.
Iminyl radical ring closures onto aromatic acceptors were also easily accomplished under MAOS conditions. Alkanal oxime ethers 57 were prepared from the corresponding carbonyl compounds and they ring closed to quinolines 58 in moderate yields when subjected to MW heating (Scheme 17a). Again, the isolated products here were fully aromatic. 6-Endocyclisations of the intermediate iminyls generated cyclohexadienyl radicals which either suffered H-atom abstraction or were oxidised to the corresponding cations which, on proton loss, yielded 58.
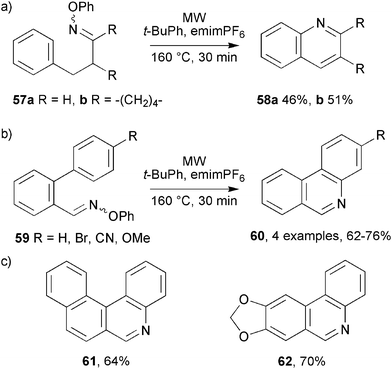 | ||
| Scheme 17 Preparations of (a) quinolines and (b,c) phenanthridines from oxime ethers.41 | ||
A set of biphenyl-containing precursors 59 was readily transformed into the corresponding 3-substituted phenanthridines 60 by MW irradiation (Scheme 17b). Yields were good for electron releasing and electron-withdrawing substituents. Analogous methodology enabled benzo[k]phenanthridine 61 and the natural producttrispheridine 62 to be assembled in good yields (Scheme 17c).
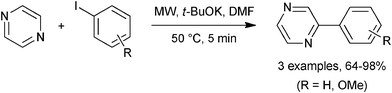
This oxime ether/MAOS methodology was readily adapted for preparations of benzo[h][1,6]naphthyridines 63 by using pyridine as the basal support for the aromatic acceptor and the iminyl radical (Scheme 18a). Curiously, the parallel sequence to benzo[c][1,7]naphthyridine, for which the oxime ether reactant contained a pyridine radical acceptor unit, failed because of sluggish iminyl attack on pyridine. In the benzothiophene-containing precursor 64 the iminyl radical and the phenyl acceptor are supported on a 5-member, rather than a 6-member ring. This did not inhibit ring closure, however, and on MW irradiation benzo[b]thieno[2,3-c]quinoline 65 was isolated in 53% yield (Scheme 18b).
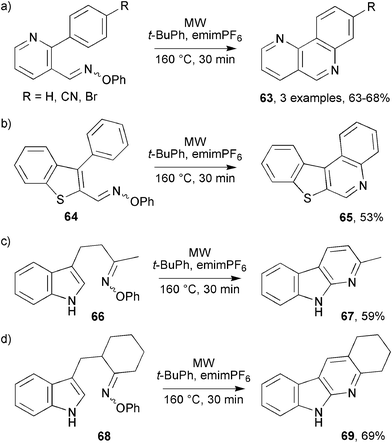 | ||
| Scheme 18 MW assisted preparations of naphthyridines, thienoquinolines and indolopyridines.41b | ||
The precursor 66, containing indole as the radical acceptor unit, was obtained via4-indolylbutan-2-one from the ZrCl4-catalysed reaction of indole with methyl vinyl ketone. That indolopyridine derivative 67 was produced, in which both the indole and pyridine moieties had become aromatic, was a striking example of the propensity for aromatisation under MW conditions (Scheme 18c). A related sequence enabled a formal synthesis of the natural product neocryptolepine to be completed. The starting ketone was obtained from reaction of gramine with 1-cyclohexenylpyrrolidine.42 MW irradiation of 68 in t-BuPh afforded tetrahydroindolo[2,3-b]quinoline 69 in 69% yield (Scheme 18d). Product 69 had previously been converted to the biologically active neocryptolepine in two steps.43
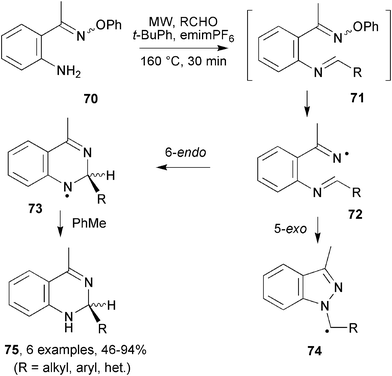
It seemed probable that other N-heterocycles could be made by using O-phenyl oximes containing different types of radical acceptors in their side chains. Radical additions to imines and related compounds are gradually emerging as useful synthetic processes;44 although imines can be difficult to handle because they are prone to hydrolysis and tautomerism. In respect of imines, MW assistance enabled preparations of diaza-heterocycles to be realised under mild and neutral conditions by use of imine functionality in the side chains of suitably functionalised iminyl radicals. The O-phenyl oximes, 70, of 2-aminoacetophenone can be made in good yields by treatment of 2-aminoacetophenone with O-phenylhydroxylamine hydrochloride. The imine precursors 71 could have been made by subsequent condensation with aldehydes in the conventional way. However, it was known that MW irradiation assists imine formation,45 and this technique forms part of a standard method for syntheses of secondary amines.46 It was proposed therefore that the imine-yielding step could be integrated with MW generation of iminyl radical 72; thus enabling the whole sequence to be combined in one pot. For radical 72, competition between the 5-exo and 6-endo ring closures onto the CN bond could lead to formation of quinazoline 73 and/or indazole 74 structures. Attack at the carbon of the CN bond has usually, though not exclusively,47 been observed for C-centred radicals.
On MW irradiation of oxime ether 70 with an aldehyde, in toluene containing emimPF6, the isolated products were actually dihydroquinazolines 75 obtained in good to excellent yields (Scheme 19).48 Reactions with aliphatic aldehydes delivered the corresponding dihydroquinazolines very efficiently. Somewhat lower yields were obtained with aromatic and heterocyclic aldehydes. The presence of unreacted starting materials suggested imine formation was not complete. In no case was any indazole detected; all the ring closures were regiospecific for quinazoline formation. It is likely that the architecture of iminyl radical 72 favours ring closure in the 6-endo mode because the resultant aminyl radical 73 will be resonance stabilized. Furthermore, the 5-exo approach of the nucleophilic iminyl radical to the nitrogen atom of the imine would be polarity mismatched.49 Together these two factors probably account for the observed regiospecificity.
It was known that MW assisted imine formation was promoted by inclusion of zinc chloride.50 Sub-molar equivalents of ZnCl2 were therefore included to promote the MAOS of 76 with less reactive carbonyl compounds.48 Excellent product yields were obtained with aliphatic, aromatic and heterocyclic aldehydes, but the surprising outcome was that quinazolines 77 were formed directly, rather than dihydroquinazolines (Scheme 20).
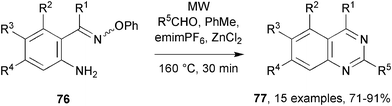 | ||
| Scheme 20 Preparations of quinazolines from oxime ethers and aldehydes.48 | ||
A mechanism in which Zn(II) coordinated to the iminyl nitrogen of an intermediate dihydroquinazoline ammonium radical cation was proposed. No doubt the Lewis acidic nature of ZnCl2 affected the pKa of the neighbouring hydrogen atom in the 2-position of the ring, facilitating deprotonation and hence aromatization to the product. The method also worked well for a variety of substituents in the anilinic part of the molecule. Similar reactions employing ketones instead of aldehydes were less efficient; though some dihydroquinazolines did form, they were accompanied by several by-products.
2.4. Oxygen centred radicals
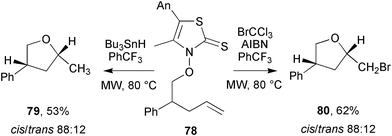
In a series of studies on the MW generation of oxygen-centred radicals, Hartung and co-workers reported on the viability of intramolecular additions, β-fragmentations and homolytic substitutions.51 MW irradiation of thiazole thione 78 in the presence of tributyltin hydride gave addition product 79 in 53% yield with only traces of β-cleavage and homolytic substitution products observed (Scheme 21). Similar yields of brominated addition product 80 were obtained when MW irradiation was performed in the presence of BrCCl3 and AIBN. In both cases the cis/trans ratios were identical, 88:12.
The intermolecular alkoxyl radical addition onto bicyclo[2.2.1]heptene was reported by Hartung and co-workers.52 Thermal activation of N-(alkoxy)thiazolethiones 81 was studied by conventional heating in the presence of AIBN as initiator and also under MW irradiation without any initiator (Scheme 22). Photochemical activation was also investigated using a reactor with irradiation at 350 nm. Whilst all experiments generated alkoxyl radicals that gave addition product 82, it was clear that conventional heating was the most successful. Dielectric heating persistently gave lower yields, but not as low as photolysis yields which were routinely the poorest.
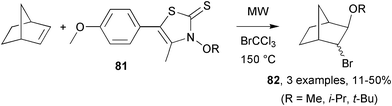 | ||
| Scheme 22 Alkoxyl radical addition to norbornene.52 | ||
2.5. Phosphorus centred radicals
Phosphorus hydrides are seen as attractive replacements for organotin hydrides in radical reductions.53 Parsons and co-workers investigated how to tune the reactivity of phosphorus hydrides by varying the phosphorus substituents.54 Typically initiators (AIBN or Et3B) were used to promote radical additions of phosphorus hydrides. Initiation by heat alone was also examined with hydrides having weak P–H bonds. Conventional heating of phosphinothioate 83a in the presence of diallyl ether 84a in refluxing cyclohexane for 60 h resulted in only a 39% yield of THF derivative 85, present as an inseparable mixture of diastereoisomers (Scheme 23). However, with the aid of Et3B as initiator, reaction time was reduced to 1 h and the yield increased to 67%. With the same substrates, MW irradiation in toluene at 150 °C for 1 h gave the THF derivative in 93% yield. Pyrrolidines (X = NBoc) were also produced using the same conditions from t-butyl diallycarbamate 84b. MW assistance was also successful with diphenylphosphane sulfide 83b which furnished cyclic products from both diallyl ether 84a and carbamate 84b. Unfortunately, the use of MAOS, whilst both improving yields and shortening reaction times, did not improve the selectivity of the reactions, similar diastereoselectivities were observed.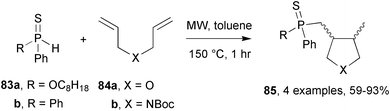 | ||
| Scheme 23 Addition/cyclisations from phosphorus hydrides and diallyl-compounds.54 | ||
2.6. Sulfur centred radicals
Zou and co-workers developed the use of manganese triacetate, an excellent one electron oxidant, as a replacement for potassium ferricyanide or bromine in the radical cyclisation of substituted thioformanilides.55 Conventional heating of arylthioformanilides 86 in acetic acid in the presence of Mn(OAc)3 gave unsatisfactory yields of benzothiazoles 87, however, dielectric heating improved yields and reduced the reaction time 60-fold. Arylthioformanilide 86 tautomerises to thioimidoyl 88 which can be oxidised by Mn(III) to release thiyl radical 89 (Scheme 24a). Ring closure gave cyclohexadienyl radical 90 which, after aromatisation, yielded 2-substituted benzothiazoles, 87. A similar triumph over conventional heating was observed for the MW-assisted Mn(OAc)3 promoted cyclisations of α-benzoylthioformanilides yielding 2-benzoylbenzothiazoles (Scheme 24b).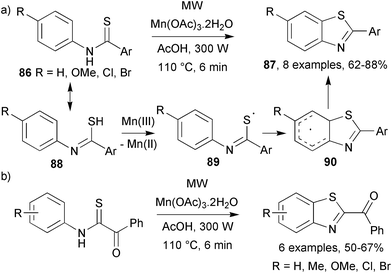 | ||
| Scheme 24 Mn(III) catalysis and benzothiazole formation.55 | ||
3. Conclusions
Building from pioneering research on MW-assisted radical dehalogenations in the 1990s, the methodology has gradually spread to embrace almost all radical reaction types. MW-assistance has proved beneficial for radical additions to, and cyclisations onto, a wide range of acceptors including alkenes, alkynes, aromatics, imines and hetero-aromatics. Furthermore, MW-assisted cyclisations were shown to work well for at least one library of polymer supported precursors. MW conditions have also proved to be advantageous for bimolecular radical phenylations as well as numerous cascade and sequential processes. Significantly, radical-MW methods have been applied to the key steps in the syntheses of several natural products.Reaction times of chain processes initiated by AIBN were, in several cases, reduced to a few minutes. Decomposition of AIBN will be extremely rapid on MW heating so fast propagation steps are needed to match this. Organic halide reductions by tin hydrides, where halogen abstraction by tin radicals and H-atom abstraction from the tin hydride by C-centred radicals are both extremely rapid propagation steps, clearly fulfil this condition. It has been found that regioselectivity in radical addition and ring closure processes is normally maintained under MW conditions. However, as might be expected from the somewhat higher temperatures, stereoselectivities were not improved by MW assistance in the few cases so far examined.
MW methods have been developed not only for the generation of C-centred radicals but also for N-centred and a few other hetero-radicals. This has resulted in many rapid and benign preparative procedures for an impressive variety of heterocycles. A feature of many such ring closures is the spontaneous aromatisation of dihydro-intermediates. Such oxidations are obviously favoured by the dielectric heating, particularly in non-H-donor solvents.
MW-methods have opened up several new and innocuous alternatives to toxic organotin reagents. In this way homolytic dissociations of several novel classes of compounds have been brought within the realm of everyday bench chemistry. For example, alkoxyamines smoothly release C-centred radicals and oxime ethers provide a variety of iminyl radicals under these conditions. In addition, several types of organo-element and organometallic precursors have yielded novel C-centred and hetero-radicals.
It is apparent that the union of MW with radical methods is set to accelerate in the immediate future resulting in a multitude of new and improved syntheses.
Acknowledgements
We thank EaStChem and the EPSRC for financial support.References
- (a) See for example P. Renaud and M. Sibi (ed.) Radicals in Organic Synthesis, Vols. 1 & 2, Wiley, Weinheim, 2001 Search PubMed; (b) S. Z. Zard, Radical Reactions in Organic Synthesis, Oxford University Press, Oxford, 2003 Search PubMed; (c) H. Togo, Advanced Free Radical Reactions for Organic Synthesis, Elsevier, Amsterdam, 2004 Search PubMed; (d) A. Gansäuer, ed. Top. Curr. Chem., 2006, 264 Search PubMed(parts I & II); (e) A. Postigo, RSC Adv., 2011, 1, 14–32 RSC.
- C. Chatgilialoglu, Organosilanes in Radical Chemistry, Wiley, Chichester, 2004 Search PubMed.
- (a) S. Caddick, Tetrahedron, 1995, 51, 10403–10432 CrossRef CAS; (b) L. Perreux and A. Loupy, Tetrahedron, 2001, 57, 9199–9223 CrossRef CAS; (c) M. Larhed, C. Moberg and A. Hallberg, Acc. Chem. Res., 2002, 35, 717–727 CrossRef CAS; (d) C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS; (e) A. de la Hoz, A. Díaz-Ortiz and A. Moreno, Chem. Soc. Rev., 2005, 34, 164–178 RSC; (f) D. Bogdal, Microwave-Assisted Organic Synthesis, Elsevier, Amsterdam and Boston, 2005 Search PubMed; (g) A. Loupy, Microwaves in Organic Synthesis 2nd ed., Wiley-VCH, Weinheim, 2006 Search PubMed; (h) M. Larhed and K. Olofsson (ed.), Microwave Methods in Organic Synthesis, Springer, Berlin, 2006 Search PubMed; (i) E. Van der Eycken and C. O. Kappe (ed.), Microwave-Assisted Synthesis of Heterocycles, Springer, Berlin and New York, 2006 Search PubMed.
- (a) C. Gabriel, S. Gabriel, E. H. Grant, B. S. J. Halstead and D. M. P. Mingos, Chem. Soc. Rev., 1998, 27, 213–223 RSC; (b) D. M. P. Mingos and D. R. Baghurst, Chem. Soc. Rev., 1991, 20, 1–47 RSC.
- D. R. Baghurst and D. M. P. Mingos, J. Chem. Soc., Chem. Commun., 1992, 674–677 RSC.
- The capacity for a given material to convert MW energy to heat is given by a loss factor expressed as a quotient tanδ = ε′′/ε′. Where ε′′ is the dielectric loss, the efficiency at which electromagnetic radiation is converted into heat energy, and ε′ is the dielectric constant, the ability of a molecule to be polarised by the electric field.
- (a) P. A. Baguley and J. C. Walton, Angew. Chem., Int. Ed., 3072–3082 Search PubMed; (b) A. Studer and S. Amrein, Synthesis, 835–849 Search PubMed; (c) V. Darmency and P. Renaud, Top. Curr. Chem., 71–106 Search PubMed; (d) S.-H. Ueng, A. Solovyev, X. Yuan, S. J. Geib, L. Fensterbank, E. Lacôte, M. Malacria, M. Newcomb, J. C. Walton and D. P. Curran, J. Am. Chem. Soc., 2009, 131, 11256–11262 CrossRef CAS.
- (a) J. I. G. Cadogan, B. A. J. Clark, D. Ford, R. J. MacDonald, A. D. MacPherson, H. McNab, I. S. Nicolson, D. Reed and C. C. Sommerville, Org. Biomol. Chem., 2009, 7, 5173–5183 RSC; (b) H. McNab, Flash Vacuum Pyrolysis, in Kirk-Othmer Encyclopedia of Chemical Technology, 5th ed., A. Seidel (Ed.), 2006, 21, 134–158 Search PubMed; (c) R. A. Aitken, N. Karodia, T. Massil and R. J. Young, J. Chem. Soc., Perkin Trans. 1, 2002, 533–541 RSC (and references cited therein); (d) H. McNab, Contemp. Org. Synth., 1996, 3, 373–396 RSC.
- A. K. Bose, M. S. Manhas, M. Ghosh, M. Shah, V. S. Raju, S. S. Bari, S. N. Newaz, B. K. Banik, A. G. Chaudhary and K. J. Barakat, J. Org. Chem., 1991, 56, 6968–6970 CrossRef CAS.
- J. A. Aimetti, E. S. Hamanaka, D. A. Johnson and M. S. Kellogg, Tetrahedron Lett., 1979, 20, 4631–4634 CrossRef.
- (a) K. Olofsson, S.-Y. Kim, M. Larhed, D. P. Curran and A. Hallberg, J. Org. Chem., 1999, 64, 4539–4541 CrossRef CAS; (b) W. Zhangin Microwave Methods in Organic Synthesis, M. Larhed and K. Olofsson (ed.), SpringerBerlin, 2006, vol. 266, pp. 145–166 Search PubMed.
- Y. Borguet, A. Richel, S. Delfosse, A. Leclerc, L. Delaude and A. Demonceau, Tetrahedron Lett., 2007, 48, 6334–6338 CrossRef CAS.
- S. Horikoshi, J. Tsuzuki, M. Kajitani, M. Abe and N. Serpone, New J. Chem., 2008, 32, 2257–2262 RSC.
- C. Wetter, K. Jantos, K. Woithe and A. Studer, Org. Lett., 2003, 5, 2899–2902 CrossRef CAS.
- (a) A. Studer, Chem. Soc. Rev., 2004, 33, 267–273 RSC; (b) A. Studer, Chem.–Eur. J., 2001, 7, 1159–1164 CrossRef CAS; (c) H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS.
- C. Wetter and A. Studer, Chem. Commun., 2004, 174–175 RSC.
- A. J. Herrera and A. Studer, Synthesis, 2005, 1389–1396 CAS.
- C. Ericsson and L. Engman, J. Org. Chem., 2004, 69, 5143–5146 CrossRef CAS.
- A. J. Clark, Chem. Soc. Rev., 2002, 31, 1–11 RSC.
- (a) J. A. Bull, M. G. Hutchings, C. Luján and P. Quayle, Tetrahedron Lett., 2008, 49, 1352–1356 CrossRef CAS; (b) J. A. Bull, M. G. Hutchings and P. Quayle, Angew. Chem., Int. Ed., 2007, 46, 1869–1872 CrossRef CAS.
- M. Lamberto, D. F. Corbett and J. D. Kilburn, Tetrahedron Lett., 2003, 44, 1347–1349 CrossRef CAS.
- A. Teichert, K. Jantos, K. Harms and A. Studer, Org. Lett., 2004, 6, 3477–3480 CrossRef CAS.
- B. Janza and A. Studer, Org. Lett., 2006, 8, 1875–1878 CrossRef CAS.
- H. Akamatsu, K. Fukase and S. Kusumoto, Synlett, 2004, 1049–1053 CAS.
- I. Prediger, T. Weiss and O. Reiser, Synthesis, 2008, 2008, 2191–2198 CrossRef.
- E. S. Sherman, P. H. Fuller, D. Kasi and S. R. Chemler, J. Org. Chem., 2007, 72, 3896–3905 CrossRef CAS.
- (a) S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673–4676 CrossRef CAS; (b) G. Deng, K. Ueda, S. Yanagisawa, K. Itami and C.-J. Li, Chem.–Eur. J., 2009, 15, 333–337 CrossRef CAS.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018–5022 CrossRef CAS.
- H. Y. Cho, A. Ajaz, D. Himali, P. A. Waske and R. P. Johnson, J. Org. Chem., 2009, 74, 4137–4142 CrossRef CAS.
- (a) M. Hasebe, K. Kogawa and T. Tsuchiya, Tetrahedron Lett., 1984, 25, 3887–3890 CAS; (b) M. Hasebe and T. Tsuchiya, Tetrahedron Lett., 1988, 29, 6287–6290 CrossRef CAS.
- (a) J. Boivin, A.-M. Schiano and S. Z. Zard, Tetrahedron Lett., 1994, 35, 249–252 CrossRef CAS; (b) J. Boivin, A.-C. Callier-Dublanchet, B. Quiclet-Sire, A.-M. Schiano and S. Z. Zard, Tetrahedron, 1995, 51, 6517–6528 CrossRef CAS; (c) M. Depature, D. Siri, J. Grimaldi, J. Hatem and R. Faure, Tetrahedron Lett., 1999, 40, 4547–4550 CrossRef CAS.
- (a) A. J. McCarroll and J. C. Walton, Chem. Commun., 2000, 351–352 RSC; (b) A. J. McCarroll and J. C. Walton, J. Chem. Soc., Perkin Trans. 2, 2000, 2399–2409 RSC; (c) R. Alonso, P. J. Campos, B. García and M. A. Rodríguez, Org. Lett., 2006, 8, 3521–3523 CrossRef CAS; (d) R. Alonso, P. J. Campos, M. A. Rodríguez and D. Sampedro, J. Org. Chem., 2008, 73, 2234–2239 CrossRef CAS.
- (a) F. Portela-Cubillo, E. M. Scanlan, J. S. Scott and J. C. Walton, Chem. Commun., 2008, 4189–4191 RSC; (b) F. Portela-Cubillo, J. Lymer, E. M. Scanlan, J. S. Scott and J. C. Walton, Tetrahedron, 2008, 64, 11908–11916 CrossRef CAS.
- (a) J. Boivin, A.-M. Schiano and S. Z. Zard, Tetrahedron Lett., 1992, 33, 7849–7852 CrossRef CAS; (b) S. Z. Zard, Synlett, 1996, 1148–1154 CrossRef CAS.
- J. C. Walton, unpublished results Search PubMed.
- F. Portela-Cubillo, B. A. Surgenor, R. A. Aitken and J. C. Walton, J. Org. Chem., 2008, 73, 8124–8127 CrossRef CAS.
- A. J. McCarroll and J. C. Walton, J. Chem. Soc., Perkin Trans. 2, 2000, 1868–1875 RSC.
- J. A. Blake, K. U. Ingold, S. Lin, P. Mulder, D. A. Pratt, B. Sheeller and J. C. Walton, Org. Biomol. Chem., 2004, 2, 415–420 CAS.
- J. A. Blake, D. A. Pratt, S. Lin, J. C. Walton, P. Mulder and K. U. Ingold, J. Org. Chem., 2004, 69, 3112–3120 CrossRef CAS.
- (a) F. Bellina and R. Rossi, Tetrahedron, 2006, 62, 7213–7256 CrossRef CAS; (b) L. Constantino and D. Barlocco, Curr. Med. Chem., 2006, 13, 65–85 CrossRef.
- (a) F. Portela-Cubillo, J. S. Scott and J. C. Walton, Chem. Commun., 2007, 4041–4043 RSC; (b) F. Portela-Cubillo, J. S. Scott and J. C. Walton, J. Org. Chem., 2008, 73, 5558–5565 CrossRef CAS.
- M. von Strandtmann, M. P. Cohen and J. Shavel, Jr., J. Org. Chem., 1965, 30, 3240–3242 CrossRef CAS.
- G. S. M. Sundaram, C. Venkatesh, U. K. Syam Kumar, H. Ila and H. Junjappa, J. Org. Chem., 2004, 69, 5760–5762 CrossRef CAS.
- K. Friestad, Tetrahedron, 2001, 57, 5461–5496 CrossRef.
- G. Vo-Thanh, H. Lahrache, A. Loupy, I.-J. Kim, D.-H. Cang and C.-H. Jun, Tetrahedron, 2004, 60, 5539–5543 CrossRef.
- P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2004, 126, 7450–7451 CrossRef CAS.
- M. J. Tomaszewski and J. Warkentin, Tetrahedron Lett., 1992, 33, 2123–2126 CrossRef CAS.
- (a) F. Portela-Cubillo, J. S. Scott and J. C. Walton, Chem. Commun., 2008, 2935–2937 RSC; (b) F. Portela-Cubillo, J. S. Scott and J. C. Walton, J. Org. Chem., 2009, 74, 4934–4942 CrossRef CAS.
- S. Kim, K. S. Yoon and Y. S. Kim, Tetrahedron, 1997, 38, 73–80 CrossRef.
- A. Pouilhès, Y. Langlois and A. Chiaroni, Synlett, 2003, 1488–1490 Search PubMed.
- J. Hartung, K. Daniel, T. Gottwald, A. Groβ and N. Schneiders, Org. Biomol. Chem., 2006, 4, 2313–2322 CAS.
- J. Hartung, N. Schneiders and T. Gottwald, Tetrahedron Lett., 2007, 48, 6027–6030 CrossRef CAS.
- (a) D. Leca, L. Fensterbank, E. Lacôte and M. Malacria, Chem. Soc. Rev., 2005, 34, 858–865 RSC; (b) T. A. Khan, R. Tripoli, J. J. Crawford, C. G. Martin and J. A. Murphy, Org. Lett., 2003, 5, 2971–2974 CrossRef CAS; (c) C. G. Martin, J. A. Murphy and C. R. Smith, Tetrahedron Lett., 2000, 41, 1833–1836 CrossRef CAS; (d) D. H. R. Barton, D. O. Jang and J. C. Jaszberenyi, J. Org. Chem., 1993, 58, 6838–6842 CrossRef CAS.
- C. M. Jessop, A. F. Parsons, A. Routledge and D. J. Irvine, Eur. J. Org. Chem., 2006, 1547–1554 CrossRef CAS.
- X.-J. Mu, J.-P. Zou, R.-S. Zeng and J.-C. Wu, Tetrahedron Lett., 2005, 46, 4345–4347 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |