High-performance aqueous supercapacitors based on hierarchically porous graphitized carbon†
Zheng
Chen
a,
Ding
Weng
a,
Hiesang
Sohn
a,
Mei
Cai
*b and
Yunfeng
Lu
*a
aDepartment of Chemical and Biomolecular Engineering, University of California, Los Angeles, CA 90095. E-mail: luucla@ucla.edu; Fax: 310-206-410; Tel: 310-794-7238
bGeneral Motor R&D Center, Warren, MI 48090, USA. E-mail: mei.cai@gm.com
First published on 11th January 2012
Abstract
Aqueous supercapacitors were fabricated using hierarchically porous graphitized carbon as the anode and metal oxide as the cathode, exhibiting high energy and high power densities for cost-effective energy storage.
Supercapacitors, a class of electrical energy storage devices with high power densities (103–104 W kg−1) and long cycling lives (>100,000 cycles), hold great promise for a broad spectrum of applications, such as hybrid electric vehicles, power tools and mobile electronic devices. However, the current applications of supercapacitors are still limited by their low energy density, and developing better electrode materials to lead to improved device energy density has been an essential but challenging topic.1–3
In this context, carbon materials such as activated carbons, mesoporous carbon and carbon nanotubes, have been extensively investigated.4–6 Activated carbons may provide capacitances up to 300 F g−1 in aqueous electrolytes or 120 F g−1 in organic electrolytes at low discharge rates. However, their storage performance radically deteriorates at high discharge rates due to the lagged transport of ions within their tortuous microporous channels.7,8 Mesoporous carbons, on the other hand, exhibit uniform pore geometry and larger pore sizes; however, they did not show exciting improvement in either energy or power density, possibly due to unfavourable ion transport within their long-range pore channels.9,10Carbon nanotubes possess excellent electronic conductivity and readily accessible external surfaces that can provide outstanding rate performance. However, carbon nanotubes also generally possess low surface areas, which provide low specific capacitances of less than 100 F g−1 in aqueous electrolytes or less than 50 F g−1 in organic electrolytes.11–13 Recently, high-surface-area microporous carbons with regulated pore channels, such as carbide-derived carbon and zeolite-templated carbon, were synthesized with capacitances up to 150 F g−1 and improved high-rate performance in organic electrolytes,8,14,15 however, their synthesis is extremely inefficient.
Recently, we developed a continuous aerosol process leading to the formation of hierarchically porous graphitized carbon particles. The synthesis of such carbon particles is quite simple and efficient.16,17 Briefly, the synthesis was started with an aqueous solution containing sucrose, nickel nitrate, colloidal silica particles and silicate clusters; an atomization process using nitrogen as the carrier gas continuously generated aerosol droplets, which were passed through a heating zone and converted to nanocomposite particles. Subsequent carbonization in the presence of the nickel moieties in situ converted the sucrose into graphitized carbon.18 Further removal of the templates resulted in graphitized, porous carbon particles with a high surface area and hierarchical pores.
Such carbon materials exhibited impressive cycling stability in organic electrolytes and significantly improved energy and power density compared with commercial activated carbons and carbon nanotubes. However, the achievement of such performance relies on utilizing high-cost electrolytes and a cell-fabrication process; moreover, the use of flammable solvents and electrolytes presents potential safety issues that may prevent their widespread applications. Herein, we explore the use of such hierarchically porous graphitized carbon for aqueous-electrolyte based devices. Owing to their unique structure, such as their high surface area, small particle size, hierarchically porous architecture and conductive graphitic framework, such graphitized carbon material may provide a large double layer area, fast ion transport and efficient charge harvesting in aqueous electrolytes. In this work, asymmetric supercapacitors with high energy density that is close to those of organic devices were successfully fabricated, offering a new method towards cost-effective energy storage.
Fig. 1A shows representative scanning electron microscope (SEM) images of the carbon particles. Such particles are polydisperse with diameters generally ranging from 50 to 300 nm, which are much smaller than those of activated carbon normally used in commercial supercapacitor devices (5–20 μm). The transmission electron microscope (TEM) image (Fig. 1B) reveals a highly porous sponge- or foam-like pore structure with interconnected mesopores and micropores. A high-resolution TEM (inset, Fig. 1B) image suggests that the “bubbles” observed around each particle are graphite shells formed by the catalytic carbonization of sucrose.18Raman spectra of the carbon particles clearly show two bands at around 1345 cm−1 (D band) and 1583 cm−1 (G band), with a relative intensity ratio (ID/IG) of ∼2, which further confirms the existence of a large amount of graphitized carbon (Fig. S1, ESI†). In addition, an interconnected mesopore and micropore structure can also be observed. Nitrogen sorption isotherms and pore size distributions of the particles clearly suggest the coexistence of micropores and mesopores (Fig. 2). The mesopores are narrowly distributed and centred at 11 nm, which is consistent with the size of the colloidal silica template. These particles exhibit a large pore volume of 2.02 cm3 g−1 and a Brunauer–Emmett–Teller (BET) surface area (SBET) of 1522 m2 g−1, of which ∼80% (1208 m2 g−1) is contributed to by the external surface, as calculated from the t-plot method. Such carbon architecture provides an ideal platform for fast ion transport and double layer formation, and thus is of great interest for capacitive energy storage.
 | ||
| Fig. 1 SEM and TEM images of the hierarchically porous aerosol-carbon particles. | ||
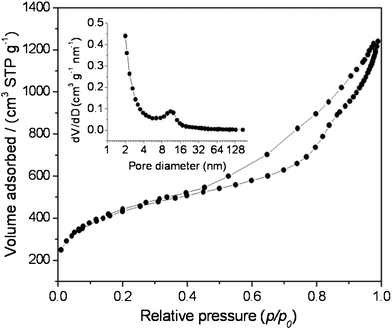 | ||
| Fig. 2 Nitrogen sorption isotherms and pore size distributions (inset) of the hierarchically porous aerosol-carbon particles. | ||
Capacitive performance of the particles was first examined in 3 M KOH, an alkaline electrolyte commonly used for carbon–NiO–based hybrid capacitors.19Fig. 3A shows their typical cyclic voltammetry (CV) curves at potential scan rates from 10 to 100 mV s−1 in a three-electrode cell using a platinum foil as the counter electrode and an Hg/HgO as the reference electrode. The cut-off potential was defined to be −0.9 and 0.1 V (vs. ref.) for stable and reversible cycling. The CV curves exhibit near-rectangular shapes even at high scan rates, suggesting a pure double-layer charge storage behavior with excellent rate- capability. The normalized specific capacitance Cs (F g−1) was calculated from the discharge curves of galvanostatic charge/discharge profiles at current densities from 0.5 to 10 A g−1 (Fig. S2, ESI†).
 | ||
| Fig. 3 Cyclic voltammograms of aerosol carbon electrodes at a potential sweep rate from 10 to 100 mV s−1 in 3 M KOH (A), 1 M Na2SO4 (B) and 1 M Li2SO4 (C); Dependence of specific capacitance and charge/discharge current density for aerosol carbon electrodes in different electrolytes (D). | ||
It was found that the electrode possesses a Cs of 220 F g−1 at a current density of 0.5 A g−1 and retains about 73% of this capacitance (160 F g−1) at a high current density of 10 A g−1. In comparison, a commercial activated carbon (SBET = 1900 m2 g−1) shows a Cs of 250 F g−1 at 0.5 A g−1 and 105 F g−1 at 10 A g−1, a moderate rate performance commonly observed for activated carbons.20 In addition, in comparison with the carbon materials reported recently, such as activated hierarchical porous carbon (SBET = 1660 m2 g−1, Cs = 180 F g−1),21 partially graphitic ordered mesoporous carbon (SBET = 1300 m2 g−1, Cs = 155 F g−1), nitrogen-enriched porous carbon (33 to 153 F g−1),22 and modified graphene (SBET = 950 m2 g−1, Cs = 101 F g−1),23 this unique carbon material also shows significantly improved performance. Such a high capacitance and high rate-capability is ascribed to the large accessible surface area and hierarchically porous structure which together provide efficient ion-adsorption and fast charge transport. Note that mesoporous carbon nanospheres (SBET = 2396 m2 g−1) can provide a similar high capacitance and rate capability in a similar testing condition, however, the synthesis is extremely inefficient.15
In addition to the alkaline electrolyte, the capacitive performance of the carbon particles was also evaluated in 1 M Na2SO4 and 1 M Li2SO4 aqueous electrolytes, the electrolytes commonly used for carbon/MnO224 and carbon/LiMn2O425 asymmetric supercapacitors. The carbon electrodes were tested in three-electrode cells using platinum foil as the counter electrode and Ag/AgCl as the reference electrode. Highly reversible charge and discharge behavior was also observed at a potential window from −0.8 to 0.6 V (vs. ref.). Similar to that in alkaline electrolytes, the electrodes show ideal CV curves with excellent rate performance in both electrolytes (Fig. 3B, C). Notably, the curvature of the CV curves in the three electrolytes shows different features, which is due to the size difference of the electrolyte ions adsorbed on the electrode surface. According to galvanostatic charge/discharge tests, the electrode shows a capacitance of 170 F g−1 at a current density of 0.5 A g−1 and 115 F g−1 at 10 A g−1 in 1 M Na2SO4, and a capacitance of 175 F g−1 at 0.5 A g−1 and 122 F g−1 at 10 A g−1 in 1 M Li2SO4, respectively (Fig. 3D). Due to a lower electrolyte concentration and larger anions, these capacitance values are relatively low compared to the KOH electrolyte. Nevertheless, the rate-capability for all electrodes is similar (∼70% of initial capacitance was retained at 10 A g−1), which suggests a rapid ion response of such carbon electrodes in all the electrolytes.
To further demonstrate their device applications, prototype coin-type devices were assembled using nickel oxide (NiO) as the positive material and the aerosol carbon particles as the negative material. In such devices, the carbon electrodes store charges by an electrical-double layer process, while the oxide electrodes are based on the redox reaction. To match the high rate-capability of the carbon electrode, a high-rate NiO electrode was also fabricated from nano-flakes of NiO (∼10 nm thick) synthesized using a chemical deposition process (Fig. S3, ESI†).26 As-synthesized NiO flakes show a surface-redox process according to the CV curve shown in Fig. 4A. The electrochemical reaction that occurs at the NiO electrode can be expressed by NiO + OH− ↔ NiOOH + e−.26,27 Two pairs of peaks appear symmetrically in the CV curve, indicating a good reversibility of this reaction. Galvanostatic charge/discharge was used to further quantify the capacitance of NiO electrode; the Cs was calculated to be ∼800 F g−1 at a potential window from 0.1 ∼ 0.5 V and a current density of 0.5 A g−1. According to a charge balance between positive and negative electrodes, an optimal negative-to-positive mass ratio was determined to be 3.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :124 in this asymmetric supercapacitor.
:124 in this asymmetric supercapacitor.
 | ||
| Fig. 4 Typical CV curves of aerosol carbon electrodes in combination of NiO (A), MnO2 (B) and LiMn2O4 (C) electrodes in different electrodes; comparison of charge/discharge potential window of different prototypes (D). | ||
Considering that the working potential for a graphitized carbon anode and NiO cathode are −0.9–0.1 V and 0–0.5 V, respectively, a maximum operation potential of the full cell was achieved at 1.4 V, which was confirmed by galvanostatic charge/discharge of the assembled prototype cells (Fig. 4A, C and Fig S4, ESI†). For a device at optimal configuration, it reached a capacitance of ∼80 F g−1 at a discharge current density of 0.25 A g−1, corresponding to an energy density of 25.0 Wh kg−1 at a power density of 180 W kg−1 (based on the total weight of cathode and anode active materials). This device could still possess an energy density of 8.5 Wh kg−1 even at a power density of 6000 W kg−1, suggesting a very high rate-capability. In addition, the long-term cycling test shows that the device still retains ∼75% of the initial capacity after 2000 cycles under a current density of 5 A g−1, suggesting a good cycling stability of the cell. (Fig S5, ESI†) Compared with a symmetric supercapacitor based on hierarchical porous carbon (Fig S6, ESI†), Maxsorb activated carbon, chemically modified graphene,23 or asymmetric capacitors based on hierarchically porous NiO and carbon,28 this asymmetric capacitor represents a significant improvement in terms of energy and power density, thus demonstrating the superiority of using our aerosol carbon for high-performance asymmetric devices.
Successful implementation of our hierarchically porous carbon electrodes in other devices was also demonstrated using manganese oxide (MnO2) and lithium manganese oxide (LiMn2O4) as the cathode materials. Nano-sized MnO2 (Fig S7, ESI†) and LiMn2O4 (Fig S8) were synthesized according to our previous work29 or the reported method with some modification.30Fig. 4B shows a typical CV plot of MnO2 in 1 M Na2SO4 together with a graphitized carbon electrode. The electrochemical reaction that occurs at the MnO2 electrode can be expressed by MnO2 + xNa+ + xe− ↔ MnOONax.31 The MnO2 electrodes show the ideal pseudocapacitive behavior with a specific capacitance of ∼150 F g−1 between a potential window from 0 to 0.8 V, which agrees well with the previous reports.
The electrochemical reaction occurs at the LiMn2O4 electrode can be expressed by LiMn2O4 + xLi+ + xe− ↔ LixMn2O4.32 Slightly different from the MnO2 electrodes, the LiMn2O4 electrodes store charge by the lithium intercalation reaction, which gives a charge capacity of 95 mAh g−1 (averaged to be 700 F g−1) at a potential between 0.6 and 1.1V (Fig. 4C). Similar to the above carbon/NiO asymmetric cells, the asymmetric devices, in combination with carbon electrodes, can provide a maximum cell voltage of 1.6 and 1.9 V when using MnO2 and LiMn2O4 as cathodes, respectively (Fig. 4D). At a slow rate (0.1 mA cm−2), optimal asymmetric carbon/MnO2 and carbon/LiMn2O4 devices provided an energy density of ∼14 and 32 Wh kg−1, respectively. The energy density of the cells decreased to 4.2 and 4.5 Wh kg−1, respectively, at a high rate (5 mA cm−1), revealing that the structure of the oxides needs to be further tuned for high-rate applications. Nevertheless, such high energy densities approach those of organic electrolyte-based devices, and are significantly higher than those of the carbon-based symmetric devices in aqueous electrolytes. These results further reveal the potential of using such aerosol carbons as the anodes for hybrid capacitive energy storage. Further effort to make high-rate MnO2 and LiMn2O4 bulk electrodes will enable us to fabricate these high-energy supercapacitors with improved power density.
In summary, we have shown the excellent capacitive performance of hierarchically porous carbon in aqueous electrolytes, where high-energy asymmetric supercapacitors were fabricated using this carbon material as anodes. The appealing capacitive performance is attributed to its unique structure that provides large double layer capacitance, fast ion transport and efficient charge harvesting capability. This new family of porous carbons offers great opportunity to fabricate high-energy and safer supercapacitor devices at low cost.
Acknowledgements
This research work was supported as part of the Molecularly Engineered Energy Materials, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under award DE-SC001342. The authors also acknowledge the support from General Motor Inc. and IMRA America Inc.References
- A. Burke, J. Power Sources, 2000, 91, 37–50 CrossRef CAS.
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS.
- J. R. Miller and A. F. Burke, Electrochem. Soc. Interface, 2008, 53–57 CAS.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- P. Simon and J. R. Miller, Electrochem. Soc. Interface, 2008, 38–43 CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373–376 CrossRef CAS.
- A. Kajdos, A. Kvit, F. Jones, J. Jagiello and G. Yushin, J. Am. Chem. Soc., 132, 3252–3253 CrossRef CAS.
- D. Carriazo, F. Picó, M. C. Gutiérrez, F. Rubio, J. M. Rojo and F. d. Monte, J. Mater. Chem., 2010, 20, 773–780 RSC.
- H. J. Liu, W. J. Cui, L. H. Jin, C. X. Wang and Y. Y. Xia, J. Mater. Chem., 2009, 19, 3661–3667 RSC.
- C. Du and N. Pan, Nanotechnology, 2006, 17, 5314–5318 CrossRef CAS.
- M. Kaempgen, J. Ma, G. Gruner, G. Wee and S. G. Mhaisalkar, Appl. Phys. Lett., 2007, 90, 264104 CrossRef.
- S. W. Lee, B. S. Kim, S. Chen, Y. Shao-Horn and P. T. Hammond, J. Am. Chem. Soc., 2009, 131, 671–679 CrossRef CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS.
- Z. Lei, N. Christov, L. L. Zhang and X. S. Zhao, J. Mater. Chem., 2011, 21, 2274–2281 RSC.
- Z. Chen, J. Wen, C. Yan, L. Rice, H. Sohn, M. Shen, M. Cai, B. Dunn and Y. Lu, Adv. Energy Mater., 2011, 1, 551–556 CrossRef CAS.
- J. E. Hampsey, Q. Hu, L. Rice, J. Pang, Z. Wu and Y. Lu, Chem. Commun., 2005, 28, 3606–3608 RSC.
- H. S. Bengaard, J. K. Nørskov, J. Sehested, B. S. Clausen, L. P. Nielsen, A. M. Molenbroek and J. R. Rostrup-Nielsen, J. Catal., 2002, 209, 365–384 CrossRef CAS.
- V. Ganesh, S. Pitchumani and V. Lakshminarayanan, J. Power Sources, 2006, 158, 1523–1532 CrossRef CAS.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- W. Xing, C. C. Huang, S. P. Zhuo, X. Yuan, G. Q. Wang, D. Hulicova-Jurcakova, Z. F. Yan and G. Q. Lu, Carbon, 2009, 47, 1715–1722 CrossRef CAS.
- D. Hulicova-Jurcakova, M. Kodama, S. Shiraishi, H. Hatori, Z. H. Zhu and G. Q. Lu, Adv. Funct. Mater., 2009, 19, 1800–1809 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS.
- V. Khomenko, E. Raymundo-Piñero and F. Béguin, J. Power Sources, 2006, 153, 183–190 CrossRef CAS.
- Y. G. Wang and Y. Y. Xia, Electrochem. Commun., 2005, 7, 1138–1142 CrossRef CAS.
- J. W. Lang, L. B. Kong, W. J. Wu, Y. C. Luo and L. Kang, Chem. Commun., 2008, 35, 4213–4215 RSC.
- J.-H. Kim, S. H. Kang, K. Zhu, J. Y. Kim, N. R. Neale and A. J. Frank, Chem. Commun., 2011, 47, 5214–5216 RSC.
- D. W. Wang, F. Li and H. M. Cheng, J. Power Sources, 2008, 185, 1563–1568 CrossRef CAS.
- Y. Peng, Z. Chen, J. Wen, Q. Xiao, D. Weng, S. He, H. Geng and Y. Lu, Nano Res., 2011, 4, 216–225 CrossRef CAS.
- K. M. Shaju and P. G. Bruce, Chem. Mater., 2008, 20, 5557–5562 CrossRef CAS.
- M. Toupin, T. Brousse and D. Belanger, Chem. Mater., 2004, 16, 3184–3190 CrossRef CAS.
- A. Eftekhari, Electrochim. Acta, 2001, 47, 495–499 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental details, XRD, SEM, CVs. See DOI: 10.1039/c2ra00887d/ |
| This journal is © The Royal Society of Chemistry 2012 |