DOI:
10.1039/C2RA01170K
(Paper)
RSC Adv., 2012,
2, 3367-3373
Synthesis of substituted pyrazolo[4,3-b]pyridines via copper-mediated intramolecular C–N cross-coupling of primary allylamines†‡
Received
23rd November 2011
, Accepted 20th January 2012
First published on 27th February 2012
Abstract
Copper-catalyzed intramolecular amination followed by in situ oxidation of primary allylamines generated from the Morita–Baylis–Hillman adducts of 4-iodopyrazolecarbaldehydes for preparing substituted pyrazolo[4,3-b]pyridines is described. The synthetic methodology is versatile and is not affected by the stereochemistry of the allylamine. In contrast, similar coupling reactions employing secondary allylamines as the starting material were unsuccessful.
Introduction
Pyrazoles and annulated-pyrazoles figure as the core unit of a variety of bioactive molecules including several drugs.1–2 This property of pyrazole derivatives provides impetus to organic chemists to continue to develop novel strategies for synthesizing them efficiently.3 In one of our research programs, we have interest in accessing diverse pyrazole-annulated compounds via transition-metal-based coupling reactions4,5 employing substrates afforded from substituted 4-iodo-3-pyrazolecarbaldehydes (I). Towards this goal, we have reported the synthesis of substituted pyrazolo[4,3-b]pyridin-5-ones (II) via an unprecedented copper-catalyzed intramolecular amidation of substituted 4-iodopyrazoles generated either via Morita–Baylis–Hillman (MBH) or Horner–Wadsworth–Emmons chemistries (Scheme 1).6 During the course of these studies our initial attempts to perform intramolecular C–N cross coupling with the secondary allylamines at C-4 prepared from the MBH acetates of acrylates were unsuccessful. Later however, we could successfully demonstrate the synthesis of pyrazole-fused benzodiazepines (III) and benzoxazoazepines (IV) via copper-mediated C–N and C–O cross-coupling reactions of 4-iodo-3-pyrazolecarbaldehydes (I) with 1,2-phenylenediamines and 2-aminophenols, respectively.7 Essentially the success of this reaction relied on the initial condensation between the formyl group of pyrazole followed by a cross-coupling reaction. In contrast, it was observed that under similar conditions the intermolecular C–N or C–O cross-coupling reactions at C-4 of 4-iodopyrazole derivatives with aniline or phenol were unsuccessful. Further, in a more recent study we reported that the reaction of 4-iodopyrazolecarbaldehydes with substituted amidines to furnish substituted pyrazolo[4,3-d]pyrimidines (V) also proceeded via initial condensation followed by intramolecular cross coupling.8 A summary of the results pertaining to the cross coupling reactions at C-4 in 4-iodopyrazoles reported by us until now is delineated in Scheme 1. These results suggested that for the success of the C–N cross coupling reaction the amino group should be present in the primary state. In lieu of this reasoning, we considered investigating the intramolecular C–N cross coupling reaction in the primary allylamine (VI) readily prepared from the MBH acetates of the 4-iodo-3-pyrazolecarbaldehydes. Whereas the success of this reaction is expected to afford pyrazolo[4,3-b]pyridines9 (VII) which is the basic scaffold of several compounds ascribed with anti-inflammatory,1b,10 CRF1-antagonist,11 and AA2-antagonist12 activities, it would enhance the substrate scope for the C–N coupling reaction at C-4 position in 4-iodopyrazoles. As a consequence, we prepared the primary allylamines from the MBH acetates of 4-iodo-3-pyrazolecarbaldehydes and investigated them for intramolecular cross coupling reactions. As expected, these primary allylamines successfully afforded the substituted pyrazolo[4,3-b]pyridines via a copper-promoted intramolecular C–N cross coupling reaction. To the best of our knowledge this report constitutes the first example of C–N cross coupling reaction between an aliphatic amino group and the C-4 of the 4-iodopyrazole derivative. Furthermore, compared to the influence of relative stereochemistry on the success of the intramolecular amidation reported by us earlier,5 we discovered that the products from the primary allylamines originating from the MBH adducts of acrylate or acrylonitrile are afforded in high yields irrespective of the stereochemistry of the reactant. These interesting results provoked us to present the extension of our work on the intramolecular C–N cross coupling reaction of 4-iodopyrazole derivatives.
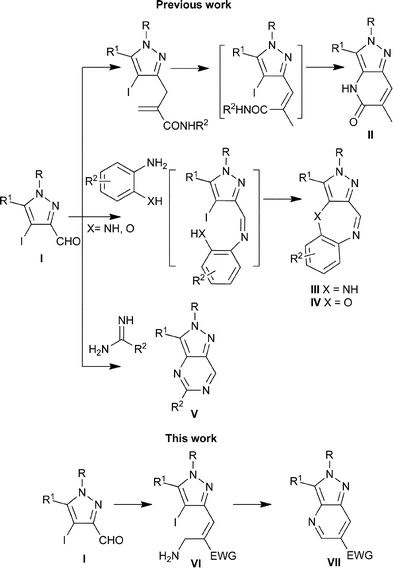 |
| | Scheme 1 Summary of C–N/O cross-coupling reactions at C-4 position in 4-iodopyrazole derivatives. | |
Results and discussion
The first objective of the study was to prepare the required allylamines. Influenced by our previous observation during the intramolecular amidation that the success of the C–N coupling relies on the E-stereochemistry of the reactant, we considered initiating our studies with primary allylamines generated from the MBH adducts of acrylate since they exist as E-isomer. Therefore in the first instance the MBH reactions of several 4-iodo-3-pyrazolecarbaldehydes (1a–k) with methyl acrylate were performed to generate the MBH adducts (2a–k) following the reported procedure (Scheme 2). Subsequent transformation of MBH adducts into respective acetates (3a–k) was readily accomplished by treating them with acetyl chloride in the presence of pyridine in methylene chloride. A reaction of these acetates with MeOH–NH3 for 30 min at room temperature gave the desired primary allylamines (4a–k) in 78–83% yields. The E-stereochemistry of the amines (4a–k) was evident from the chemical shift of the allylic proton.13 With the primary allylamine in hand, we proceeded with optimization of the intramolecular cross coupling reaction employing 4a as the model substrates. Initially, the reaction of 4a was investigated with CuI in the presence of K3PO4 in DMSO under ligand-free conditions. Fortunately, the reaction was successful to afford a product in 46% yield which was spectrally characterized to be the required methyl 2,3-diphenyl-2H-pyrazolo[4,3-b]pyridine-6-carboxylate 5a. This implied that during the reaction not only the expected cyclization takes place, but simultaneous oxidation of pyrazole-fused-dihydropyridine which may have initially formed occurs. In subsequent examination the reaction was performed in the presence of ligands such as L-proline, N,N-dimethylethylenediamine and ethylenediamine. The use of ligand improved the yields of the isolated products considerably. As illustrated in Table 1, the use of L-proline as ligand resulted in the best yields of 5a (88%, entry 2).14 Next keeping the ligand as constant, other variants including copper salt, base and solvent were assessed to discover the most suitable condition. As evident from Table 1, most of the copper salts and bases tested were found to be effective for the purpose. Amongst solvents though DMSO was the optimal solvent, DMF was also successful to give products albeit in lower yields. However the reaction was sluggish in toluene to give the product in 13% yields only (entry 7). The temperature of 80 °C was observed to be best suited for the success of the reaction. It may be worthwhile to mention that the reaction was unsuccessful in the absence of a copper source (entry 12).
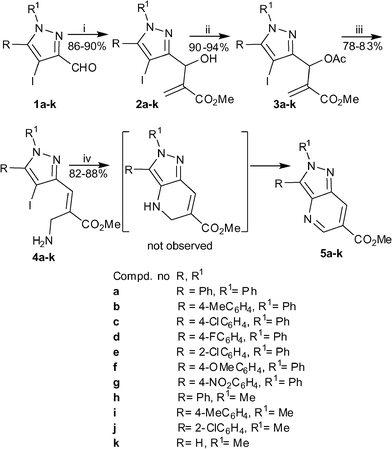 |
| | Scheme 2
Reagents and conditions: (i) methyl acrylate (1.1 eq), neat, rt, 36–48 h; (ii) AcCl (1.5 eq), Py (1.3 eq), CH2Cl2, 0 °C-rt, 1 h; (iii) NH3–MeOH, rt, 30 min; iv) CuI (0.1 eq), L-proline (0.2 eq), K3PO4 (2.0 eq), DMSO, 80 °C, 12 h. | |
Table 1 Optimization of the conditions for intramolecular amination with substrate 4aa
| Entry |
Catalyst |
Ligand |
Base |
Solvent |
Yieldb 5a (%) |
|
Reaction conditions: under nitrogen atmosphere, 4a (0.74 mmol), Cu salt (0.074 mmol), ligand (0.15 mmol), base (1.48 mmol), solvent (2 mL), temperature (80 °C), reaction time (12 h).
Isolated yields.
|
| 1 |
CuI |
— |
K3PO4 |
DMSO |
46 |
| 2 |
CuI |
L-Proline |
K3PO4 |
DMSO |
88 |
| 3 |
CuI |
DMEDA |
K3PO4 |
DMSO |
74 |
| 4 |
CuI |
EDA |
K3PO4 |
DMSO |
72 |
| 5 |
CuI |
L-Proline |
Cs2CO3 |
DMSO |
65 |
| 6 |
CuI |
L-Proline |
K2CO3 |
DMSO |
69 |
| 7 |
CuI |
L-Proline |
K3PO4 |
Toluene |
13 |
| 8 |
CuI |
L-Proline |
K3PO4 |
DMF |
73 |
| 9 |
Cu2O |
L-Proline |
K3PO4 |
DMSO |
75 |
| 10 |
CuCl |
L-Proline |
K3PO4 |
DMSO |
72 |
| 11 |
CuBr |
L-Proline |
K3PO4 |
DMSO |
76 |
| 12 |
— |
L-Proline |
K3PO4 |
DMSO |
— |
Next the scope of the methodology was tested with other primary allylamines using optimized conditions of 10 mol% of CuI as the copper-source, 20 mol% of L-proline, 2 equiv of K3PO4 as the base, and DMSO as the solvent at 80 °C. It was satisfying to note that all primary allylamines (4b–k) furnished the corresponding annulated pyrazoles (5b–k) in 82–88% yields. Changing the substituents phenyl to methyl at 2-position of the pyrazole does not alter the outcome of the reaction. Similarly, leaving 5-position of pyrazole unsubstituted did not have any influence on the yield of the reaction.
In order to increase the diversity of annulated pyrazoles, we next investigated the coupling reaction of primary allylamines afforded from MBH reaction of 4-iodo-5-pyrazolecarbaldehydes. The primary allylamines 9a–c,l were synthesized from 6a–c,lvia an identical set of reactions (Scheme 3). Treating 9a–c,l with CuI under optimized conditions resulted in the formation of pyrazolo[4,3-b]pyridines 10a–c,l in 84–86% yield.
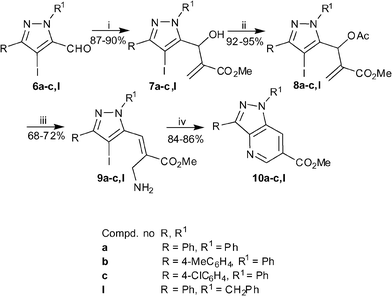 |
| | Scheme 3
Reagents and conditions: (i) methyl acrylate (1.1 eq), neat, rt, 40–58 h; (ii) AcCl (1.5 eq), Py (1.3 eq), CH2Cl2, 0 °C-rt, 1 h; (iii) NH3–MeOH, rt, 30 min; (iv) CuI (0.1 eq), L-proline (0.2 eq), K3PO4 (2.0 eq), DMSO, 80 °C, 12 h. | |
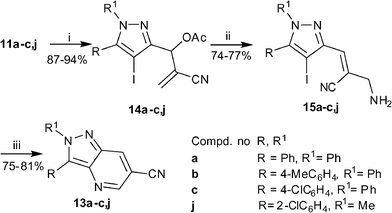
After successful formation of pyrazolo[4,3-b]pyridines from the primary allylamines afforded from the MBH adducts of methyl acrylate, we turned our attention to the MBH adducts of acrylonitrile. It is already established that the primary allylamine afforded from the MBH adduct of acrylonitrile exists as Z-isomer and we have reported that Z-isomer in case of acrylamide does not undergo an effective cross-coupling reaction.5 Henceforth, we decided to formulate an alternative route for preparing substituted 2H-pyrazolo[4,3-b]pyridine-6-carbonitriles wherein we envisaged to perform the Michael addition with MeOH–NH3 at the MBH adduct stage followed by C–N coupling and subsequent dehydration and oxidation. In order to optimize the strategy 11a was selected as the model substrate. The Michael reaction of 11a with MeOH–NH3 afforded the aminoalcohol 12a in 90% yields (Scheme 4). Subsequent intramolecular C–N coupling under the optimized conditions afforded a product which could be isolated in 39% yields only. Unexpectedly, spectroscopic characterization of the product revealed it to be the required 2,3-biphenyl 2H-pyrazolo[4,3-b]pyridine-6-carbonitrile 13a. This result reflected that during the reaction a domino process occurs wherein cyclization is followed by dehydration and oxidation. In addition, during the column chromatographic purification a significant amount of the corresponding aldehyde 1a was recovered. However this observation did not come as a surprise to us, since we have experienced earlier too that 1,3-aminoalcohols containing a nitrile group in 2-position undergo elimination of the benzylic part as aldehyde under basic medium.15 Nevertheless, the success of the protocol provided an impetus to examine its scope with other MBH adducts. Therefore MBH adducts 11b–c were prepared and subjected to an analogous set of reactions. Although these substrates were successful in furnishing the respective products 13b–c, their isolated yields were poor.
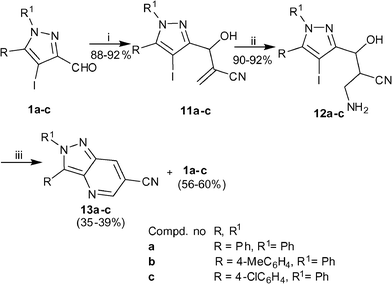 |
| | Scheme 4
Reagents and conditions: (i) acrylonitrile (1.1 eq), neat, rt, 24 h; (ii) NH3–MeOH, rt, 6–8 h; (iii) CuI (0.1 eq), L-Proline (0.2 eq), K3PO4 (2.0 eq), DMSO, 80 °C, 12 h. | |
In order to provide an experimental evidence that the C–N coupling was feasible only with the derivatives bearing E-stereochemistry it was considered essential to examine the coupling reaction in the allylamine originating from the MBH acetate of acrylonitrile which would exist as Z-isomer. Therefore, in a representative study the allylamine 15a was prepared from the MBH acetate 14a which in turn was derived from 11a (Scheme 5). The Z-stereochemistry of the amine was evident by the presence of an allylic proton at 7.07 ppm in the 1H–NMR spectrum. Subsequently 15a was subjected to the coupling reaction under the optimized conditions. The reaction was complete in 12 h to yield a product in 80% yields. To our surprise the spectral data of this product corresponded to that of 13a. This result implied that the Z-stereochemistry did not have a role in the success of the coupling reaction for these primary allylamines. To substantiate this finding the protocol was extended to more substrates 15b–c,j which could be readily generated from the respective MBH adducts 11b–c,j. It was a delight to note that all reactants produced the respective products in 75–81% yields. We speculated that during the heating of the allylamine in the presence of a base the Z-isomer isomerizes to E-isomer which takes part in the reaction to deliver the product. In order to provide an experimental evidence of this assumption, in a representative example the amine 15a bearing Z-stereochemistry was heated with K3PO4 in DMSO for 12 h. This resulted in a complete transformation of Z-15a to E-15a, which was confirmed by the change in the chemical shifts of the signals for ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH and –CH2NH2 protons.
CH and –CH2NH2 protons.
 |
| | Scheme 5
Reagents and conditions: (i) AcCl (1.5 eq), Py (1.3 eq), CH2Cl2, 0 °C-rt, 1 h; (ii) NH3–MeOH, rt, 30 min; (iii) CuI (0.1 eq), L-proline (0.2 eq), K3PO4 (2.0 eq), DMSO, 80 °C, 12 h. | |
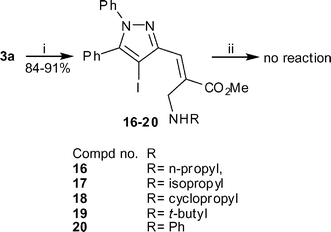
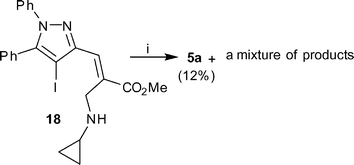
After the success of the C–N cross-coupling reaction with primary allylamine, in order to validate our previous findings, we were tempted to reinvestigate the cross coupling reaction with secondary allylamine obtained from an MBH adduct of 4-iodo pyrazolecarbaldehyde. As a result several amines such as n-propylamine, isopropylamine, cyclopropylamine, t-butylamine, and aniline were reacted with 3a in methanol to produce secondary allylamines 16–20 as E-isomer exclusively (Scheme 6). Similar CuI-mediated cross coupling reactions under standardized conditions were however found to be unsuccessful and the starting materials were recovered unreacted. Although no reason could be assigned to such observation, we decided to investigate the reaction of secondary amine employing 18 as a representative compound under different conditions of temperature, base and ligand. During the study it was observed that changing the base to Cs2CO3 (2.0 eq) and performing the reaction at 140 °C for 24 h afforded a mixture of products from which 5a was isolated in 12% yields (Scheme 7). This implied that though the coupling reaction was successful, the cyclopropyl group was eliminated, leading to an aromatized system.
 |
| | Scheme 6
Reagents and conditions: (i) RNH2, MeOH, rt, 30 min; (ii) CuI (0.1 eq), L-proline (0.2 eq), K3PO4 (2.0 eq), DMSO, 90 °C, 12 h. | |
 |
| | Scheme 7
Reagents and conditions: (i) CuI (0.1 eq), L-proline (0.2 eq), Cs2CO3 (2.0 eq), DMSO, 140 °C, 24 h. | |
Conclusions
In summary, we have reported for the first time a copper-mediated intramolecular C–N cross coupling reaction involving an aliphatic amino group and the iodine present at C-4 in MBH derivatives of 4-iodopyrazolecarbaldehydes. Importantly, the methodology works nicely with MBH adducts of acrylates and acrylonitrile thereby making the protocol versatile and independent of stereochemical implications. Further work is underway in our laboratory to enhance the scope of C–N coupling at the 4-position of the pyrazole ring to generate a diverse pyrazole-annulated system.
Experimental
General
Melting points are uncorrected and were determined in capillary tubes on a Precision melting point apparatus containing silicon oil. IR spectra were recorded using a Perkin Elmer's RX I FTIR spectrophotometer. 1H NMR and 13C NMR spectra were recorded either on a Bruker DPX-200 FT or Bruker Avance DRX-300 spectrometer, using TMS as an internal standard (chemical shifts in δ). The ESMS were recorded on MICROMASS Quadro-II LCMS system. The HRMS spectra were recorded as ESI-HRMS on an Agilent system or as DART-HRMS (recorded as ES+) on a JEOL-AccuTOF JMS-T100 LC Mass spectrometer having DART (Direct Analysis in Real Time) source using 19F and 35Cl for the mass calculation. Elemental analyses were performed on a Carlo Erba's 108 or an Elementar's Vario EL III microanalyzer. The room temperature varied between 21 °C and 35 °C. All solvents and reagents were used as supplied by the vendors without any further purification.
General procedure for the preparation of compounds 5a–k, 10a–c,l and 14a–c,j as exemplified 5a.
To a solution of compound 4a (250 mg, 0.545 mmol) in DMSO (4 mL), CuI (10 mg, 0.0545 mmol), L-proline (13 mg, 0.109 mmol) and K3PO4 (245 mg, 1.09 mmol) were added and the reaction mixture was heated at 80 °C for 12 h under a nitrogen atmosphere. Thereafter, water (50 mL) and EtOAc (25 mL) were added and the reaction mass was passed through a Celite bed and the layers were separated. The aqueous layer was further extracted with ethyl acetate (2 × 20 mL) and the collected organic layer was washed with brine, dried over anhydrous Na2SO4 and concentrated under vacuum. Column chromatography of the crude product over silica gel furnished the pure 5a as a white solid (ethyl acetate/hexanes, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9; yield: 158 mg, 88%).
:9; yield: 158 mg, 88%).
Methyl 2,3-diphenyl-2H-pyrazolo[4,3-b]pyridine-6-carboxylate (5a).
Mp 109–110 °C; Rf 0.47 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1721 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 4.02 (s, 3H), 7.40–7.56 (m, 10H), 8.84 (d, 1H, J = 1.4 Hz), 9.23 (d, 1H, J = 1.5 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 52.7, 124.4, 126.1, 128.1, 129.0, 129.1, 129.3, 129.4, 129.5, 130.1, 136.6, 139.2, 140.3, 141.0, 148.8, 166.3 ppm; ESIMS (m/z) 330.3 [M+H]+; ESI-HRMS Calcd. for C20H16N3O2 [MH]+: 330.1243. Found: 330.1248.
Methyl 3-(4-methylphenyl)-2-phenyl-2H-pyrazolo[4,3-b]pyridine-6-carboxylate (5b).
85% as a white solid (154 mg from 250 mg); mp 112–113 °C; Rf 0.49 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1722 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 2.38 (s, 3H), 4.02 (s, 3H), 7.22 (d, 2H, J = 8.0 Hz), 7.41–7.50 (m, 7H), 8.82 (d, 1H, J = 1.8 Hz), 9.21 (s, 1H, J = 1.8 Hz) ppm; 13C NMR (50 MHz, CDCl3): δ 21.6, 52.7, 124.3, 125.1, 126.1, 129.3, 129.5, 129.7, 129.9, 136.7, 139.2, 140.4, 140.9, 148.6, 166.3 ppm; ESIMS (m/z) 344.3 [M+H]+; DART-HRMS Calcd. for C21H18N3O2 [MH]+: 344.1399. Found: 344.1402.
Methyl 3-(4-chlorophenyl)-2-phenyl-2H-pyrazolo[4,3-b]pyridine-6-carboxylate(5c).
83% as a white solid (153 mg from 250 mg); mp 114–115 °C; Rf 0.51 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1718 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 4.02 (s, 3H), 7.38–7.49 (m, 9H), 8.84 (s, 1H), 9.23 (s, 1H) ppm; 13C NMR (75 MHz, CDCl3): δ 52.7, 124.5, 126.0, 126.5, 129.2, 129.4, 129.58, 129.62, 131.2, 135.2, 135.3, 139.1, 140.0, 141.0, 149.0, 166.1 ppm; ESIMS (m/z) 364.3 [M+H]+; ESI-HRMS Calcd. for C20H15ClN3O2 [MH]+: 364.0853. Found: 364.0836.
Methyl 3-(4-fluorophenyl)-2-phenyl-2H-pyrazolo[4,3-b]pyridine-6-carboxylate (5d).
86% as a white solid (156 mg from 250 mg); mp 120–121 °C; Rf 0.48 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1717 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 4.02 (s, 3H), 7.12 (t, 2H, J = 8.7 Hz), 7.48–7.56 (m, 7H), 8.84 (d, 1H, J = 1.7 Hz), 9.23 (d, 1H, J = 1.7 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 52.7, 116.1, 116.3, 124.1, 124.4, 126.0, 129.4, 129.6, 131.9, 132.0, 135.5, 139.0, 140.0, 140.9, 148.8, 161.4, 166.1 ppm; ESIMS (m/z) 348.2 [M+H]+; ESI-HRMS Calcd. for C20H15FN3O2 [MH]+: 348.1148. Found: 348.1143.
Methyl 3-(2-chlorophenyl)-2-phenyl-2H-pyrazolo[4,3-b]pyridine-6-carboxylate (5e).
86% as a white solid (158 mg from 250 mg); mp 181–182 °C; Rf 0.50 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1721 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 4.02 (s, 3H), 7.38–7.43 (m, 5H), 7.44–7.49 (m, 4H), 8.88 (d, 1H, J = 1.7 Hz), 9.21 (d, 1H, J = 1.7 Hz) ppm; 13C NMR (50 MHz, CDCl3): δ 52.6, 124.3, 124.9, 127.3, 127.8, 129.1, 129.2, 129.5, 130.4, 131.3, 133.0, 134.3, 134.8, 139.9, 140.2, 140.7, 149.0, 166.1 ppm; ESIMS (m/z) 364.2 [M+H]+; ESI-HRMS Calcd. for C20H15ClN3O2 [MH]+: 364.0853. Found: 364.0858.
Methyl 3-(4-methoxyphenyl)-2-phenyl-2H-pyrazolo[4,3-b]pyridine-6-carboxylate (5f).
84% as a white solid (154 mg from 250 mg); mp 164–165 °C; Rf 0.48 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1721 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 3.83 (s, 3H), 4.01 (s, 3H), 6.94 (d, 2H, J = 8.8 Hz), 7.46–7.54 (m, 7H), 8.81 (d, 1H, J = 1.8 Hz), 9.20 (d, 1H, J = 1.7 Hz) ppm; 13C NMR (50 MHz, CDCl3): δ 52.6, 55.5, 114.5, 120.4, 124.3, 126.0, 129.2, 129.3, 129.5, 131.4, 136.5, 139.0, 140.3, 140.9, 148.4, 160.2, 166.3 ppm; ESIMS (m/z) 360.3 [M+H]+; ESI-HRMS Calcd. for C21H18N3O3 [MH]+: 360.1348. Found: 360.1387.
Methyl 3-(4-nitrophenyl)-2-phenyl-2H-pyrazolo[4,3-b]pyridine-6-carboxylate (5g).
83% as a yellow solid (154 mg from 250 mg); mp 184–186 °C; Rf 0.46 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1723 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 4.03 (s, 3H), 7.50–7.53 (m, 5H), 7.77 (d, 2H, J = 8.9 Hz), 8.26 (d, 2H, J = 8.9 Hz), 8.87 (d, 1H, J = 1.7 Hz), 9.27 (d, 1H, J = 1.7 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 52.8, 124.1, 124.9, 126.1, 129.6, 129.9, 130.1, 130.6, 133.8, 134.4, 139.5, 139.9, 141.2, 147.6, 149.8, 165.9 ppm; ESIMS (m/z) 375.2 [M+H]+; ESI-HRMS Calcd. for C20H15N4O4 [MH]+: 375.1093. Found: 375.1101.
Methyl 2-methyl-3-phenyl-2H-pyrazolo[4,3-b]pyridine-6-carboxylate (5h).
87% as a white solid (146 mg from 250 mg); mp 128–129 °C; Rf 0.34 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1716 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 4.00 (s, 3H), 4.31 (s, 3H), 7.49–7.54 (m, 1H), 7.57–7.62 (m, 2H), 7.70 (d, 2H, J = 7.0 Hz), 8.75 (d, 1H, J = 1.7 Hz), 9.16 (d, 1H, J = 1.7 Hz) ppm; 13C NMR (50 MHz, CDCl3): δ 40.0, 52.5, 123.7, 127.9, 128.5, 129.2, 129.4, 129.9, 137.0, 139.0, 139.8, 148.1, 166.4 ppm; ESIMS (m/z) 268.2 [M+H]+; ESI-HRMS Calcd. for C15H14N3O2 [MH]+: 268.1086. Found: 268.1084.
Methyl 2-methyl-3-(4-methylphenyl)-2H-pyrazolo[4,3-b]pyridine-6-carboxylate (5i).
85% as a white solid (145 mg from 250 mg); mp 98–99 °C; Rf 0.35 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1723 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 2.46 (s, 3H), 4.00 (s, 3H), 4.29 (s, 3H), 7.40 (d, 2H, J = 7.9 Hz), 7.59 (d, 2H, J = 8.1 Hz), 8.74 (d, 1H, J = 1.8 Hz), 9.14 (d, 1H, J = 1.7 Hz) ppm; 13C NMR (50 MHz, CDCl3): δ 21.5, 39.9, 52.5, 123.6, 124.9, 128.4, 129.8, 129.9, 137.1, 139.0, 139.6, 139.7, 147.9, 166.4 ppm; ESIMS (m/z) 282.3 [M+H]+; ESI-HRMS Calcd. for C16H16N3O2 [MH]+: 282.1243. Found: 282.1240.
Methyl 3-(2-chlorophenyl)-2-methyl-2H-pyrazolo[4,3-b]pyridine-6-carboxylate (5j).
84% as a white solid (147 mg from 250 mg); mp 109–110 °C; Rf 0.36 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1722 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 4.01 (s, 3H), 4.15 (s, 3H), 7.49–7.55 (m, 3H), 7.61–7.64 (m, 1H), 8.79 (d, 1H, J = 1.7 Hz), 9.15 (d, 1H, J = 1.7 Hz) ppm; 13C NMR (50 MHz, CDCl3): δ 39.6, 52.6, 123.7, 127.2, 127.5, 128.8, 130.4, 131.6, 133.0, 134.7, 135.1, 139.6, 139.8, 148.4, 166.3 ppm; ESIMS (m/z) 302.2 [M+H]+; ESI-HRMS Calcd. for C15H13ClN3O2 [MH]+: 302.0696. Found: 302.0691.
Methyl 2-methyl-2H-pyrazolo[4,3-b]pyridine-6-carboxylate (5k).
86% as a white solid (128 mg from 250 mg); mp 140–141 °C; Rf 0.35 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1720 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 3.99 (s, 3H), 4.31 (s, 3H), 8.23 (s, 1H), 8.73 (s, 1H), 9.15 (d, 1H, J = 1.3 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 41.6, 52.5, 123.3, 125.2, 128.7, 140.6, 141.1, 148.2, 166.3 ppm; ESIMS (m/z) 192.2 [M+H]+; ESI-HRMS Calcd. for C9H10N3O2 [MH]+: 192.0773. Found: 192.0779.
Methyl 1,3-diphenyl-1H-pyrazolo[4,3-b]pyridine-6-carboxylate (10a).
86% as a white solid (154 mg from 250 mg); mp 156–157 °C; Rf 0.53 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1720 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 4.02 (s, 3H), 7.42–7.47 (m, 2H), 7.52–7.64 (m, 4H), 7.80 (d, 2H, J = 7.6 Hz), 8.60 (d, 2H, J = 7.2 Hz), 8.71 (d, 1H, J = 1.7 Hz), 9.31 (d, 1H, J = 1.6 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 52.8, 120.5, 122.8, 123.4, 127.6, 128.9, 129.0, 130.0, 131.6, 132.5, 139.4, 143.2, 145.1, 146.8, 166.1 ppm; ESIMS (m/z) 330.1 [M+H]+; ESI-HRMS Calcd. for C20H16N3O2 [MH]+: 330.1243. Found: 330.1426.
Methyl 3-(4-methylphenyl)-1-phenyl-1H-pyrazolo[4,3-b]pyridine-6-carboxylate (10b).
85% as a white solid (154 mg from 250 mg); mp 160–161 °C; Rf 0.55 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1721 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 2.44 (s, 3H), 4.02 (s, 3H), 7.35 (d, 2H, J = 8.0 Hz), 7.44 (t, 1H, J = 7.4 Hz), 7.60 (t, 2H, J = 7.8 Hz), 7.79 (d, 2H, J = 7.7 Hz), 8.49 (d, 2H, J = 8.1 Hz), 8.70 (d, 1H, J = 1.6 Hz), 9.30 (d, 1H, J = 1.6 Hz) ppm; 13C NMR (50 MHz, CDCl3): δ 21.7, 52.8, 120.4, 122.8, 123.4, 127.6, 128.7, 129.6, 130.0, 132.5, 139.0, 139.5, 143.2, 145.2, 146.7, 166.2 ppm; ESIMS (m/z) 344.1 [M+1]+; DART-HRMS Calcd. for C21H18N3O2 [MH]+: 344.1399. Found: 344.1406.
Methyl 3-(4-chlorophenyl)-1-phenyl-1H-pyrazolo[4,3-b]pyridine-6-carboxylate (10c).
84% as a white solid (155 mg from 250 mg); mp 194–195 °C; Rf 0.54 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1726 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 4.03 (s, 3H), 7.44–7.53 (m, 3H), 7.62 (t, 2H, J = 7.8 Hz), 7.78 (d, 2H, J = 7.7 Hz), 8.58 (d, 2H, J = 8.5 Hz), 8.73 (s, 1H), 9.32 (s, 1H) ppm; 13C NMR (50 MHz, CDCl3): δ 52.7, 120.5, 122.6, 123.4, 127.7, 128.7, 128.9, 129.8, 129.9, 132.4, 134.7, 139.1, 142.8, 143.8, 146.7, 165.8 ppm; ESIMS (m/z) 364.2 [M+H]+; ESI-HRMS Calcd. for C20H15ClN3O2 [MH]+: 364.0853. Found: 364.0841.
Methyl 1-benzyl-3-phenyl-1H-pyrazolo[4,3-b]pyridine-6-carboxylate (10l).
82% as a white solid (149 mg from 250 mg); mp 184–186 °C; Rf 0.48 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 1722 (CO2Me) cm−1; 1H NMR (300 MHz, CDCl3): δ 3.99 (s, 3H), 5.69 (s, 2H), 7.27–7.38 (m, 5H), 7.42 (d, 1H, J = 7.3 Hz), 7.52 (t, 2H, J = 7.5 Hz), 8.33 (d, 1H, J = 1.7 Hz), 8.53 (d, 2H, J = 7.1 Hz), 9.23 (d, 1H, J = 1.7 Hz) ppm; 13C NMR (50 MHz, CDCl3): δ 52.8, 54.0, 119.4, 122.8, 127.4, 128.4, 128.7, 128.9, 129.1, 132.0, 133.1, 136.0, 142.2, 143.6, 146.2, 166.2 ppm; ESIMS (m/z) 344.2 [M+H]+; ESI-HRMS Calcd. for C21H18N3O2 [MH]+: 344.1399. Found: 344.1387.
2,3-Diphenyl-2H-pyrazolo[4,3-b]pyridine-6-carbonitrile (13a).
81% as a white solid (141 mg from 250 mg); mp 184–185 °C; Rf 0.57 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 2228 (CN) cm−1; 1H NMR (300 MHz, CDCl3): δ 7.42–7.52 (m, 10H), 8.54 (s, 1H), 8.76 (s, 1H) ppm; 13C NMR (75 MHz, CDCl3): δ 107.4, 117.7, 126.0, 127.5, 129.1, 129.5, 129.6, 129.7, 130.0, 132.4, 137.3, 138.3, 139.6, 139.9, 147.9 ppm; ESIMS (m/z) 297.3 [M+H]+; ESI-HRMS Calcd. for C19H13N4 [MH]+: 297.1140. Found: 297.1145.
3-(4-Methylphenyl)-2-phenyl-2H-pyrazolo[4,3-b]pyridine-6-carbonitrile (13b).
80% as a white solid (141 mg from 250 mg); mp 173–174 °C; Rf 0.55 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 2229 (CN) cm−1; 1H NMR (300 MHz, CDCl3): δ 2.39 (s, 3H), 7.24 (d, 2H, J = 6.6 Hz), 7.40 (d, 2H, J = 6.6 Hz), 7.48 (s, 5H), 8.53 (s, 1H), 8.75 (s, 1H) ppm; 13C NMR (75 MHz, CDCl3): δ 21.5, 107.2, 117.7, 124.5, 125.9, 126.8, 129.5, 129.7, 129.8, 130.5, 132.3, 137.4, 138.2, 139.5, 139.6, 139.9, 147.6 ppm; ESIMS (m/z) 311.3 [M+H]+; ESI-HRMS Calcd. for C20H15N4 [MH]+: 311.1297. Found: 311.1291.
3-(4-Chlorophenyl)-2-phenyl-2H-pyrazolo[4,3-b]pyridine-6-carbonitrile (13c).
78% as a white solid (140 mg from 250 mg); mp 174–175 °C; Rf 0.56 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 2227 (CN) cm−1; 1H NMR (300 MHz, CDCl3): δ 7.40 (d, 2H, J = 8.6 Hz), 7.46–7.52 (m, 7H), 8.54 (d, 1H, J = 1.7 Hz), 8.76 (d, 1H, J = 1.7 Hz) ppm; 13C NMR (50 MHz, CDCl3): δ 107.5, 117.5, 126.0, 129.4, 129.8, 129.9, 131.2, 132.5, 135.8, 136.0, 138.2, 139.6, 139.7, 148.1 ppm; ESIMS (m/z) 331.1 [M+H]+; ESI-HRMS Calcd. for C19H12ClN4 [MH]+: 331.0750. Found: 331.0754.
3-(2-Chlorophenyl)-2-methyl-2H-pyrazolo[4,3-b]pyridine-6-carbonitrile (13j).
75% as a white solid (126 mg from 250 mg); mp 160–161 °C; Rf 0.49 (hexanes: EtOAc, 80:20, v/v); IR (KBr): νmax 2218 (CN) cm−1; 1H NMR (300 MHz, CDCl3): δ 4.16 (s, 3H), 7.46–7.57 (m, 3H), 7.62–7.64 (m, 1H), 8.48 (d, 1H, J = 1.7 Hz), 8.68 (d, 1H, J = 1.7 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 39.8, 106.7, 117.7, 126.6, 127.6, 130.5, 131.8, 131.9, 132.9, 135.1, 135.6, 138.5, 138.8, 147.7 ppm; ESIMS (m/z) 269.2 [M+H]+; ESI-HRMS Calcd. for C14H10ClN4 [MH]+: 269.0594. Found: 269.0593.
Acknowledgements
One of the authors (M.N.) gratefully acknowledges financial assistance in the form of a fellowship from CSIR, New Delhi. The authors acknowledge the SAIF Division for providing the spectroscopic and analytical data.
References
-
(a)
L. Yet, in Comprehensive Heterocyclic Chemistry, Vol.4, 4.01, ed. A. R. Katrizky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Pergamon, London, 2008, p 1 Search PubMed
 ;
(b) T. V. Herk, J. Brussee, A. M. C. H. van den Nieuwendijk, P. A. M. van der Klein, A. P. IJzerman, C. Stannek, A. Burmeister and A. Lorenzen, J. Med. Chem., 2003, 46, 3945 CrossRef ;
(c) T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Cater, P.W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Vennhuizen, Y. Y. Zhang and P. Isakson, J. Med. Chem., 1997, 40, 1347 CrossRef CAS ;
(d) S. Kortagere, W. J. Welsh, J. M. Morrisey, T. Daly, I. Ejigiri, P. Sinnis, A. B. Vaidya and L. W. Bergman, J. Chem. Inf. Model., 2010, 50, 840 CrossRef CAS ;
(e) R. L. Dow, P. A. Carpino, J. R. Hadcock, S. C. Black, P. A. Iredale, P. DaSilva-Jardine, S. R. Schneider, E. S. Paight, D. A. Griffith, D. O. Scott, R. E. O'Connor and C. I. Nduaka, J. Med. Chem., 2009, 52, 2652 CrossRef CAS ;
(f) G. Ouyang, X.-J. Cai, Z. Chen, B.-A. Song, P. S. Bhadury, S. Yang, L.-H. Jin, W. Xue, D.-Y. Hu and S. Zeng, J. Agric. Food Chem., 2008, 56, 10160 CrossRef CAS ;
(g) M. Herdemann, I. Heit, F.-U. Bosch, G. Quintini, C. Scheipers and A. Weber, Bioorg. Med. Chem. Lett., 2010, 20, 6998 CrossRef CAS ;
(h) P. Giovanni, G. Luca, B. Francesca, T. Giorgio, M. Patrizia, G. Grazia, G. Fabrizio, C. Massimo, G. Orlando and C. Paolo, Org. Biomol. Chem., 2007, 5, 2567 RSC ;
(i) N. K. Terret, A. S. Bell, D. Brown and P. Ellips, Bioorg. Med. Chem. Lett., 1996, 6, 1819 CrossRef ;
(j) M. Rinaldi-Carmona, F. Barth, M. Heaulme, D. Shire, B. Calandra, C. Congy, S. Martinez, J. Maruani, G. Neliat, D. Caput, P. Ferrara, P. Soubrie, J. C. Breliere and G. Le Fur, FEBS Lett., 1994, 350, 240 CrossRef CAS ;
(k) H. A. De Wald, S. Lobbestael and B. P. H. Poschel, J. Med. Chem., 1981, 24, 982 CrossRef CAS .
;
(b) T. V. Herk, J. Brussee, A. M. C. H. van den Nieuwendijk, P. A. M. van der Klein, A. P. IJzerman, C. Stannek, A. Burmeister and A. Lorenzen, J. Med. Chem., 2003, 46, 3945 CrossRef ;
(c) T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Cater, P.W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Vennhuizen, Y. Y. Zhang and P. Isakson, J. Med. Chem., 1997, 40, 1347 CrossRef CAS ;
(d) S. Kortagere, W. J. Welsh, J. M. Morrisey, T. Daly, I. Ejigiri, P. Sinnis, A. B. Vaidya and L. W. Bergman, J. Chem. Inf. Model., 2010, 50, 840 CrossRef CAS ;
(e) R. L. Dow, P. A. Carpino, J. R. Hadcock, S. C. Black, P. A. Iredale, P. DaSilva-Jardine, S. R. Schneider, E. S. Paight, D. A. Griffith, D. O. Scott, R. E. O'Connor and C. I. Nduaka, J. Med. Chem., 2009, 52, 2652 CrossRef CAS ;
(f) G. Ouyang, X.-J. Cai, Z. Chen, B.-A. Song, P. S. Bhadury, S. Yang, L.-H. Jin, W. Xue, D.-Y. Hu and S. Zeng, J. Agric. Food Chem., 2008, 56, 10160 CrossRef CAS ;
(g) M. Herdemann, I. Heit, F.-U. Bosch, G. Quintini, C. Scheipers and A. Weber, Bioorg. Med. Chem. Lett., 2010, 20, 6998 CrossRef CAS ;
(h) P. Giovanni, G. Luca, B. Francesca, T. Giorgio, M. Patrizia, G. Grazia, G. Fabrizio, C. Massimo, G. Orlando and C. Paolo, Org. Biomol. Chem., 2007, 5, 2567 RSC ;
(i) N. K. Terret, A. S. Bell, D. Brown and P. Ellips, Bioorg. Med. Chem. Lett., 1996, 6, 1819 CrossRef ;
(j) M. Rinaldi-Carmona, F. Barth, M. Heaulme, D. Shire, B. Calandra, C. Congy, S. Martinez, J. Maruani, G. Neliat, D. Caput, P. Ferrara, P. Soubrie, J. C. Breliere and G. Le Fur, FEBS Lett., 1994, 350, 240 CrossRef CAS ;
(k) H. A. De Wald, S. Lobbestael and B. P. H. Poschel, J. Med. Chem., 1981, 24, 982 CrossRef CAS .
-
(a)
A. Kleemann, J. Engel, B. Kutscher and D. Reichert, in Pharmaceutical Substances, Thieme, Stuttgart, New York, 1999, 3rd edn, p. 58, p. 1725 Search PubMed ;
(b) M. Boollel, S. Gepi-Attee, J. C. Gingell and M. J. Allen, Br. J. Urol., 1996, 7, 257 Search PubMed ;
(c) P. Pacher, A. Nivorozhkin and C. Szabo, Pharmacol. Rev., 2006, 58, 87 CrossRef CAS ;
(d) J. Mittendorf, S. Weigand, C. Alonso-Alija, E. Bischoff, A. Feurer, M. Gerisch, A. Kern, A. Knorr, D. Lang, K. Muenter, M. Radtke, H. Schirok, K. H. Schlemmer, E. Stahl, A. Straub, F. Wunder and J. P. Stasch, ChemMedChem, 2009, 4, 853 CrossRef CAS ;
(e) R. Elie, E. Ruther, I. Farr, G. Emilien and E. Salinas, J. Clin. Psychiatry, 1999, 60, 536 CrossRef CAS .
-
(a) S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984 CrossRef CAS ;
(b)
L. Yet, Five-Membered Ring Systems: With More than One N Atom, in: Progress in Heterocyclic Chemistry, ed. G. W. Gribble and J. A. Joule), Elsevier, 2009, Vol. 21, pp 224 Search PubMed ;
(c) G. Vilkauskaite, A. Sackus and W. Holzer, Eur. J. Org. Chem., 2011, 5123 CrossRef CAS ;
(d) W. Jin, H. Yu and Z. Yu, Tetrahedron Lett., 2011, 52, 5884 CrossRef CAS ;
(e) J. Z. Chandanshive, B. F. Bonini, W. Tiznado, C. A. Escobar, J. Caballero, C. Femoni, M. Fochi and M. C.Franchini, Eur. J. Org. Chem., 2011, 4703 Search PubMed ;
(f) S. Lee and S. B. Park, Org. Lett., 2009, 11, 5214 CrossRef CAS ;
(g) S. Guillou and Y. L. Janin, Chem.–Eur. J., 2010, 16, 4669 CrossRef CAS ;
(h) C. Almansa, M. Virgili, E. Carceller and P. Grima-Poveda, Heterocycles, 2008, 75, 1695 CrossRef CAS ;
(i) A. F. A. Harb, H. H. Abbas and F. H. Mostafa, Chem. Pap., 2005, 59, 187 CAS .
-
(a) For reviews on Copper-mediated cross couplings- S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS ;
(b) I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef CAS ;
(c) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS ;
(d) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2008, 47, 3096 CrossRef CAS ;
(e) M. Carril, R. SanMartin and E. Dominguez, Chem. Soc. Rev., 2008, 37, 639 RSC ;
(f) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954 CrossRef CAS ;
(g) D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13 RSC ;
(h) H. Rao and H. Fu, Synlett, 2011, 745 CAS ;
(i) Y. Liu and J.-P. Wan, Org. Biomol. Chem., 2011, 9, 6873 RSC .
-
(a) For reviews on Palladium-mediated cross couplings- L. Jiang and S. L. Buchwald, Palladium-Catalyzed Aromatic Carbon-Nitrogen Bond Formation.In Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere, F. Diederich, Wiley-VCH: Weinheim, 2nd edn 2004, pp 699–760 Search PubMed ;
(b)
A. R. Muci and S. L. Buchwald, Practical Palladium Catalysts for C-N and C-O Bond Formation. In Topics in Current Chemistry; N. Miyaura, Ed.; Springer-Verlag: Berlin, 2002, Vol. 219, pp 131–209 Search PubMed ;
(c)
J. F. Hartwig, In Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, Wiley-Interscience: New York, 2002, pp 1051–1096 Search PubMed ;
(d) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534 CrossRef CAS ;
(e) J. F. Hartwig, Angew. Chem., 1998, 110, 2154 CrossRef ; Angew. Chem. Int. Ed., 1998, 37, 2046;
(f) J. P. Wolfe, S. Wagaw, J.-F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805 CrossRef CAS ;
(g) J. F. Hartwig, Acc. Chem. Res., 1998, 31, 852 CrossRef CAS ;
(h) C. G. Frost and P. Mendonca, J. Chem. Soc., Perkin Trans. 1, 1998, 2615 RSC .
- S. Nag, M. Nayak and S. Batra, Adv. Synth. Catal., 2009, 351, 2715 CrossRef CAS , and references cited therein..
- M. Nayak and S. Batra, Adv. Synth. Catal., 2010, 352, 3431 CrossRef CAS , and references cited therein..
- M. Nayak, N. Rastogi and S. Batra, Eur. J. Org. Chem., 2012, 1360 CrossRef CAS .
-
(a) H. M. Ibrahim, H. Behbehani, S. Makhseed and M. H. Elnagdi, Molecules, 2011, 16, 3723 CrossRef CAS ;
(b) B. Avila, D. M. Solano, M. J. Haddadin and M. J. Kurth, Org. Lett., 2011, 13, 1060 CrossRef CAS ;
(c)
M. Mochizuki, T. Imaeda, WO 2010140339, 2010, Chem. Abst., 154, 30328 Search PubMed ;
(d)
J. D. Ha, J. O. Lee, J. U. Ryu, S. Y. Cho, H. J. Jung, J. G. Lee, S. Y. Han, KR 2010057433, 2010, Chem. Abst., 153, 87776 Search PubMed ;
(e) T. Tite, Ni. Lougiakis, V. Myrianthopoulos, P. Marakos, E. Mikros, N. Pouli, R. Tenta, E. Fragopoulou and T. Nomikos, Tetrahedron, 2010, 66, 9620 CrossRef CAS ;
(f) C. Nyffenegger, E. Pasquinet, F. Suzenet, D. Poullain, C. Jarry, J.-M. Léger and G. Guillaumet, Tetrahedron, 2008, 64, 9567 CrossRef CAS ;
(g) M. Béres, G. Hajós, Z. Riedl, T. Soós, G. Timári and A. Messmer, J. Org. Chem., 1999, 64, 5499 CrossRef .
-
R. E. Markwell, R. W. Ward and C. R. De Mello, US Pat. 4833136, 1989, Chem. Abst.87, 578 Search PubMed .
-
(a) K. Wilcoxen, C. Q. Huang, J. R. McCarthy, D. E. Grigoriadis and C. Chen, Bioorg. Med. Chem. Lett., 2003, 13, 3367 CrossRef CAS ;
(b) R. S. Gross, Z. Guo, B. Dyck, T. Coon, C. Q. Huang, R. F. Lowe, D. Marinkovic, M. Moorjani, J. Nelson, S. Zamani-Kord, D. E. Grigoriadis, S. R. J. Hoare, P. D. Crowe, J. Han Bu, M. Haddach, J. McCarthy, J. Saunders, R. Sullivan, T. Chen and J. P. Williams, J. Med. Chem., 2005, 48, 5780 CrossRef CAS .
- G. Piersanti, L. Giorgi, F. Bartoccini, G. Tarzia, P. Minetti, G. Gallo, F. Giorgi, M. Castorina, O. Ghirardi and P. Carminati, Org. Biomol. Chem., 2007, 5, 2567 CAS .
- R. Pathak, V. Singh, S. Nag, S. Kanojiya and S. Batra, Synthesis, 2006, 813 CAS .
- D. Ma and Q. Cai, Acc. Chem. Res., 2008, 41, 1450 CrossRef CAS , and references cited therein..
- S. Nag, R. Pathak, M. Kumar, P. K. Shukla and S. Batra, Bioorg. Med. Chem. Lett., 2006, 16, 3824 CrossRef CAS .
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures for 4-iodo pyrazolecarbaldehydes, MBH adducts, MBH acetates and allylamines and corresponding characterization data along with copies of 1H and 13C-NMR spectra. See DOI: 10.1039/c2ra01170k |
| ‡ CDRI Communication No. 8200 |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.