DOI:
10.1039/C1RA00926E
(Paper)
RSC Adv., 2012,
2, 1047-1054
Synthesis of Pd nanoparticles on heteropolyacid-supported silica by a photo-assisted deposition method: an active catalyst for the direct synthesis of hydrogen peroxide
Received
20th October 2011
, Accepted 20th October 2011
First published on 7th December 2011
Abstract
A simple and unique methodology to synthesize active Pd nanoparticles using Cs2.5H0.5PW12O40 heteropolyacid-supported silica (CsHPA/SiO2) under UV-light irradiation has been developed. By the photo-assisted deposition (PAD) method, a Pd2+ precursor in aqueous solution can be deposited and easily reduced by the [PW12O40]4− species generated by photolysis of [PW12O40]3− species in the presence of iPrOH as a sacrificial reagent, to afford nanosized Pd0 metal particles (Pd/CsHPA/SiO2). No direct Pd metal deposition was observed on either pure SiO2 without the CsHPA phase under UV-light irradiation or on CsHPA/SiO2 without UV-light irradiation. The catalytic activity for the direct synthesis of hydrogen peroxide (H2O2) using H2 and O2 gases under atmospheric pressure was strongly dependent on the preparation conditions, variation of CsHPA content, and the type of catalyst support. In addition, the Pd/CsHPA/SiO2 catalyst prepared by the PAD method exhibited significantly better activity than the conventionally prepared impregnation catalyst. The Pd surface area from CO adsorption measurements, and also the suppression of undesired side reactions were determined as the crucial factors to achieve efficient catalytic performance.
1. Introduction
Palladium is one of the most precious catalyst metals and is frequently employed in a wide variety of industrial catalytic processes, especially for the removal of emissions from the automobiles (e.g., three-way catalyst), in addition to functional group transformation (e.g., Wacker reaction) and carbon–carbon bond formation (e.g., Mizoroki–Heck and Suzuki–Miyaura coupling reactions) in organic synthesis.1 Other examples of Pd metal applications are in electronics, dentistry, medicine, hydrogen purification, and clean hydrogen production, including fuel cells. The versatile applicability of Pd metal has resulted in progress toward the manufacture of highly active catalysts with reliable durability in order to minimize the quantity consumed.
Until now, the most convenient methods for the preparation of supported-metal catalysts have been the incipient wetness technique and the ion-exchange method, the former of which a support material is impregnated with metal precursors in the solution phase, followed by the thermal treatment and/or reduction with H2 to generate metal nanoparticles.2 This process can be further sophisticated by post-synthesis modification of the support materials, which stimulates the interaction of the metal precursor with the surface.3 This process is effective for the large scale production of catalysts due to its essential practicality; however, unfortunately, it does not offer sufficiently small nanoparticles.
As an alternative, we anticipate that the photo-assisted deposition (PAD) method is a promising technique for the synthesis of nanosized metal particles on solid surfaces.4 In the case of semiconductor materials, exemplified by TiO2, photochemical excitation leads to charge separation, in which electrons (e−) are promoted to the conduction band and holes (h+) are left in the valence band (eqn 1).5 Thus, irradiation of a mixture of semiconductor materials and metal precursor ions initiates reduction by the conduction band electrons and leads to the formation of metal nanoparticles (eqn 2). Considering this, our previous works have also proposed a novel PAD method combined with isolated and tetrahedrally-coordinated Ti-containing zeolite and mesoporous silica as photo-responsive catalyst supports.6 Photoexcitation causes electron transfer from the oxygen to Ti4+ ions within the silica framework, resulting in the formation of pairs of trapped hole centers (O−) and electron centers (Ti3+), which serve as anchors for the deposition of metal precursors, such as precursors based on Pt, Pd, and PdAu.
| | | e− + Mn+ → M0colloidal | (2) |
In the present study, we have successfully utilized Cs2.5H0.5PW12O40 heteropolyacid (HPA)-supported silica (CsHPA/SiO2) as a platform to produce highly dispersed Pd nanoparticles under UV-light irradiation. HPAs are precisely defined anionic metal-oxygen clusters with diverse and unique physicochemical properties.7 HPAs perform as both catalytic reducing reagents and as stabilizers for the synthesis of colloidal Ag, Au, Pd, and Pt nanoparticles upon illumination with UV/near-vis light.8 During the process, HPAs absorb and abstract electrons from oxidizable organic substrates (S) as sacrificial reagents, for instance iPrOH (eqn 3). The photochemically reduced HPAs subsequently reduce metal ions to the corresponding metal nanoparticles with reasonably good dispersity, according to eqn 4. Recently, it has been demonstrated that the size and shape of Ag nanoparticles can be controlled by varying operational parameters such as the [Mn+]/[HPA] ratio, pH, or the reaction temperature.9 Although previous studies on the utilization of HPAs have mainly been focused on soluble systems in solution, to the best of our knowledge, application to the synthesis of nanosized metal particles on solid surfaces have not been investigated to date. The present paper describes the synthesis and characterization of Pd catalysts obtained and evaluation of their prominent catalytic activity for the direct synthesis of hydrogen peroxide (H2O2) from H2 and O2 under atmospheric pressure.
| | | HPA + S + hν → HPA(e−) + Sox | (3) |
| | | HPA(e−) +Mn+ → HPA + M0colloidal | (4) |
2. Experimental
2.1. Materials
PdCl2 was purchased from Wako Pure Chemical Ind., Ltd. H3PW12O4, Cs2CO3, and fumed SiO2 (particle size: 0.007 μm) were obtained from Aldrich Chemical Co. γ-Al2O3 (JRC-ALO-8) was supplied from the Japan Catalysis Society. USY-6 was supplied by Union Syowa K.K. Mesoporous silica MCM-41 was prepared via the surfactant self-assembly approach. Solvents and all commercially-available organic compounds for catalytic reactions were purified using standard procedures.
2.2. Preparation of catalyst
The preparation procedure is illustrated in Fig. 1. SiO2 (5.0 g) was mixed with a 5 mL H3PW12O4 HPA aqueous solution (0.035 M) and 5 mL Cs2CO3 aqueous solution (0.044 M) and stirred at room temperature for 1 h. The suspension was evaporated under vacuum, and the obtained powder was calcined at 673 K for 2 h in air, giving CsHPA/SiO2 as a white powder. In this way, the CsHPA/SiO2 samples with different CsHPA loadings (5, 10, 20, and 50 wt%) were prepared. CsHPA/MCM-41, CsHPA/Al2O3, and CsHPA/USY-6 (SiO2/Al2O3 = 6) were also synthesized according to this procedure.
Pd
nanoparticles were deposited by photo-assisted methods. CsHPA/SiO2 (0.2 g) was mixed with a 0.81 mL aqueous PdCl2 solution (11.6 mM) in a quartz vessel at room temperature for 1 h with vigorous stirring under UV-light irradiation using a 100 W high pressure Hg lamp. The obtained precipitate was centrifuged, washed repeatedly with deionized water, and dried at room temperature under vacuum, giving Pd/CsHPA/SiO2 (Pd: 0.5 wt%). In comparison, catalysts denoted imp-Pd/CsHPA/SiO2 and imp-Pd/SiO2 were prepared by the conventional impregnation method.
2.3. Characterization
Brunauer–Emmett–Teller (BET) surface area measurements were conducted at 77 K using a BEL-SORP max (Bel Japan, Inc.) apparatus. The sample was degassed under vacuum at 473 K prior to data collection. Inductively coupled plasma measurements were performed using a Nippon Jarrell-Ash ICAP-575 Mark II instrument. CO pulse adsorption was performed to measure the surface area of metal using a BEL-METAL-1 (Bel Japan, Inc) instrument. Pd K-edge extended X-ray absorption fine structure (EXAFS) spectra were recorded using a fluorescence-yield collection technique at the beam line 01B1 station with an attached Si(311) monochromator at SPring-8 of the Japan Synchrotron Radiation Research Institute (JASRI), (prop. No. 2011A1092, 2010B1098). The EXAFS data were normalized by fitting the background absorption coefficient, around the energy region higher than the edge of approximately 35–50 eV, with the smoothed absorption of an isolated atom. The EXAFS data were examined using the Rigaku EXAFS analysis program. Fourier transformation (FT) of k3-weighted normalized EXAFS data was performed over the 3.5 < k/Å−1 < 11 range to obtain the radial structure function. CN (coordination number of scatters), R (distance between an absorbing atom and scatterer), and the Debye–Waller factor were estimated by curve-fitting analysis with the inverse FT of the 1.6 < R/Å < 3.0 range assuming single scattering.
2.4. Direct synthesis of H2O2 from H2 and O2
The catalyst (0.05 g) and 0.01 M HCl aqueous solution (20 mL) were placed into a reaction vessel (50 mL). The resulting mixture was stirred under bubbling of gaseous H2 and O2 (20 mL min−1, H2:O2 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 303 K for 6 h with magnetic stirring. The amount of H2O2 formed was monitored using a hydrogen peroxide counter (HP-300, Hiranuma).
:1) at 303 K for 6 h with magnetic stirring. The amount of H2O2 formed was monitored using a hydrogen peroxide counter (HP-300, Hiranuma).
3. Results and discussion
Most HPAs are highly soluble in polar solvents and have low surface areas (<10 m2 g−1). Cs-exchanged HPA (Cs2.5H0.5PW12O40) was employed due to its insolubility, high surface area, and porous structure when forming a tertiary structure.10 The Cs:H ratio of 2.5:0.5 was selected to take advantage of the high surface area and high acidity.11 The synthesis of CsHPA on SiO2 was performed by an incipient wetness impregnation method using a mixture of H3PW12O4 HPA and Cs2CO3 aqueous solution with variation of the Cs2.5H0.5PW12O40 content (5, 10, 20 and 50 wt%) to afford CsHPA/SiO2 as white powders. CsHPA/MCM-41, CsHPA/Al2O3, and CsHPA/USY-6 with 10 wt% CsHPA loading were also synthesized according to this procedure. The CsHPA content was measured by ICP analysis. The BET surface area and pore volume were slightly decreased with the increase in the CsHPA content (Table 1).
Table 1 Textural properties of CsHPA-supported catalysts
| Sample |
HPA/wt (%) |
SBET/m2 g−1 |
Pore volume/cm3 g−1 |
|
CsHPA/SiO2 |
5 |
286 |
1.30 |
| 10 |
276 |
1.18 |
| 20 |
237 |
1.13 |
| 50 |
194 |
0.86 |
|
CsHPA/Al2O3 |
10 |
139 |
0.67 |
|
CsHPA/MCM-41 |
10 |
864 |
0.68 |
|
CsHPA/USY-6 |
10 |
417 |
0.33 |
Fig. 2(A) shows XRD patterns of CsHPA/SiO2 with different amounts of CsHPA. For the 5 and 10 wt% samples, only a characteristic diffraction peak at 26φ, due to the well-defined Keggin structure of CsHPA, was observed, which suggests that the CsHPAs are highly dispersed on the silica support at these loading levels. The peak intensity increased when the amount of CsHPA loading increased, which also indicates the agglomeration of CsHPA crystals. XRD patterns of Pd/CsHPA(10 wt%)/support for various types of catalyst supports are also depicted in Fig. 2(B). The formation of CsHPA was observed on CsHPA/SiO2 and CsHPA/USY-6. However, CsHPA/MCM-41 and CsHPA/Al2O3 exhibited very weak intensity, which suggests the formation of a low crystalline and/or amorphous phase of CsHPA.
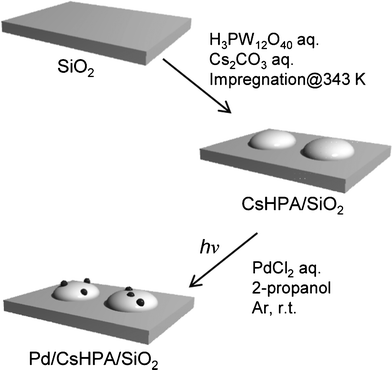 |
| | Fig. 1 Schematic illustration of Pd/CsHPA/SiO2 synthesis by the PAD method under UV-light irradiation. | |
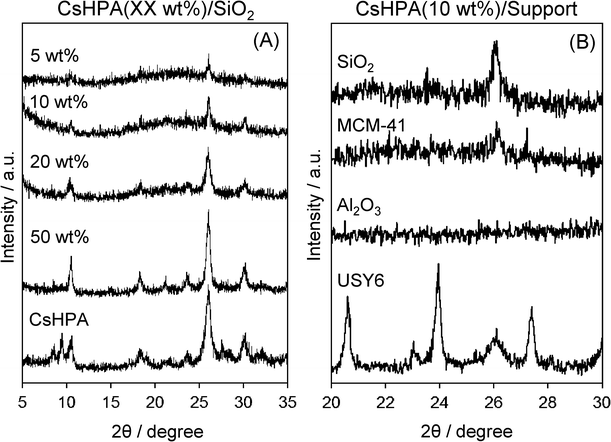 |
| | Fig. 2
XRD pattern of (A) CsHPA/SiO2 with different amounts of CsHPA and (B) CsHPA(10 wt%)/support with different catalyst supports | |
Fig. 3A shows UV-vis spectra of the CsHPA-supported SiO2, which exhibits an absorption band around 200–390 nm. No absorption was observed in the absence of the CsHPA, which indicates the presence of fine particles of the photo-responsive CsHPA. The larger particles of crystalline CsHPA usually exhibit an absorption band in the longer wavelength region. The absorption band was slightly shifted to the longer wavelength region with increasing CsHPA content, which indicates an increase in the particle size of crystalline CsHPA. Comparison of catalyst supports revealed the absorption edge of CsHPA/SiO2 was observed in the longer wavelength region than those of CsHPA/MCM-41, CsHPA/Al2O3 and CsHPA/USY-6 (Fig. 3B). This result corresponds with the XRD pattern intensity, which suggests that the formation of crystalline CsHPA is suppressed on such catalyst supports.
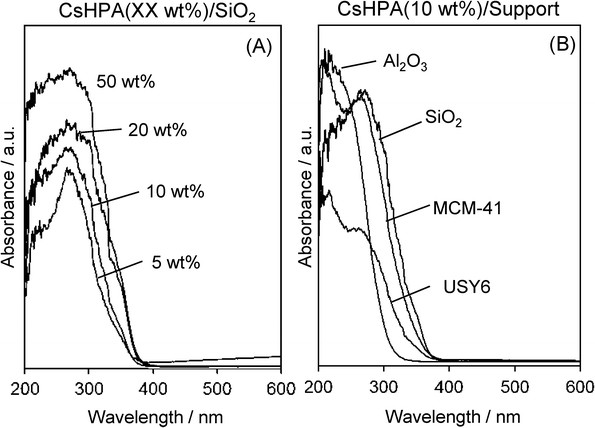 |
| | Fig. 3
UV-vis spectra of (A) CsHPA/SiO2 with different amounts of CsHPA and (B) CsHPA(10 wt%)/support with different catalyst supports. | |
Under UV-light irradiation of a slurry of CsHPA/SiO2 in an aqueous PdCl2 solution, the Pd2+ precursor can be successfully reduced to Pd0 metal on its surface. The color change of samples in the slurry gradually changed from white to gray and the color density increased with increasing irradiation time. No direct Pd metal deposition was observed, on either the pure silica without CsHPA phase under UV-light irradiation or on CsHPA/SiO2 without UV-light irradiation. The addition of iPrOH as a sacrificial reagent was necessary to achieve reduction of the Pd2+ precursor. The results indicate that the Pd2+ precursor underwent reduction by the photochemically generated [PW12O40]4− species from [PW12O40]3− and subsequently transformed into nanosized Pd0 metal particles. The reduction power of the electrons is sufficient to drive reduction of the Pd2+ precursor, because the reduction potential of Pd2+ ions is far more positive than that of HPA (E0(Pd2+/Pd0) = + 0.63 V, E0([PW12O40]3−/[PW12O40]4−) = + 0.22 V vs.NHE).12 The XRD peaks attributable to fcc Pd metal and/or oxide could not be confirmed, due to the small size of the nanoparticles dispersed on the silica surface. There was no significant difference in the specific surface area of CsHPA/SiO2 before and after the photo-deposition process.
3.2. Direct synthesis of H2O2
In an effort to investigate the potential catalytic activities of the Pd catalysts prepared by the PAD method, the synthesis of hydrogen peroxide (H2O2) from H2 and O2 was conducted under atmospheric pressure at room temperature. H2O2 is increasingly used in industry, because the only by-product of its reaction is water. This chemical is currently produced by the anthraquinone method, which involves sequential hydrogenation and oxidation of an alkyl anthraquinone as an intermediate.13 This indirect process requires the use of toxic solvents and is energy intensive, resulting in a high overall cost for industry. Recently, the direct synthesis of H2O2 from H2 and O2 over supported Pd catalysts has attracted much attention, because it leads to fewer side products and is significantly cheaper.14 In preliminary experiments, the highest activity was obtained under bubbling of gaseous H2 and O2 (20 mL min−1, H2:O2 = 1:1). The low flow rate and different H2 and O2 ratios decreased catalytic activity, and thus employed the above conditions in the following catalytic experiments.
Fig. 4 and 5 show the results of H2O2 production using Pd/CsHPA(10 wt%)/SiO2 catalysts prepared under various PAD conditions. The photo-irradiation time has a significant influence on the catalytic activities; a relatively short time (0.5–2 h) is desirable to achieve high H2O2 concentration, while gradual decreases were observed over 2 h (Fig. 4). Additionally, The Pd/CsHPA/SiO2 catalyst prepared by the PAD method exhibits higher activity than that prepared by the conventional impregnation technique by a factor of ca. 6. The average Pd diameters were determined to be 2.0 and 8.4 nm for PAD and imp methods, respectively, according to CO adsorption measurements. The higher dispersion of Pd metal particles is preferable for the formation of H2O2; therefore, this result supports the usefulness of the PAD method to produce nanosized Pd metal by a simple photo-irradiation process. The catalytic activity also varies according to the type of sacrificial reagent employed in the PAD method; the H2O2 concentration increases in the order of MeOH < EtOH < iPrOH as sacrificial reagents (Fig. 5). This tendency corresponds well with the increasing hydrogenation enthalpies of the corresponding aldehydes15; oxidation of iPrOH is favorable compared to MeOH and EtOH, which produce more [PW12O40]4− species by the photolysis of [PW12O40]3− according to eqn 3. The formation of [PW12O40]4− may play a crucial role in determining the particle size, because rapid reduction generates more nuclei for Pd metal particles in a shorter period and efficiently suppresses the growth of metal nanoparticles.16
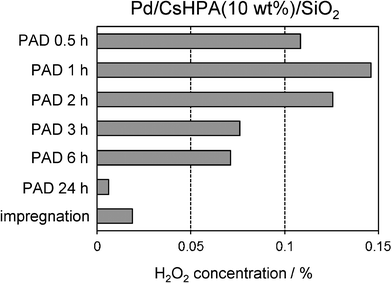 |
| | Fig. 4 Effect of photo-irradiation time in the PAD method on the concentration of generated H2O2. | |
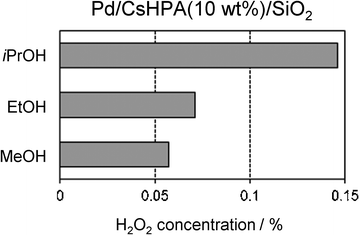 |
| | Fig. 5 Effect of sacrificial agents used in the Pd deposition on the concentration of generated H2O2. | |
The effect of the CsHPA loading amount on the concentration of generated H2O2 is shown in Fig. 6. The activity increased with increasing CsHPA content and was maximized at 10 wt%. On the other hand, the catalysts with CsHPA loading greater than 10 wt% exhibited a gradual decrease in the H2O2 concentration. The catalytic activity of pure Pd/SiO2 (CsHPA: 0 wt%) prepared by the conventional impregnation method was extremely low under identical reaction conditions. The enhancement effect was almost negligible when a physical mixture of Pd/SiO2 and CsHPA was employed. These catalytic efficiencies are strongly dependent on the Pd surface area, as determined by CO adsorption analysis. Pd/CsHPA(10 wt%)/SiO2 with the largest Pd surface area had the highest activity for the synthesis of H2O2, while the catalytic activity of Pd/CsHPA(50 wt%)/SiO2, which had the smallest Pd surface area, was substantially less effective (Fig. 7). These results indicate that the CsHPA phase influences the preparation of active Pd nanoparticles by photo-irradiation as well as the catalytic activity for the synthesis of H2O2.
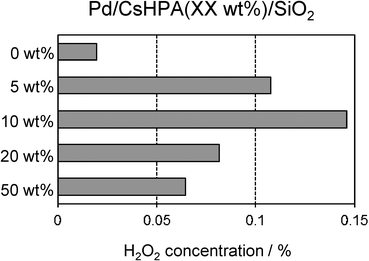 |
| | Fig. 6 Effect of CsHPA amount in Pd/CsHPA/SiO2 on the concentration of generated H2O2. | |
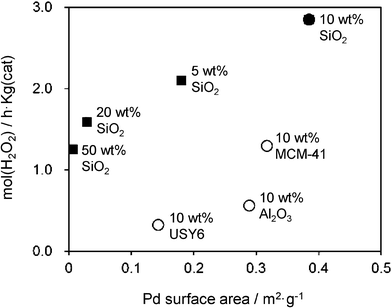 |
| | Fig. 7 Correlation of H2O2 productivity with the Pd surface area determined by CO adsorption for (■) Pd/CsHPA/SiO2 with different amounts of CsHPA and (○) Pd/CsHPA(10 wt%)/support with different catalyst supports. | |
The support material was also found to be an important factor in attaining efficient catalytic activity, as shown in Fig. 8. Among the supports investigated, amorphous SiO2 was the most suitable, and was significantly better than mesoporous silica MCM-41 and zeolite USY-6 (SiO2/Al2O3 = 6) with higher BET surface areas and Al2O3. The crystal size of CsHPA (10–50 nm) is larger than the pore size of MCM-41 and zeolite USY-6; therefore, the CsHPA phase is deposited on external surfaces, which may not have a significant impact on the catalytic activity. The main reason for the differences in catalytic activity can also be explained with respect to the Pd surface area (determined by CO adsorption); the use of MCM-41, Al2O3, and zeolite USY substantially decreased the Pd surface area and thereby decreased the catalytic activity (Fig. 7).
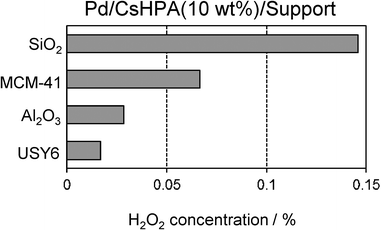 |
| | Fig. 8 Effect of support on the concentration of generated H2O2. | |
In addition, the state of the Pd species also accounts for the differences in catalytic activity. To elucidate such phenomena, XAFS measurements were conducted. Fourier transforms of the Pd K-edge EXAFS spectra and the results of curve-fitting analysis of Pd/CsHPA/support are shown in Fig. 9 and Table 2, respectively, together with that for PdO and Pd foil as reference samples. The Pd/CsHPA/SiO2 and Pd/CsHPA/MCM-41 exhibited a single peak at approximately 2.7–2.8 Å, which could be assigned to the contiguous Pd–Pd bond in the metallic form. This spectrum is similar to that for Pd foil. The intensity of the Pd–Pd peak of the Pd/CsHPA/SiO2 catalyst is lower than that of Pd foil and Pd/CsHPA/MCM-41. Indeed, curve-fitting analysis revealed that the CN value of the Pd/CsHPA/SiO2 was smaller than the CN value of Pd foil and Pd/CsHPA/MCM-41, which suggests the formation of smaller Pd nanoparticles. On the other hand, the Pd/CsHPA/Al2O3 and Pd/CsHPA/USY-6 samples showed an additional peak at around 1.6 Å, attributable to the Pd–O bond, which was observed in PdO, together with the Pd–Pd bond in the metallic form at around 2.7–2.8 Å. This result suggests that the complete reduction of the Pd2+ species cannot be performed on such catalyst supports. The crystallinity of the CsHPA phase on MCM-41, Al2O3, and zeolite USY-6, as observed by XRD, is low compared to that of SiO2, which may have an influence on the reduction process of the Pd2+ precursor into active Pd0 nanoparticles.
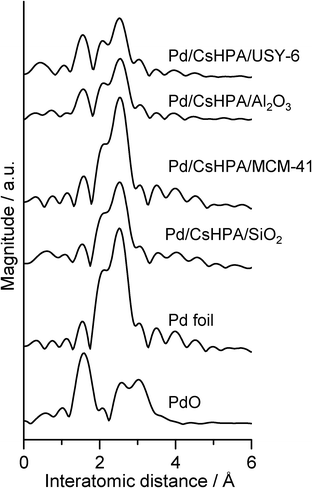 |
| | Fig. 9 FT-EXAFS spectra of Pd/CsHPA(10 wt%)/support with different catalyst supports and reference samples. | |
Table 2 Results of curve-fitting analysis
| Sample |
Shell |
C.N
|
R/Å |
Δσ2/Å2 |
|
PdO
|
Pd–O |
4 |
2.02 |
— |
|
Pd foil |
Pd–Pd |
12 |
2.75 |
— |
|
Pd/CsHPA/SiO2 |
Pd–Pd |
10.6 |
2.75 |
0.007 |
|
Pd/CsHPA/MCM-41 |
Pd–Pd |
11.9 |
2.76 |
0.005 |
|
Pd/CsHPA/Al2O3 |
Pd–O |
1.8 |
2.02 |
0.006 |
| |
Pd–Pd |
7.4 |
2.75 |
0.012 |
|
Pd/CsHPA/USY-6 |
Pd–O |
2.5 |
2.02 |
0.005 |
| |
Pd–Pd |
7.2 |
2.75 |
0.023 |
In the direct synthesis of H2O2, several undesirable reactions occur simultaneously, including the formation of water, and hydrogenation and decomposition of H2O2, which are thermodynamically favorable and highly exothermic.17 It is well known that acids inhibit the decomposition of H2O2, while halides prevent the formation of water.17 We attempted an investigation of the acid and/or halide effect in the present catalytic system, the results are summarized in Fig. 10. The use of H3PO4 and HNO3 instead of HCl as a reaction medium in the absence of any externally added halide significantly decreased the H2O2 concentration. On the other hand, the H2O2 concentration was appreciably improved when Cl− was added to H3PO4. The addition of Br− also increased the H2O2 concentration, but was found to be less effective than Cl−. A similar phenomenon was also observed in the case of HNO3 as a reaction medium. These results are in contrast to those of other catalytic systems, in which Br− is more effective than Cl−.18 The presence of I− was found to have no effect on the H2O2 concentration. This is attributed to the strong affinity of I−, which blocks the catalytic sites and thereby inhibits accessibility of the reactants.
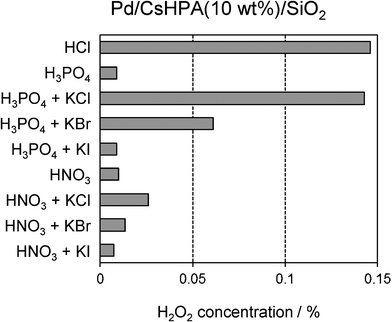 |
| | Fig. 10 Effect of solvent on the concentration of generated H2O2. | |
It is known that CsHPA has strong acidic properties. Therefore, such acidic properties of the catalysts should have a significant impact on their catalytic activities. For example, the Pd/CsHPA/SiO2 catalyst performed significantly better than Pd/SiO2, as mentioned before. In order to elucidate the role of CsHPA, the H2O2 concentrations in the decomposition (H2O2 → H2O + 1/2O2) and hydrogenation of H2O2 (H2O2 + H2 → 2H2O) over Pd/CsHPA/SiO2 and Pd/SiO2 were conducted, and the results are shown in Fig. 11. H2O2 could be almost recovered in the presence of Pd/CsHPA/SiO2, while Pd/SiO2 led to the smooth decomposition of H2O2. This implies that the presence of acid CsHPA inhibits the decomposition of H2O2. In the case of the hydrogenation of H2O2, the activity over Pd/SiO2 was much higher than that over Pd/CsHPA/SiO2. In the separation experiment, the average diameter of Pd nanoparticles on Pd/CsHPA/SiO2 and Pd/SiO2 was determined (by CO adsorption) to be 2 and 6 nm, respectively. These results indicate that the relatively larger Pd nanoparticles are highly active for the hydrogenation of H2O2 compared to the smaller particles. Therefore, it can be concluded that the CsHPA-supported silica acts as an effective host material, preventing both the decomposition and hydrogenation of H2O2.
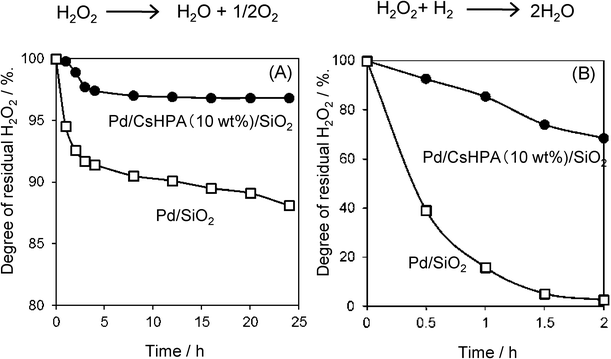 |
| | Fig. 11 Degree of residual H2O2 in the (A) decomposition of H2O2 and (B) hydrogenation of H2O2. | |
The lifetime and leaching of active metal species into solution are important points to consider when using heterogeneous catalysts, particularly for industrial and pharmaceutical applications. The spent Pd/CsHPA(10 wt%)/SiO2 catalyst could be recovered by simple filtration from the reaction mixture. ICP analysis of the filtrate confirmed that the Pd content was below the detection limit. XAFS and CO adsorption analyses of the recovered catalyst revealed that the particle size remained virtually unchanged even after the catalytic reaction. Moreover, the recovered catalyst can be reused without significant loss of catalytic activity.
4. Conclusions
CsHPA-supported silica enables the deposition and reduction of a Pd precursor in an aqueous solution by the [PW12O40]4− species generated from [PW12O40]3− species under UV-light irradiation in the presence of a sacrificial reagent to afford Pd0 metal nanoparticles. The catalytic activity for the direct synthesis of H2O2 using H2 and O2 gases were varied according to the preparation conditions, variation of HPA content, and the type of catalyst support; the Pd/CsHPA/SiO2 (CsHPA: 10 wt%) catalyst prepared by the PAD method in the presence of iPrOH exhibited higher activity (by a factor of ca. 6) than that prepared by the conventional impregnation technique. This study verified a proficient methodology for the synthesis of supported metal catalysts, and we believe that this synthetic approach will be extended to the deposition of other metals and bimetallic nanoparticles as unique catalysts and/or photocatalysts.
Acknowledgements
This work was financially supported by the Industrial Technology Research Grant Program in 2007 from the New Energy and Industrial Technology Development Organization (NEDO) of Japan. XAFS spectra were recorded at the beam line 01B1 station in SPring-8, JASRI, Harima, Japan (prop. No. 2011A1092, 2010B1098).
References
-
(a)
J. W. Geus, J. A. R. van Ween, Catalysis, An Integrated Approach to Homogeneous and Heterogeneous and Industrial Catalysis; J. A. Moulijin, P. W. N. M. van Leeuwen, van R. A. Santen, ed.; Elsevier: Amsterdam, 1993 Search PubMed;
(b)
R. F. Heck, Palladium Reagents in Organic Syntheses, Academic Press, London, 1985 Search PubMed;
(c)
J. Tsuji, Palladium Reagents and Catalysts, Wiley, Chichester, 1995 Search PubMed;
(d) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS;
(e) Applied Homogeneous Catalysis with Organometallic Compounds; B. CornilsW. A. Herrmann, ed., VCH, Weinheim, 1996.
-
(a) K. Hayek, R. Kramer and Z. Paál, Appl. Catal., A, 1997, 162, 1 CrossRef CAS;
(b) C.-M. Yang, P.-H. Liu, C.-Y. Chiu and K.-J. Chao, Chem. Mater., 2003, 15, 275 CrossRef CAS.
- A. Fukuoka, H. Araki, Y. Sakamoto, N. Sugimoto, H. Tsukada, Y. Kumai, Y. Akimoto and M. Ichikawa, Nano Lett., 2002, 2, 793 CrossRef CAS.
-
(a) F. Zhang, J. Chen, X. Zhang, W. Gao, R. Jin and N. Guan, Catal. Today, 2004, 93–95, 645 CrossRef CAS;
(b) C. Pacholski, A. Kornowski and H. Weller, Angew. Chem., Int. Ed., 2004, 43, 4774 CrossRef CAS;
(c) S. C. Chan and M. A. Barteau, Langmuir, 2005, 21, 5588 CrossRef CAS;
(d) T. Sano, S. Kutsuna, N. Negishi and K. Takeuchi, J. Mol. Catal. A: Chem., 2002, 189, 263 CrossRef CAS;
(e) K. Mori, T. Araki, T. Takasaki, S. Shironita and H. Yamashita, Photochem. Photobiol. Sci., 2009, 8, 652 RSC.
-
P. Pichat and M. A. Fox, in Photoinduced Electron Transfer, ed, M. A. Fox and M. Chanon, Elsevier, Amsterdam, 1988 Search PubMed.
-
(a) S. Shironita, K. Mori, T. Shimizu, T. Ohmichi, N. Mimura and H. Yamashita, Appl. Surf. Sci., 2008, 254, 7604 CrossRef CAS;
(b) K. Mori, T. Araki, S. Shironita, J. Sonoda and H. Yamashita, Catal. Lett., 2009, 131, 337 CrossRef CAS;
(c) K. Mori, Y. Miura, S. Shironita and H. Yamashita, Langmuir, 2009, 25, 11180 CrossRef CAS;
(d) K. Mori and H. Yamashita, Phys. Chem. Chem. Phys., 2010, 12, 14420 RSC.
-
(a)
M. T. Pope, Heteropoly, and Isopoly Oxometalates, Springer-Verlag, New York, 1983 Search PubMed.
-
(a) A. Troupis, A. Hiskita and E. Papaconstantinou, Angew. Chem., Int. Ed., 2002, 41, 1911 CrossRef CAS;
(b)
E. Papaconstantinou, A. Hiskia, in Polyoxometalate Molecular Science, ed.: J. J. Borrás-Almenar, E. Coronado, A. Müller, M. Pope, Kluwer Academic Publishers: The Netherlands, 2003 Search PubMed;
(c) B. Keita, T. Liu and L. Nadjo, J. Mater. Chem., 2009, 19, 19 RSC;
(d) T. Hsu-Yao, K. P. Browne, N. Honesty and Y. J. Tong, Phys. Chem. Chem. Phys., 2011, 13, 7433 RSC.
-
(a) L. Yang, Y. Shen, A. Xie, J. Liang, S. Li and Q. Zhang, Eur. J. Inorg. Chem., 2006, 4658 CrossRef CAS;
(b) A. Troupis, T. Triantis, A. Hiskia and E. Papaconstantinou, Eur. J. Inorg. Chem., 2008, 5579 CrossRef CAS.
- T. Okuhara, N. Mizuno and M. Misono, Adv. Catal., 1996, 41, 113 CrossRef CAS.
- R. Gao, H. Chen, Y. Le, W.-L. Dai and K. Fan, Appl. Catal., A, 2009, 352, 61 CrossRef CAS.
- A. Troupis, A. Hiskia and E. Papaconstantinou, New J. Chem., 2001, 25, 361 RSC.
-
G. Goor, W. Kunkel, O. Weiberg, in Ulman's Encyclopedia of Industrial Chemistry, VHS, Weinheim, 1989, A13, 443–466 Search PubMed.
-
(a) S. -E. Park, L. Huang, C. W. Lee and J. -S. Chang, Catal. Today, 2000, 61, 117 CrossRef CAS;
(b) J. H. Lunsford, J. Catal., 2003, 216, 455 CrossRef CAS;
(c) V. R. Choudhary, C. Samanta and T. V. Choudhary, Appl. Catal., A, 2006, 308, 128 CrossRef CAS;
(d) Q. Liu, J. C. Baur, R. E. Schaak and J. H. Lunsford, Angew. Chem., Int. Ed., 2008, 47, 6221 CrossRef CAS.
- T. Teranishi, M. Hosoe, T. Tanaka and M. Miyake, J. Phys. Chem. B, 1999, 103, 3818 CrossRef CAS.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657 CrossRef CAS.
-
(a) J. N. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962 CrossRef CAS;
(b) C. Samanta, Appl. Catal., A, 2008, 350, 133 CrossRef CAS;
(c) J. K. Edward and G. H. Hutchings, Angew. Chem., Int. Ed., 2008, 47, 9192 CrossRef.
- C. Samanta and V. R. Choudhary, Catal. Commun., 2007, 8, 2222 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.