Dynamic emission quenching of a novel ruthenium(II) complex by carbon dioxide in solution
Eri
Sakuda
*a,
Mai
Tanaka
b,
Akitaka
Ito
a and
Noboru
Kitamura
*ab
aDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo, 060-0810, Japan. E-mail: kitamura@sci.hokudai.ac.jp; Fax: +81-11-706-4630; Tel: +81-11-706-2697
bDepartment of Chemical Sciences and Engineering, Graduate School of Chemical Sciences and Engineering, Hokkaido University, Sapporo, 060-0810, Japan. E-mail: kitamura@sci.hokudai.ac.jp; Fax: +81-11-706-4630; Tel: +81-11-706-2697
First published on 22nd December 2011
Abstract
Emission from the tris(1,10-phenanthroline)ruthenium(II) complex (Ru(phen)32+) bearing a (dimesityl)boryldurylethynyl (DBDE) charge transfer unit at the 4-position of one of the three phen ligands ([Ru(phen)2(4-DBDE-phen)] = 4BRu2+) was quenched by carbon dioxide in solution with the rate constant of ∼106 M−1 s−1.
We reported quite recently that a novel ruthenium(II) complex (4BRu2+) showed intense emission at the maximum wavelength of 681 nm in CH3CN at 298 K with the quantum yield (Φem) and lifetime (τem) being 0.11 and 12 μs, respectively.1 It is worth emphasizing that, since the Φem and τem values of [Ru(phen)3]2+ in CH3CN at 298 K are 0.045 and 0.42 μs, respectively,1 the values of 4BRu2+ are extraordinarily large and long, respectively, compared with the relevant value of [Ru(phen)3]2+. Such emission characteristics of 4BRu2+ are attributed to participation of the synergistic interactions between Ru(II)-to-phen charge transfer (MLCT) and CT from the π–orbital of the durylethynyl moiety (π(aryl)) to the vacant p-orbital on the boron atom (p(B)) in the 4-DBDE-phen ligand: synergistic MLCT/π(aryl)-p(B) CT. In practice, our TD-DFT calculations indicate that the highest-energy occupied molecular orbital (HOMO) and HOMO-1 in 4BRu2+ are characterized by the electron densities on the Ru(II) atom and the pyridine ring in the durylethynyl-phen system without any contribution of p(B) to the HOMOs, while the excited electron in the lowest-energy unoccupied MO (LUMO) distributes to both p(B) and the durylethynyl system.1 Owing to MLCT/π(aryl)-p(B) CT nature and the very long τem value, the excited electron primarily localized on p(B) in 4BRu2+ would react and/or interact with an electrophile. Among various electrophiles, carbon dioxide is of primary importance in respect to the role in earth's climate warming and, thus, its chemical fixation/reduction is one of the urgent issues to be solved. In this communication, we report here that the excited triplet state of 4BRu2+ interacts with gaseous CO2 in solution at room temperature as demonstrated by emission quenching of 4BRu2+ by CO2.
4BRu2+ used in the present study was essentially the same with that reported previously.1Acetonitrile and propylene carbonate (PC) for spectroscopic/photophysical measurements, purchased from Wako Pure Chemicals Ind., were purified by the accepted procedures.2 The purity of CO2 gas (Air Water Co. Ltd.) was 99.8%. Absorption and emission spectra of 4BRu2+ were recorded on a Hitachi U-3300 spectrophotometer and a multichannel photodetector (PMA-11, Hamamatsu Photonics), respectively. Emission lifetime measurements were conducted by the system reported previously at 532 nm excitation.1
Fig. 1 shows the absorption spectrum of an N2–gas purged acetonitrile solution of 4BRu2+ (2.7 × 10−6 mol dm−3 (= M), shown by black curve), while the red curve corresponds to the spectrum observed for the same sample solution after purging CO2 gas for 20 min. The inset in Fig. 1 displays the relevant emission spectrum. As seen clearly in Fig. 1, although the absorption spectra of 4BRu2+ in N2 and CO2 gas atmosphere were essentially the same with each other, the emission from 4BRu2+ is quenched in the presence of CO2 without any change in the spectral band shape. Fig. 2 shows the emission decay profile of 4BRu2+ in the N2 (shown by black) or CO2 atmosphere (shown by red). In both systems, the emission decay profiles were fitted by single exponential functions and the τem value in the N2 (τem(N2)) or CO2 atmosphere (τem(CO2)) was determined to be 12.3 or 6.3 μs, respectively. The emission quenching efficiency, defined as τem(N2)/τem(CO2), was 2.0, whose value was almost comparable to the value determined by the emission intensity in Fig. 1:1.9. Therefore, it is concluded that the MLCT/π(aryl)-p(B) CT excited state of 4BRu2+ is dynamically quenched by CO2, probably through the interaction between the excited electron (charge) on p(B) and CO2.
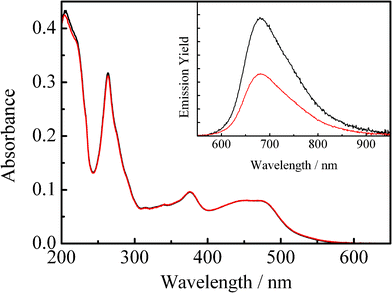 | ||
| Fig. 1 Absorption spectra of 4BRu2+ in acetonitrile under N2 (black curve) and CO2 gas atmosphere (red curve). The inset shows the relevant emission spectrum. | ||
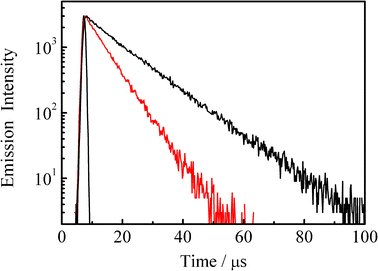 | ||
| Fig. 2 Emission decay profiles of 4BRu2+ in the N2 (black) and CO2 atmospheres (red) in acetonitrile at 298 K. | ||
In order to accumulate further experimental evidence, we studied a CO2–gas pressure (PCO2) dependence of the emission lifetime of 4BRu2+. A CH3CN solution of 4BRu2+ (2.7 × 10−6 M) in a glass tube equipped with a pressure-tight bulb was deaerated by three freeze-pump-thaw cycles and, subsequently, CO2 gas was introduced to the tube in an ice-cooled bath at a given PCO2. Emission lifetime measurements were then conducted at room temperature. The emission decay profiles of 4BRu2+ at PCO2 = 0, 4, 20, and 1013 hPa were determined in both CH3CN and PC solutions at room temperature. The emission decay profile was fitted by a single exponential function irrespective of PCO2 and a solvent, and the τem values determined were summarized in Table 1. The τem values in CH3CN and PC were ranged in 13.2–6.3 and 11.3–8.0 μs, respectively, while those of [Ru(phen)3]2+ in CH3CN and PC were 0.4 and 0.6 μs, respectively, irrespective of PCO2 (experimental data are not shown here). It is worth emphasizing that the emission quenching efficiency, τem(N2)/τem(CO2), is linearly correlated with the log PCO2 value irrespective of a solvent as seen in Fig. 3. When the log PCO2 value is correlated linearly with the molar CO2 concentration ([CO2]) in each solvent, the linear relations in Fig. 3 should be analyzed by a Stern–Volmer type equation: τem0/τem(CO2) = 1 + kqτem0[CO2] where τem0 is that at PCO2 = 0 and kq is the bimolecular quenching rate constant of the excited state of 4BRu2+ by CO2. Unfortunately, however, there is no available relation between log PCO2 and [CO2] in solution. Instead of analyzing the linear relation in Fig. 3, therefore, we evaluated kq based on the data at PCO2 = 1013 hPa. The [CO2] value in CH3CN or PC evaluated by the weight method was 0.071 or 0.056 M, respectively.3 The τem0 and τem (PCO2 = 1013 hPa) demonstrate that the kq value in CH3CN or PC is 1.2 × 106 or 0.74 × 106M−1s−1, respectively.
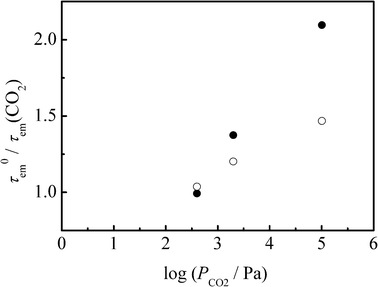 | ||
| Fig. 3 CO2–gas pressure (PCO2) dependency of the emission lifetimes of 4BRu2+ in acetonitrile (closed circles) and PC (open circles) at 298 K. | ||
| τ em/μs | ||||
|---|---|---|---|---|
| CO2–gas pressure/hPa | 0 | 4 | 20 | 1013 |
| Acetonitrile | 13.2 | 13.3 | 9.6 | 6.3 |
| PC | 11.3 | 10.9 | 9.4 | 8.0 |
Emission quenching of 4BRu2+ by CO2 is worth discussing in term of the Gibbs free energy change for photoreduction of CO2 by the excited state of 4BRu2+: ΔG‡. As reported previously, 4BRu2+ shows four consecutive reduction waves at −1.09, −1.41, −1.75, and −2.12 V (vs.SCE in CH3CN) and the three reduction waves observed more positive than −2.0 V are ascribed to consecutive one-electron reductions of the three phen ligands (E(Ru2+/+), E(Ru+/0), and E(Ru0/−)), while the fourth wave at −2.12 V is responsible for the reduction of the 4-DBDE group in the complex: electron occupation by p(B) at E(4-DBDE0/−).1 Since the excited state of 4BRu2+ is characterized by synergistic MLCT/π(aryl)-p(B) CT interactions as mentioned above, we suppose that the first reduction potential observed electrochemically (E(Ru2+/+)) for the ground state of 4BRu2+ (i.e., −1.09 V) will not correspond to the reduction potential in the excited state of the complex, E(Ru*2+/+): E(Ru*2+/+) ≠ E(Ru2+/+) + E0,0, where E0,0 is the excited state energy of 4BRu2+ (E00 = 1.91 eV at 80 K). When one assumes that CO2 interacts with p(B) in the excited state of 4BRu2+, the E(Ru*2+/+) value would be rather similar to E(4-DBDE0/−) = −2.12 V. If this is the case, the ΔG‡ value for emission quenching of the excited state of 4RuB2+ by CO2 is evaluated to be −0.02 V through the one-electron reduction potential of CO2 being −2.1 V (vs.SCE).4,5 The above discussions indicate that photoreduction of CO2 by the excited state of 4BRu2+ is thermodynamically possible but is not necessarily favourable enough, if one adopts the present quenching reaction to the Rehm-Weller type kqvs.ΔG‡ relationship.6 This may explain that the present system shows very small kq value: ∼106 M−1 s−1.
It is worth noting that [Ru(phen)2(5-DBDE-phen)] (= 5BRu2+) which is the 5-position analogue of 4BRu2+ shows E(5-DBDE0/–) at −1.89 V (vs.SCE).1 The reduction potential of 5BRu2+ is more positive than E(4-DBDE0/−) in 4BRu2+ and the excited state lifetime of 5BRu2+ is much shorter (τem = 1.2 μs in CH3CN at 298 K) than that of 4BRu2+.1 Thus, the excited state of 5BRu2+ is unfavourable for emission quenching by CO2. In practice, the emission from 5BRu2+ is not quenched by CO2 under analogous conditions to those for 4BRu2+. We suppose that the long excited state lifetime and the reduction potential negative enough for the interaction with CO2 are the necessary conditions for emission quenching by CO2.
Photoreduction/photofixation of CO2 has been hitherto explored on the basis of both molecular4,7–11 and semiconductor systems.12 In molecular systems, Lehn et al.7 and Ishitani et al.4,8 reported that CO2 was reduced photochemically to COvia a CO2–Re(CO)3LX adduct (L and X were diimine and halogen/monodentate ligands, respectively) in the presence of an electron donor. Alternatively, Tazuke et al. reported carboxylation of an aromatic hydrocarbon (ArH) through the electrophilic attack of CO2 to the anion radical of ArH produced by photoinduced electron transfer with an aromatic amine as an electron donor.9 The present emission quenching of 4BRu2+ by CO2via the electronic interaction between the excited electron/charge on p(B) in the complex and CO2 might be understood by the mechanism similar to the electrophilic attack of CO2 to the anion radical of ArH mentioned above and, thus, we suppose that the present system would produce the anion radical of CO2. To the best of our knowledge, this is the first experimental observation of direct emission quenching of a molecule by gaseous CO2 in solution. Furthermore, it is worth noting that the present system does not require an electron donor or sacrificial agent and, therefore, is very unique compared with other molecular CO2 photoreduction/photofixation systems. To confirm the quenching mechanism, we are performing product analysis of the system, which will be reported in a forthcoming paper.
We thank the Global COE program of Hokkaido University (Catalysis as the Basis for Materials Innovation) for a postdoctoral fellowship (2009) and a research fellowship (2008–2010) to E. S. and A. I., respectively. E. S. also thanks the F3 Project of MEXT, Japan (2010–).
References
- E. Sakuda, Y. Ando, A. Ito and N. Kitamura, Inorg. Chem., 2011, 50, 1603 Search PubMed.
- D. D. Perrin, A. L. Armargo and D. R. Perrin, Purification of Laboratory Chemicals, 2nd edn, Pergamon Press, New York, 1980 Search PubMed.
- The [CO2] value in CH3CN or PC at PCO2 = 1013 hPa was evaluated as follows. A 10 mL solution of CH3CN or PC in a glass vessel cooled in an ice-water bath was purged by a CO2 gas stream for 40 min. The solution was then allowed to room temperature under a CO2 gas stream flowing above the solution in the vessel and sealed with a stop-cock. By measuring the weights of the solution before and after CO2 gas purging, we evaluated [CO2] solublized in the solution. The average weight increment of a CH3CN or PC solution (10 mL) before and after CO2 gas purging was 0.0312 or 0.0363 g, respectively, corresponding to [CO2] to be 0.071 or 0.056 M, respectively.
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346 CrossRef CAS.
- In the cyclic voltammogram of 4BRu2+ in a CH3CN – 0.1 M tetra-n-butylammonium hexafluorophosphate solution, the electrode current at around E(DBDE0/−) = −2.1 V increased in the presence of CO2 compared with that under Ar-gas atmosphere, suggesting reduction of CO2 through that of the DBDE group in 4BRu2+. However, since the direct reduction current of CO2 flew in the same potential range, we could not distinguish two contributions to the observed current.
- (a) D. Rehm and A. Weller, Ber. Bunsen-Ges. Phys. Chem., 1969, 73, 834 CAS; (b) Isr. J. Chem. 1970 8 259 Search PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990 CrossRef CAS.
- (a) H. Hori, K. Koike, M. Ishizuka, K. Takeuchi, T. Ibusuki and O. Ishitani, J. Organomet. Chem., 1997, 530, 169 CrossRef CAS; (b) B. Gholamakhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326 CrossRef; (c) H. Tsubaki, A. Sekine, Y. Ohashi, K. Koike, H. Takeda and O. Ishitani, J. Am. Chem. Soc., 2005, 127, 15544 CrossRef CAS; (d) S. Sato, K. Koike, H. Inoue and O. Ishitani, Photochem. Photobiol. Sci., 2007, 6, 454 RSC; (e) K. Koike, S. Naito, S. Sato, Y. Tamaki and O. Ishitani, J. Photochem. Photobiol., A, 2009, 207, 109 CrossRef CAS.
- (a) S. Tazuke and H. Ozawa, J. Chem. Soc., Chem. Commun., 1975, 237 RSC; (b) S. Tazuke, S. Kazama and N. Kitamura, J. Org. Chem., 1986, 51, 4548 CrossRef CAS.
- (a) H. Ishida, K. Tanaka and T. Tanaka, Chem. Lett., 1985, 405 CAS; (b) H. Ishida, K. Tanaka and T. Tanaka, Organometallics, 1987, 6, 181 CrossRef CAS; (c) H. Ishida, T. Terada, K. Tanaka and T. Tanaka, Inorg. Chem., 1990, 29, 905 CrossRef CAS.
- (a) J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536 RSC; (b) R. Ziessel, J. Hawecker and J.-M. Lehn, Helv. Chim. Acta, 1986, 69, 1065 CAS; (c) D. Mandler and I. Willner, J. Chem. Soc., Perkin Trans. 2, 1988, 997 RSC; (d) I. Willner, R. Maidan, D. Mandler, H. Dürr and K. Zengerle, J. Am. Chem. Soc., 1987, 109, 6080 CrossRef CAS.
- (a) T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637; (b) K. Teramura, T. Tanaka, H. Ishikawa, Y. Kohno and T. Funabiki, J. Phys. Chem. B, 2004, 108, 346 CrossRef CAS; (c) W. Y. Lin and H. Frei, J. Am. Chem. Soc., 2005, 127, 1610 CrossRef CAS; (d) K. Teramura, H. Tsuneoka, T. Shishido and T. Tanaka, Chem. Phys. Lett., 2008, 467, 191 CrossRef CAS; (e) S. Qin, F. Xin, Y. Liu and W. Ma, J. Colloild Intface Sci., 2011, 356, 257 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |