β-Lactam-host defence peptide conjugates as antibiotic prodrug candidates targeting resistant bacteria†
Stéphane
Desgranges
a,
Carol C.
Ruddle
a,
Liam P.
Burke
b,
Tara M.
McFadden
ab,
John E.
O'Brien
c,
Deirdre
Fitzgerald-Hughes
b,
Hilary
Humphreys
bd,
Timothy P.
Smyth
e and
Marc
Devocelle
*a
aCentre for Synthesis and Chemical Biology, Department of Pharmaceutical & Medicinal Chemistry, Royal College of Surgeons in Ireland, 123, St. Stephen's Green, Dublin 2, Ireland. E-mail: mdevocelle@rcsi.ie; Fax: +353 (0)1 4022168; Tel: +353 (0)1 4022176
bDepartment of Clinical Microbiology, Royal College of Surgeons in Ireland, Dublin 9, Ireland
cSchool of Chemistry, Trinity College Dublin, Dublin 2, Ireland
dDepartment of Microbiology, Beaumont Hospital, Dublin 9, Ireland
eDepartment of Chemical & Environmental Sciences, University of Limerick, Ireland
First published on 12th January 2012
Abstract
The first hybrid molecule of a β-lactam antibiotic and a host defence peptide and a method for the preparation of this type of molecule are reported. Conjugation of an antimicrobial peptide to a cephalosporin, through a cleavable linker, reversibly masks one of the activity determinants of the peptide. Its release from the β-lactam core can be selectively triggered by bacterial enzymes (β-lactamases) which mediate resistance to β-lactam agents. A prototypical cephalothin-bactenecin candidate was synthesised, using a copper(I)-catalysed azide–alkyne cyclo-addition reaction for the conjugation step. Enzymatic hydrolysis assays of this candidate were initially performed with a purified β-lactamase to confirm that the peptide can be released from the cephalosporin. The antimicrobial activity of the conjugate was then assessed against representative strains of bacteria and compared to the activities of its parent β-lactam and peptide components and to those of two analogous conjugates based on non-cleavable linkers. The results of these assays indicate that the conjugate has an activity distinct from its separate constituents and that the release of the peptide from the cephalosporin may contribute to its mechanism of action. Furthermore, the results of antimicrobial assays performed with an isogenic strain of bacteria expressing or not an extended-spectrum β-lactamase, suggest that antimicrobial peptide prodrug candidates targeting resistant bacteria could be generated from these hybrid antibiotics.
1. Introduction
Host defence peptides (HDPs) are multifunctional molecular effectors of innate immunity and are the first line of defence against infection in multicellular organisms. Their multiple functions in host defences, associated with their low susceptibility to classical mechanisms of drug resistance and low propensity to select resistant mutants, support the rationale of developing novel peptide-based therapeutics harnessing the effector mechanisms of innate immunity.1 The development of systemic therapies based on antimicrobial peptides requires primarily solutions addressing the question of possible toxicity.2 Several strategies have been explored to exploit the therapeutic potential of HDPs, including methods to improve their pharmacokinetic and/or pharmacodynamic properties, delivery or production.3 A prodrug approach has also been proposed as a promising strategy to achieve clinical success with antimicrobial peptides.4 This approach can indeed increase the therapeutic index of a pharmacologically active agent and overcome a barrier to drug targeting.5 A prodrug modification is commonly performed by conjugating a promoiety to the parent drug through one of its activity determinants.6 If the promoiety is eliminated in vivo by a chemical or enzymatic reaction only occurring at the target body site, selective delivery of the active agent can then be achieved.The synthesis and activity of a β-lactam-HDP conjugate is reported herein. This dual-pharmacophore molecule constitutes the first reported candidate of a bioreversible derivative of a HDP which can be activated by a bacterial enzyme of antibiotic resistance.
2. Results and discussion
2.1. Prodrug design
HDPs are highly diverse in size and secondary structure. Their sequences are characterised by a net excess of positively charged residues (cationic peptides), a hydrophobic amino acid content approximating 50% and consequently by their ability to adopt amphipathic secondary structures.2 The hydrophobicity and net charge of these peptides are important determinants of their antimicrobial activity and structure–activity relationships can be described in term of numbers of positive charges and lipophilic units and of sequence patterns.7,8 The Nα-amino terminus is commonly free in natural and man-made HDPs and contributes to the net charge of the peptide sequence. Masking this group and reducing the net charge of the peptide is generally associated with a loss of activity in endogenous HDPs and their synthetic analogues.9 Transiently modifying the N-terminus of a HDP with a negatively charged promoiety can therefore generate prodrug candidates of the parent peptide.β-Lactams agents such as cephalosporins meet the requirements of a promoiety in prodrug design.6β-Lactam antibiotics have been in clinical use for more than 65 years and have proven to be safe, as well as their degradation products. They also allow the bioreversible modification of a pharmacologically active agent by conjugation of one of its functional groups at the 3′-position of the cephem core. Release of the 3′-substituent, i.e. the parent drug, is selectively triggered by cleavage of the β-lactam bond by bacterial enzymes.10 They consist essentially of the enzymes of resistance to β-lactam antibiotics (β-lactamases) and include potentially the enzymes of bacterial cell-wall biosynthesis (penicillin-binding proteins or PBPs). The expression of β-lactamases is the most commonly acquired mechanism of antibiotic resistance, particularly in Gram-negative pathogens. It is conserved in multi-drug resistant (MDR) organisms which can be resistant against multiple agents, including those used as the last line of effective antibiotic treatment.11β-Lactamases, in particular extended-spectrum β-lactamases (ESBLs), are highly efficient catalysts and attractive enzymes for the activation of antibiotic prodrugs targeting MDR pathogens.10
Conjugation at a cephalosporin's 3′-position of a HDP through its Nα-amino terminus was therefore proposed to generate β-lactamase-dependent prodrugs of antimicrobial peptides. This conjugation masks an important pharmacophoric element of the peptide, but also reduces its net positive charge by 2 units, due to the presence of a carboxylate at the 4-position of the cephem nucleus. The structure of these prodrug candidates included also a self-immolative carbamate linker between the cephalosporin and peptide moieties, to facilitate the elimination of the 3′-primary amine substituent upon hydrolysis of the β-lactam bond (Fig. 1).
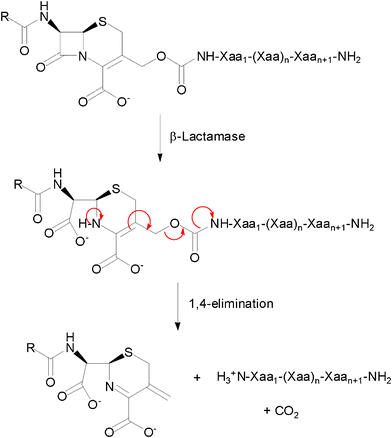 | ||
| Fig. 1 General structure and reaction of a cephalosporin-host defence peptide conjugate as a β-lactamase-dependent prodrug. | ||
2.2. Conjugate synthesis
The synthesis of a cephalosporin-peptide conjugate is a priori based on a convergent synthetic route in which the β-lactam and peptidic components are prepared separately. A conventional approach involves their conjugation by selective formation of a carbamate linkage between the cephalosporin's 3′-hydroxyl and the peptide's Nα-amino terminus, requiring protection of all the other functional groups and therefore a final deprotection step. These steps have to be performed under conditions which meet the requirements of both the β-lactam and peptide chemistries. On one hand, a variety of α-amino acid side-chain functionalities are present in peptides. The guanidine and indole groups in particular are among the most common functional groups in HDPs, the optimisation of their antimicrobial activities generally yielding sequences enriched in arginine and tryptophan, respectively. Arginine is one of the most demanding residues in terms of acid concentration and reaction time for the deprotection of its guanidino group.12 This reaction is in addition performed in the presence of nucleophilic reagents commonly named scavengers. On the other hand, the β-lactam ring of penicillins and cephalosporins is particularly labile in the presence of nucleophiles and at alkaline, but also acidic pHs.13The cephalosporin and peptide candidates selected for the synthesis of a prototypical conjugate were cephalothin and Bac8c, respectively. Cephalothin 1, containing a 3′-acetate substituent and a thienyl side-chain, is a first generation cephalosporin with a broad spectrum of activity. Bac8c is a peptide amide of sequence RIWVIWRR-NH2, obtained by optimisation of the bovine dodecapeptide bactenecin.14 The asset of this short 8-mer candidate is an activity against both Gram-negative and Gram-positive bacteria in the low micromolar range. Also, its number of residues, which remains close to the minimal length of a continuous epitope for an antigen,15 should prevent its immunogenicity and limit its production cost. To impart metabolic stability to this candidate as well, preliminary studies were conducted to assess the antimicrobial activity of its enantiomeric sequence D-Bac8c. Typical Minimum Inhibitory Concentrations (MICs) of D-Bac8c-NH2 against well characterised laboratory strains of Gram-negative Escherichia coli and Gram-positive Staphylococcus aureus were of 4.2 μM (Table 1), slightly higher than the reported MICs of 1.7 μM for L-Bac8c-NH2 against different strains of the same organisms.14 Homologous substitution of D-isoleucine with D-leucine was also attempted for economical reasons, yielding the amidated peptide 2 of sequence rlwvlwrr, named D-Bac8c(Leu2,5). The 2 E. coli reference strains selected previously had similar susceptibilities to both all-D peptides (4.2 μM), while S. aureus (8325–4) showed higher susceptibility to D-Bac8c(Leu2,5) (2.1 μM) than to D-Bac8c (Table 1). The activity of 2 against E. coli was also verified against 10 clinical isolates of ESBL-positive E. coli. A MIC of 4.2 μM was confirmed in 8 of these strains, whereas MICs of 2.1 μM and 8.4 μM were achieved against 2 individual strains.
After selection of the β-lactam and peptide components, the cephalosporin synthon was assembled first and functionalised with an active carbonate at the C-3′ position of the cephem core for reaction with the Nα-amino terminus of a peptide sequence (Scheme 1). It was synthesised from commercially available 7-aminocephalosporanic acid (7-ACA), by 3′-deacetylation, followed by protection of the 7-amino and 4-carboxyl by thienylation and esterification by diphenyldiazomethane, respectively. The order of these protection steps was imposed by the lack of solubility of the zwitterionic deacetylated intermediate 7-AHCA 3 in organic solvents. Attempts to esterify the 4-carboxyl first resulted in the lactonisation of the 3-hydroxymethyl and 4-carboxyl groups or Δ3/Δ2 isomerisation of the cephem double bond. Functionalisation with a 3′-p-nitrophenyl- or 3′-tetrachloroethyl-carbonate was finally performed by reaction of 4 with the corresponding chloroformates.
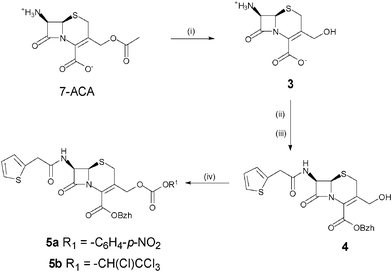 | ||
| Scheme 1 Synthesis of the cephalosporin synthon. (i) TBAOH, Et3N, MeOH, H2O; (ii) 2-thienylacetyl chloride, NaOH, H2O, acetone; (iii) diphenyldiazomethane, AcOEt; (iv) p-nitrophenylchloroformate, pyridine, DMAP, DCM for 5a; tetrachloroethylchloroformate, pyridine, DMAP, DCM for 5b. | ||
The formation of a carbamate linkage between a cephalosporin and a peptide sequence was initially investigated by assessing the displacement of the active carbonate intermediates of cephalothin with a single α-amino acid. The methyl ester of alanine was reacted with 5a/5b, with or without addition of 1 equivalent of pyridine and a catalytic or stoichiometric quantity of DMAP. In the absence of additives, partial formation of a carbamate product was only observed from the tetrachloroethyl-carbonate after extended reaction times (60% conversion in 24 h). In the presence of a catalytic quantity of DMAP, conversion to the desired carbamate occurred rapidly from the tetrachloroethyl-carbonate (100% conversion in 3 h), but only marginally from the p-nitrophenyl-carbonate even after 12 h of reaction. In both cases, the use of stoichiometric quantities of DMAP (1–2 equivalents) were associated with a degradation of the cephalosporin's β-lactam core.
Displacement of the tetrachloroethyl-carbonate was then attempted directly with a model 6-mer, fully deprotected, peptide. Reaction of 5b with the amidated sequence RWRWRW-NH2, in the presence of 1 equivalent of pyridine and a catalytic amount of DMAP, induced however the degradation of the β-lactam ring without formation of the carbamate product. This was also the case when the single amino acid arginine was used as a nucleophile candidate to displace the tetrachloroethyl-carbonate. On the other hand the carbamates of the sterically unhindered amino acids glycine and alanine, protected as methyl or allyl esters, could be formed. In the latter case, deprotection of the α-carboxyl groups was performed with conservation of the β-lactam core's integrity. The combination of a catalytic quantity of Pd(PPh3)4 with 1 equivalent of toluene sulfinic acid in THF:H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was found to provide the optimal deprotection conditions for this reaction. Finally, to complete the assembly of the peptide sequence by an approach similar to segment condensation, coupling to the 3′-carbamoylalanine derivative of cephalothin of a side-chain protected peptide was attempted in solution. A model sequence (H–Ahx–Ala–Gly–Arg(Pbf) –Pro–Val–Asp(O–t–Bu) –NH2) containing common amino acids and protecting groups and modified with a N-terminal amino-hexanoyl spacer to reduce steric hindrance at the ligation site was assembled and cleaved protected from a Sieber resin. Coupling to the cephalosporin-amino acid conjugate was successfully carried out with DCC/HOBt coupling chemistry, as confirmed by mass spectrometry and 1H NMR analyses. However, the final product could not be retrieved after attempting the side-chain deprotections by treatment with a cocktail of triisopropylsilane (5.5%), thioanisole (10.5%) and TFA (84%). Additional, independent, assays involving the treatment of cephalothin and a protected peptide with cleavage cocktails of varied composition, containing or not trifluoroacetic anhydride as a drying agent, also failed to identify a mixture of reagents which allowed at the same time complete deprotection of the arginine's guanidino group and maintenance of the β-lactam's integrity. Other protections of the guanidino group, requiring milder reaction conditions for their elimination, were therefore evaluated. The bis-allyloxycarbonyl (Alloc)-protection of arginine could be successfully removed from an isolated arginine, but yielded partially protected peptides for arginine-rich sequences such as Bac8c, even after repetitive treatment of the resin-bound peptide with tetrakis-(triphenylphosphine)-palladium(0) catalyst, acetic acid and morpholine. As the semi-permanent protection of arginine with super acid-sensitive groups is not currently available, the synthesis of a 4-methoxytrityl (Mmt) derivative of arginine was investigated. Starting from Fmoc–Arg–OH, preliminary protection of the carboxyl group as an allyl ester was required to selectively perform the bis-alkylation of the guanidino group with 4-methoxytriphenylmethyl chloride. The allyl ester was finally deprotected with palladium on charcoal to provide the precursor Fmoc–Arg(Mmt)2–OH. The application of this reagent in solid phase peptide synthesis was initially performed with a simple model sequence (Arg–Gly–Asp–Ser), assembled from a Sieber resin. However, coupling of this sterically hindered amino acid required its activation as an acid fluoride by reaction with TFFH, as even the PyBroP coupling chemistry remained inefficient. Furthermore, concentrations of TFA as high as 25%, incompatible with the stability of the β-lactam core, were required to deprotect the guanidino group.
:1) was found to provide the optimal deprotection conditions for this reaction. Finally, to complete the assembly of the peptide sequence by an approach similar to segment condensation, coupling to the 3′-carbamoylalanine derivative of cephalothin of a side-chain protected peptide was attempted in solution. A model sequence (H–Ahx–Ala–Gly–Arg(Pbf) –Pro–Val–Asp(O–t–Bu) –NH2) containing common amino acids and protecting groups and modified with a N-terminal amino-hexanoyl spacer to reduce steric hindrance at the ligation site was assembled and cleaved protected from a Sieber resin. Coupling to the cephalosporin-amino acid conjugate was successfully carried out with DCC/HOBt coupling chemistry, as confirmed by mass spectrometry and 1H NMR analyses. However, the final product could not be retrieved after attempting the side-chain deprotections by treatment with a cocktail of triisopropylsilane (5.5%), thioanisole (10.5%) and TFA (84%). Additional, independent, assays involving the treatment of cephalothin and a protected peptide with cleavage cocktails of varied composition, containing or not trifluoroacetic anhydride as a drying agent, also failed to identify a mixture of reagents which allowed at the same time complete deprotection of the arginine's guanidino group and maintenance of the β-lactam's integrity. Other protections of the guanidino group, requiring milder reaction conditions for their elimination, were therefore evaluated. The bis-allyloxycarbonyl (Alloc)-protection of arginine could be successfully removed from an isolated arginine, but yielded partially protected peptides for arginine-rich sequences such as Bac8c, even after repetitive treatment of the resin-bound peptide with tetrakis-(triphenylphosphine)-palladium(0) catalyst, acetic acid and morpholine. As the semi-permanent protection of arginine with super acid-sensitive groups is not currently available, the synthesis of a 4-methoxytrityl (Mmt) derivative of arginine was investigated. Starting from Fmoc–Arg–OH, preliminary protection of the carboxyl group as an allyl ester was required to selectively perform the bis-alkylation of the guanidino group with 4-methoxytriphenylmethyl chloride. The allyl ester was finally deprotected with palladium on charcoal to provide the precursor Fmoc–Arg(Mmt)2–OH. The application of this reagent in solid phase peptide synthesis was initially performed with a simple model sequence (Arg–Gly–Asp–Ser), assembled from a Sieber resin. However, coupling of this sterically hindered amino acid required its activation as an acid fluoride by reaction with TFFH, as even the PyBroP coupling chemistry remained inefficient. Furthermore, concentrations of TFA as high as 25%, incompatible with the stability of the β-lactam core, were required to deprotect the guanidino group.
As the synthetic routes requiring a final deprotection step were associated with an aminolysis of the β-lactam ring and/or limited conversions and reliability, an approach allowing the unequivocal attachment of a fully deprotected peptide to a cephalosporin, preserving the integrity of the β-lactam core and compatible with any peptidic functional group, was developed. The copper(I)-catalysed azide–alkyne cyclo-addition is an archetypal reaction of a click chemistry approach allowing the quantitative and selective ligation of two abiotic functional groups.16 Applied to a cephalosporin as an acylated dipeptide mimic and a peptide, this reaction offers the additional advantage of producing a peptide bond isostere, the triazole ring, between these two ligated moieties.17 To produce β-lactam-peptide conjugates, the roles of azide and alkyne partners in the cyclo-addition reaction were assigned to the peptide and the cephalosporin, respectively (Scheme 2). Substitution of the peptide's Nα-amino terminus with an azido group and functionalisation of the cephalosporin with a 3′-propargyl-carbamate group yield a 1,3-dipolar cycloaddition product in which the peptide component is modified with a N-terminal triazole-ε2-amino acid. Release of the peptide from the cephalosporin upon hydrolysis of the β-lactam unmasks a primary amino group and restores the net charge of the native peptide sequence (Fig. 2).
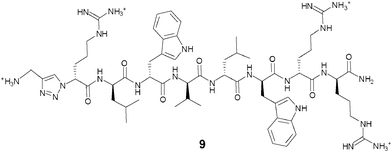 | ||
| Fig. 2 Structure of D-Arg1-triazole-ε2- D-Bac8c(Leu2,5). | ||
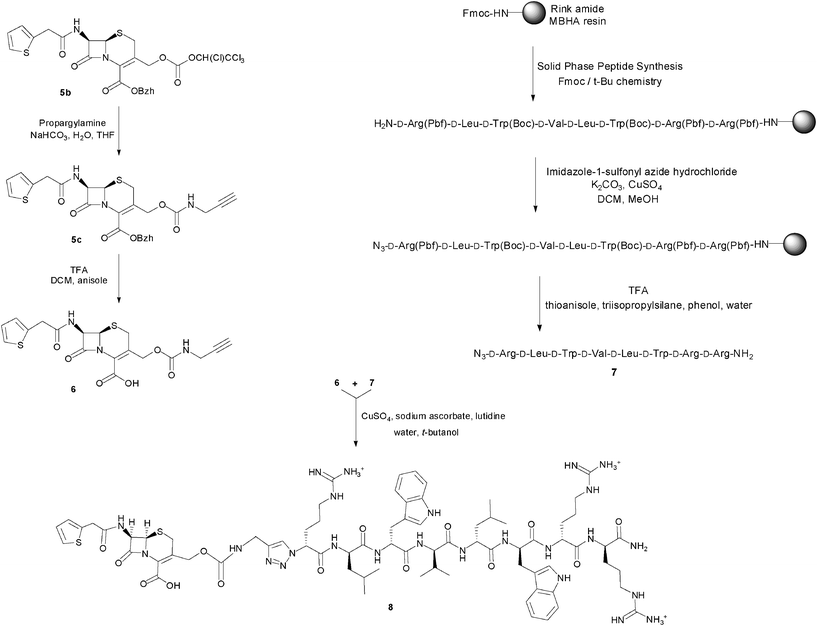 | ||
| Scheme 2 Synthesis of the prodrug candidate cephalothin-D-Bac8c(Leu2,5) 8. | ||
The alkyne-modified cephalosporin 6 was prepared by reaction of the active tetrachloroethyl-carbonate 5b with propargylamine, followed by deprotection of the benzhydryl ester under mild acidic conditions. For the azido-peptide, a simplified Bac8c sequence containing only one side-chain functionalised amino acid was initially selected to investigate its synthesis. Homologous substitution of tryptophan with 2-naphtylalanine was therefore performed at the positions 3 and 6 of the Bac8c sequence. Also, to limit the cost of these synthetic feasibility studies, Bac8c(2-Nal3,6) was assembled from L-amino acids. The sequence was elongated by Solid Phase Peptide Synthesis (SPPS)18 and N-terminally modified by a DiazoTransfer reaction directly applied on the polymer-supported peptide. The azido-peptide was then cleaved from the resin and deprotected by treatment with a cleavage cocktail composition of which was adapted to prevent reduction of the azide. This solid phase synthesis strategy for the preparation of the peptide fragment proved to be more expedient and efficient than a hybrid solid-solution phase approach, performed by converting the N-terminal α-amino acid (H-Arg(Pbf)-OH) into the corresponding α-azido acid and coupling the latter to the penultimate residue of a resin-bound peptide sequence. Finally, the copper(I)-catalysed cycloaddition of the alkyne-modified cephalosporin with the azido-peptide allowed the conjugation of the β-lactam and peptide fragments through a carbamate-1,4-triazole linkage. Ultimately, this synthetic method was successfully applied to the synthesis of the azido-D-Bac8c(Leu2,5) 7 and its conjugation to the 3′-propargyl-carbamate cephalosporin 6, to produce the prototypical prodrug candidate cephalothin-D-Bac8c(Leu2,5) 8 (Scheme 2). Characterisation of this conjugate was performed by ESI-MS and its integrity, namely the absence of Δ3/Δ2 isomerisation, was confirmed by 1H NMR. Also, the characteristic coupling constants of the geminal protons at the 2-position were verified by C–H correlation 2-D NMR (ESI Fig. S1–S3†).
The active parent peptide of this prodrug candidate was synthesised separately as a Bac8c sequence modified with a N-terminal D-Arg-triazole-ε2-amino acid 9 (Fig. 2). Two independent synthetic routes, based on solid phase synthesis or on a hybrid solution/solid-phase synthesis were evaluated for this triazole-modified peptide. In the former case, a cyclo-addition of propargylamine and a resin-bound azido-peptide was performed. In the latter case, the synthesis and coupling of a Fmoc-protected D-Arg-triazole-ε2-amino acid to a peptide sequence assembled up to the penultimate residue was carried out. Higher purities of the active parent peptide were obtained by the hybrid solution/solid-phase method as the solid phase approach yielded some unreacted peptide which could not be separated from the peptide triazole by RP-HPLC.
2.3. Activation assays and antimicrobial activity
Chemical and enzymatic hydrolysis assays of cephalothin-D-Bac8c(Leu2,5) were first undertaken to verify that the active parent peptide could be released from this prodrug candidate upon cleavage of the β-lactam bond. These experiments were carried out by monitoring the disappearance of the cephalosporin's β-lactam bond UV absorption19 at 260 nm, in a sodium hydroxide solution for the base-promoted hydrolysis assay, or with a purified P99 enzyme from Enterobacter cloacae for the β-lactamase-mediated activation assay. No background hydrolysis of cephalothin-D-Bac8c(Leu2,5) was observed in buffer alone. The results of the assays with NaOH and P99 (ESI Fig. S4–S7†) show that the kinetics of the base-promoted and enzymatic hydrolyses are slower for the prodrug candidate than for cephalothin.Also, the relative reductions in the absorbance upon hydrolysis were significantly lower for the prodrug candidate than for cephalothin, complete disappearance of the UV absorption being only obtained in the latter case. This was attributed to a residual absorption of the peptide at 280 nm and confirmed by performing wave-scans of cephalothin and the prodrug candidate 8 before and after (chemical) hydrolysis, and compared to the wave-scan of the peptide triazole 9 (ESI Fig. S8–S12†). While the absorption at 260 nm is completely abolished after hydrolysis of cephalothin, persistence of an absorption in the same region is observed with the prodrug candidate. This absorption is of similar intensity and wavelength than the one recorded in the wave-scans of the peptide triazole and is most likely associated with the absorption of the indole rings. Together, the results of these experiments indicate that the prodrug candidate can release its peptide component upon cleavage of the β-lactam ring.
The antimicrobial activity of the prodrug candidate 8 was then assessed against the strains of E. coli previously selected and against two strains of Methicillin-Resistant Staphylococcus aureus (MRSA). They were compared with the activities of its parent components, i.e. cephalothin 1 and D-Bac8c(Leu2,5) 2 and with the active parent peptide of the prodrug 9. The Minimum Inhibitory Concentration (MIC) results presented in Table 2 show that the activities of the prodrug candidate against the strains selected are distinct from those of its parent cephalosporin and peptide components. In all cases, the parent peptide remained the most active of these 3 candidates. The MICs of the prodrug candidate were generally higher with the Gram-negative strains, although 8 can have MICs as low as 3 μM against clinical isolates expressing ESBLs (data not shown). Comparing the results of the prodrug candidate and its active parent peptide showed that E. coli had higher susceptibilities to the triazole-modified peptide 9 than to its cephalosporin conjugate 8, while S. aureus had similar susceptibilities to both agents. Collectively, these results can be explained by a reduced uptake of the conjugated peptide through the outer bacterial membrane of Gram-negative organisms, as a consequence of a hindered N-terminus9 which impedes the access of the conjugate to the β-lactamases. These enzymes are indeed expressed extracellularly in Gram-positive bacteria, but within the periplasmic space in Gram-negative bacilli.
| Strain | Cephalothin 1 | D-Bac8c (Leu2,5) 2 | Prodrug 8 | Triazole peptide 9 | Control 10 | Control 11 |
|---|---|---|---|---|---|---|
| a MIC experiments were carried out in triplicate. b MRSA NCTC strain. c β-Lactamase-negative ATCC strain. d β-Lactamase-positive ATCC strain. e Highest concentration tested. f Not determined. | ||||||
| S. aureus 12493b | 2.0 | 2.1 | 3.0–8.0 | 4.0–5.0 | 25.6 | > 24.8e |
| S. aureus 44330b | 2.0 | 1.0 | 1.5 | 2.0–4.0 | 12.8 | > 24.8e |
| E. coli 25922c | 31.6 | 2.1 | 12.0 | 5.0 | > 25.6 | > 24.8e |
| E. coli 35218c | 119.0 | 1.0 | 8.0-16.0 | 32.0 | ≥ 25.6 | > 24.8e |
| E. coli BL21 blaCTX-M-15 − | 12.6 | 2.1 | 6.1 | ndf | 25.6 | nd |
| E. coli BL21 blaCTX-M-15 + | > 404.0 | 1.1 | 3.0 | nd | 25.6 | nd |
To assess the specificity of the activity of the prodrug candidate, analogs which recapitulate the hydrophobicity and net charge determinants of the antimicrobial activities of 8 were designed as controls.
In the first control 10 (Fig. 3), the separation of the peptide component from the β-lactam moiety cannot be triggered by the reaction of the latter with β-lactamases. The results presented in Table 2 show that this control has reduced antimicrobial activities against both organisms, in particular S. aureus, indicating that the release of the peptide component from the cephalosporin may contribute to the mechanism of action of the prodrug candidate. Another control 11 (Fig. 3) only differing from 8 by the introduction of a stable oxime linker in place of the self-immolative carbamate linker also had reduced antimicrobial activities against both organisms (Table 2).
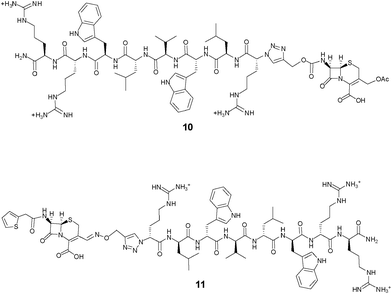 | ||
| Fig. 3 Structures of the cephalosporin-D-Bac8c(Leu2,5) conjugates based on non-cleavable linkers. In 10, the peptide is conjugated at the 7-position of the cephem core; in 11, the peptide is conjugated at the 3′-position of the cephem core through a stable oxime linker. | ||
Finally, to further investigate the mechanism of action of the cephalosporin-peptide conjugate 8, isogenic strains of E. coli expressing or not the ESBL CTX-M-1520 were created. The use of strains differing only by the presence of an ESBL gene directly indicates the contribution of the β-lactamase to the activity of a prodrug candidate.21 The results reported in Table 2 show an activity differential of 8 against these isogenic strains consistent with a β-lactamase-dependent prodrug activity. Although these isogenic strains present different inherent susceptibilities to the parent peptide, the MICs of the non-cleavable control conjugate 10 containing the same peptide were identical against these strains, regardless of their β-lactamase status.
3. Conclusions
Prodrug candidates of HDPs activated by the main mechanism of antibiotic resistance can be synthesised by conjugation of non-ribosomal (β-lactam) and ribosomal (host defence) peptides through a carbamate-1,4-triazole linker. The results of the activation assays, antimicrobial testing and control experiments substantiate a β-lactamase-dependent prodrug activity of the cephalothin-D-Bac8c(Leu2,5) candidate, but also indicate a contribution of the intact conjugate to its overall activity. This could be due to an insufficient modification by the cephalosporin of the net charge and/or hydrophobicity of the parent peptide. Shorter peptide sequences and/or sequences of lower net charges could be selected as active parent peptides to generate prodrug candidates with greater activity differentials between ESBL-positive and ESBL-negative strains. Alternatively, introducing a positive charge in the side-chain of the cephalosporin could be performed to reduce the hydrophobicity of the conjugate and potentially overcome its reduced uptake through the outer bacterial membrane of Gram-negative bacteria by restoring a cationic N-terminal end. Even if the parent peptide remains more active than its prodrug candidate, the latter possesses an activity which can potentially be controlled to prevent toxicity issues in systemic therapies and to target resistant Gram-negative Enterobacteriaceae producing ESBLs. These pathogens have been listed among the top 6 organisms to which novel antibiotics are urgently needed.224. Experimental
4.1. General methods
Material and reagents were purchased from commercial suppliers and used without further purification, unless stated otherwise. Protected amino acids were obtained from Novabiochem (Merck Biosciences, Hohenbrunn, Germany), Iris Biotech GmbH (Marktredwitz, Germany) and Senn chemicals (Dielsdorf, Switzerland). HBTU and resins for SPPS were purchased from Novabiochem; HOBt and PyBrop were from Iris Biotech GmbH. Solvents (NMP, DCM) for the peptide synthesiser were purchased from Applied Biosystems (Warrington United Kingdom). Other reagents and solvents were obtained from Sigma-Aldrich Ireland.NMR spectra were recorded on a BRUCKER Avance 400 spectrometer. Samples were prepared in CDCl3 (referenced to 7.26 ppm for 1H and 77.0 for 13C), DMSO-d6 (referenced to 2.52 for 1H and 40 for 13C), DMF-d7 (referenced to 2.74 ppm for 1H and 75.46 for 13C), MeOD (referenced to 3.31 ppm for 1H and 49.00 for 13C), CDCl3/MeOD (referenced specified for each compound), D2O (referenced to 4.79 ppm for 1H) or CD3CN (referenced to 1.94 ppm for 1H and 118.26 for 13C); normal TFA was used as reagent grade and referenced to 11 ppm for 1H and 40 for 13C. Infrared spectra (IR) were recorded as KBr discs using a Bruker Tensor27 FT-IR instrument. Absorption maximum (υmax) was recorded in wave numbers (cm−1) and only selected peaks are reported. Ultraviolet spectra (UV) were recorded in quartz cuvettes of 1cm length using abiochrom Libra S22 instrument. Electrospray ionization mass spectra were recorded on a Waters Micro mass Quattro LCMS for High Resolution Mass Spectrometry (HRMS) and on a Waters Micro mass LCT for Low Resolution Mass Spectrometry (LRMS) at 80 eV. Flash chromatography was performed using silica gel 60 (0.040–0.063 mm, 230–400 mesh) or alumina oxide, activated, basic Brockmann 1, standard grade ≈ 150 mesh, 50 Å.
Peptide sequences were assembled from a Rink Amide MBHA resin, unless stated otherwise, by standard SPPS according to the Fmoc/t-Bu strategy with either HBTU/HOBt/DIEA coupling chemistry in NMP solvent (automated synthesis), or PyBrop/DIEA coupling chemistry in DMF solvent (manual synthesis). Automated syntheses were performed on an Applied Biosystems 433A synthesiser (Warrington, UK), on a 2.5 × 10−4 mol scale, using single coupling cycles with a 4-fold excess of Fmoc-amino acid derivatives to resin-bound peptide. Manual syntheses were carried out in a 5 mL syringe fitted with a teflon frit and a stopcock, on a 1.0 × 10−4 mol scale and a 3.5-fold excess of Fmoc-amino acid derivatives to resin-bound peptide. Chain elongation was monitored by the qualitative Kaiser Test and multiple coupling procedures were applied until a negative test was obtained. Coupling reactions were performed for 30 to 150 min. Nα–Fmoc-protecting groups were removed by treatment with 25% piperidine in DMF (5 mL) for 10 min followed by two additional treatments of 5 min each. The resin was then washed three times with DMF (5 mL, 3 min each).
Following chain assembly, the dry resin was treated with a cleavage cocktail consisting of 90% TFA, 2.5% triisopropylsilane, 2.5% thioanisole, 2.5% phenol and 2.5% water, unless stated otherwise. The reaction time was adjusted to the number of arginine residues present in the sequence, from a minimum reaction time of 2 h, incremented by 30 min for each arginine residue present in the sequence. After eliminating the resin by filtration, the peptide was precipitated from the filtrate with the minimum amount required of cold diethyl ether and isolated by centrifugation for five minutes at 2.8 × 103 rpm. The peptide pellet was resuspended in diethyl ether and centrifuged twice. The peptide was allowed to dry before being dissolved in water for lyophilisation.
Chromatographic analyses and purifications were performed on a BioCAD SPRINT Perfusion Chromatography Workstation (PerSeptive Biosystems) using Gemini columns (Phenomenex, 110 Å, 5μ, C18, 4.6 mmd/250 mmL or 10 mmd/250 mmL, for the analytic or semi-preparative columns, respectively). Buffers used were mobile phase A: 0.1% TFA in water; mobile phase B: 0.1% TFA in acetonitrile, with a gradient of 2 to 65% B in 18 column volumes (analytical) or 5 column volumes (semi-preparative) with a flow rate of 1 ml min−1 (analytical) or 4 ml min−1 (semi-preparative) and single wavelength detection at 214 nm.
Mass spectrometry analyses performed by Matrix Assisted Laser Desorption Ionization - Time of Flight (MALDI-TOF) were obtained on a Reflex Bruker spectrometer. Two type of matrix were used, 2,5-dihydroxybenzoic acid and α-cyano-4-hydroxy-cinnamic acid, dissolved in 30% acetonitrile and 70% water at a concentration of 5 mg mL−1 and 6 mg mL−1, respectively.
4.2. Synthesis of the cephalosporin derivatives
IR: νmax (KBr)/cm−1: 3376, 3184, 1798, 1614, 1548, 1410, 1348; δH (400 MHz, TFA), 3.08 (s, 1H, H-3′A), 3.25 (s, 1H, H-3′B), 4.41 (s, 2H, H-2), 4.67 (s, 1H, H-6), 4.81 (d, 1H, H-7); δC (100.6 MHz, TFA), 21.57, 52.50, 59.81, 72.09 (C-2, C-6, C-7, C-3′), 122.23, 143.21 (C-3, C-4), 157.50, 170.53 (2 × C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). LRMS m/z: 229.05 (100%, M-H+) .
O). LRMS m/z: 229.05 (100%, M-H+) .
IR: νmax (KBr)/cm−1: 3493, 3288, 1755, 1725, 1667, 1521, 1376, 1221; δH (400 MHz, CDCl3), 2.62 (m, J = 9.6 and 4.4 Hz, 1H, OH), 3.54 (s, 2H, H-2), 3.85 (s, 2H, CH2 thienyl side-chain), 3.90 (dd, J = 12.8 and 9.6 Hz, 1H, H-3′A), 4.39 (dd), J = 13.00 and 4.80 Hz, 1H, H-3′B), 4.93 (d, J = 4.80 Hz, 1H, H-6), 5.89 (m, J =9.2 and 4.80 Hz, 1H, H-7), 6.46 (d, J = 9.2 Hz, 1H, N-H), 6.89 (s, 1H, CH(Ar)2), 6.96-7.01 (m, 2H, Ar-H thiophene), 7.26-7.36 (m, 10H, Ar-H, benzhydryl), 7.42- 7.45 (m, 1H, Ar–H thiophene); δC (100.6 MHz, CDCl3) 27.64, 37.10, 56.99, 59.00, 62.08, (C-2, C-3′, C-6, C-7, CH2 thienyl side-chain), 80.13 (CH(Ar)2), 124.89, 126.14, 127.06, 127.48, 127.60, 127.96, 128.27, 128.49, 128.68, 133.32, 134.64 (C-3, C-4, (4 x C-Ar thiophene), (10 x C-Ar benzhydryl)), 138.92, 139.14 (2 × C-Ar benzhydryl), 161.72, 164.99, 170.09 (3× CO). HRMS m/z: found 519.1034 [M-H+], calcd for C27H23N2O5S2 519.1049; LRMS m/z: 519.12 (100%, M-H+).
:30, hexane:ethyl acetate to yield a pale yellow solid (100 mg, 0.152 mmol, 51%).
δ
H (270 MHz, CDCl3) 3.45 (d, J = 18.56 Hz, 1H, H-2A), 3.65 (d, J = 18.80 Hz, IH, H-2B), 3.89 (s, 2H, CH2 cephalothin), 4.99 (d, J = 13.36 Hz, 1H, H-3′A), 5.02 (d, J = 4.95 Hz, 1H, H-6), 5.25 (d, J = 13.11 Hz, 1H, H-3′B), 5.87 (m, J = 4.95 and 9.15 Hz, 1H, H-7), 6.56 (d, J = 8.91 Hz, 1H, N-H), 6.94 (s, 1H, CH(Ar)2), 6.82–7.04 (m, 2H, 2 Ar-H thiophene) overlapping with (m, 2H, Ar-NO2), 7.22–7.42 (m, 10H, Ar-H), overlapping with (m 1H, Ar-H thiophene) and 8.10–8.32 (m, 2H, Ar-H-NO2); δC (75.47 MHz, CDCl3) 26.45, 36.96, 57.19, 59.17, 67.33 (C-2, C-3′, C-6, C-7, -CH2 cephalothin), 80.24 (CH(Ar)2), 121.60, 124.99, 125.34, 126.37, 126.96, 127.64, 127.68, 128.19, 128.23, 128.36, 128.49, 129.59, 133.97 (C-3, C-4, 4 × (C-Ar thiophene), 8 × (C-Ar), 6 × (C-Ar ArNO2), 138.65, 138.81 (2 × C-Ar), 145.45, 152.02, 155.17, 160.43, 164.36, 171.15 (4 × CO).
IR: νmax (KBr)/cm−1: 3365, 1776, 1727, 1669, 1523, 1233; δH (400 MHz, CDCl3), 3.37 (d, J = 18.56 Hz, 1H, H-2A), 3.56 (d, J = 18.56 Hz, IH, H-2B), 3.86 (s, 2H, CH2 thienyl side-chain), 4.96–5.00 (m, 2H, H-3′A overlapping with H-6), 5.24–5.31 (m, 1H, H-3′B), 5.89 (m, 1H, H-7), 6.28 (d, J = 9.2 Hz, 1H, N–H), 6.64 (s, 1H, CHCl), 6.93 (s, 1H, CH(Ar)2), 6.97–7.02 (m, 2H, Ar–H thiophene), 7.27–7.36 (m, 10H, Ar–H benzhydryl; 1H, Ar–H thiophene); δC (100.6 MHz, CDCl3) 26.17, 37.11, 57.35, 59.16, 68.14, 68.21 (C-2, C-3′, C-6, C-7, CH2 thienyl side-chain), 80.17 (CH(Ar)2), 91.06 (CHCl), 96.89 (CCl3), 124.22, 124.26, 126.51, 126.56, 127.02, 127.69, 128.06, 128.23, 128.35, 128.52, 128.64, 134.50 (C-3, C-4, (4 × C-Ar thiophene), (10 × C-Ar benzhydryl)), 138.76, 138.90 (2 × C–Ar benzhydryl), 151.54, 160.35, 164.65, 169.98 (4 × CO). Found C, 49.10; H, 3.45; N, 3.13%. C30H24Cl4N2O7S2 requires C, 49.33; H, 3.31; N, 3.84%. LRMS m/z: 311 (100%, M-H+).
IR: νmax (KBr)/cm−1: 3314, 3291, 2913, 1791, 1695, 1645, 1538, 1250; δH (400 MHz, CDCl3), 2.26 (t, J = 2.4 Hz, 1H, CH alkyne), 3.35 (d, J = 18.4 Hz, 1H, H-2 A), 3.53 (d, J = 18.4 Hz, 1H, H-2 B), 3.85 (s, 2H, CH2 thienyl side-chain), 3.92 (m , 2H, CH2 alkyne), 4.77 (d, J = 13.6 Hz, 2H, H-3′A overlapping with N–H), 4.96 (d, J = 4.8 Hz,1H, H-6), 5.03 (J = 13.6 Hz, 1H, H-3′B), 5.87 (m, 1H, H-7), 6.34 (d, J = 9.2 Hz, 1H, N-H), 6.97 (s, 1H, CH(Ar)2), 7.01 (m, 2H, Ar-H thiophene), 7.24–7.41 (m, 11H, 1 Ar–H thiophene, 10 × Ar–H benzhydryl); δC (100.6 MHz, CDCl3) 26.40, 30.92, 37.11, 57.23, 59.07, 63.53 (C-2, CH2 alkyne, CH2 thienyl side-chain, C-6, C-7, C-3′),71.93, 79.70, (C-H alkyne, C-alkyne) 126.20, 126.46, 126.55, 127.17, 127.60, 127.64, 127.69, 128.02, 128.17, 128.25, 128.47, 128.53, 128.60, 134.59, 138.96, 139.19, 143.81 (C-3, C-4, (10 × C–Ar benzhydryl), (4 × C–Ar thiophene)),155.35, 160.69, 164.78, 170.07 (4 × CO). Found: C, 62.48; H, 4.98; N, 5.95%. C31H27N3O6S2 requires C, 61.88; H, 4.52; N, 6.98%.
IR: νmax (KBr)/cm−1: 3285, 1746, 1679, 1656, 1600, 1532, 1402, 1250; δH (400 MHz, CDCl3/MeOD (0.450/0.3), (MeOD referenced to 3.49 ppm for 1H and 48.17 ppm for 13C). 2.53 (m, 1H, CH alkyne), 3.59 (d, J = 18.4 Hz 1H, H-2 A), 3.74 (d, J = 18.4 Hz, 1H, H-2 B), 3.97 (m, J = 20.8 and 15.6 Hz, 2H, CH2 alkyne), 4.05 (s, 2H, CH2 thienyl side-chain), 5.04 (d, J = 13.6 Hz, 1H, H-3′A), 5.15 (m, 1H, H-6), 5.31 (d, J = 13.6, 1H, H-3′B), 5.89 (s, 1H, H-7), 7.09 (m, 2H, Ar-H thiophene), 7.38 (m, 1H, Ar-H thiophene); δC (100.6 MHz, CDCl3), 25.61, 29.98, 35.99, 57.23, 58.99, 63.32 (C-2, CH2 thienyl side-chain, CH2 alkyne, C-6, C-7, C-3′), 70.86, 79.24, (C-H alkyne, C-alkyne), 124.73, 125.32, 126.96 126.54, 135.26 (C-3, C-4, (4 × C-Ar thiophene)),156.28, 163.01, 164.55, 171.49 (4 × CO). HRMS m/z: found 434,0473 [M-H+], calcd for C18H16N3O6S2 434.0481; LRMS m/z: 434.03 (100%, M-H+).
7-ACA (500 mg, 1.83 mmol) was dissolved in 4 mL of acetone and 2.5 mL of H2O and the mixture was cooled to 0 °C. N,N-Diisopropylethylamine (300 μL, 0.22 mmol) was then added to the solution, followed by propargylchloroformate (214 μL, 0.22 mmol) dropwise. The pH was kept between 7.5 and 8.5 by addition of a molar solution of NaOH. After 1 h, the mixture was acidified with HCl 1 N, before extracting the product with diethyl ether and ethyl acetate. The combined organic phases were dried and evaporated yielding a white solid (400 mg, 1.13 mmol, 61.7%).
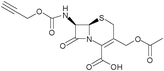
δ
H (400 MHz, DMSO), 2.04 (s, 3H, CH3), 3.50 (d, J = 18.4 Hz, 1H, H-2A), 3.56 (t, J = 2,4 Hz, 1H, CH alkyne) 3.63 (d, J = 18.4 Hz, 1H, H-2B), 4.69 (m, 3H, H-3′A overlapping with CH2 propargyl), 4.99 (d, J = 12.4 Hz, 1H, H-3′B), 5.10 (d, J = 4.8 Hz, 1H, H-6) , 5.56 (dd, J = 4.8 Hz 1H, H-7), 8.58 (d, J = 11 Hz 1H, N–H), 13.85 (s, 1H, COOH). δC (100.6 MHz, CDCl3) 20.55, 25.53, 52.35, 57.49, 60.87, 62.67, 77.61, 78.63 (CH3, C-2, CH2, C-6, C-7, C-3′, CH, CH2–C), 123.63, 126.30, (C-3, C-4), 155.09, 162.83, 164.25, 170.19 (4 × CO).
4.3. Synthesis of the peptides and their conjugates
Synthesis by DiazoTransfer reaction with imidazole-1-sulfonyl azide hydrochloride. – Imidazole-1-sulfonyl azide hydrochloride
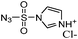
IR: νmax (KBr)/cm−1: 3111, 2502, 2169, 1915, 1588, 1414, 1160; δH (400 MHz, D2O), 7.55 (s, 1H, H-2), 7.97 (s,1H, H-4), 9.27 (s,1H, H-5); δC (100.6 MHz, D2O), 119.3, 124.08, 137.66 (C-3, C-2, C-5).
– DiazoTransfer reaction
0.33 mmol of resin-bound peptide (fully assembled sequence) was placed in a syringe and swelled during one hour in DCM/MeOH (4/1). Imidazole-1-sulfonyl azide hydrochloride (42 mg, 0.165 mmol) was then added, with K2CO3 (51.75 mg, 0.375 mmol) and some traces of CuSO4, in a mixture of DCM/MeOH (4/1) and the mixture was agitated for 6 h. The resin was then isolated by filtration and washes were performed with DMF, DCM and MeOH. The reaction was repeated in THF/MeOH (4/1) as the solvent and the resin washed with DMF, DCM and THF after filtration. The reaction was monitored by the Kaiser test.
Synthesis by coupling of the α-azido acid of arginine. – Synthesis of azido arginine25
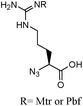
H-Arg(Pbf)-OH (1.18 g, 2.78 mmol) or H-Arg(Mtr)-OH (1.07 g, 2.78 mmol) was dissolved in a mixture of water (9 mL) and MeOH (18 mL) in presence of K2CO3 (577.5 mg, 4.19 mmol) and CuSO4, 5H2O (6.98 mg, 27.9 μmol). The triflyl azide solution (15 mL) was then added and the mixture stirred overnight at ambient temperature. After evaporation of the organic solvent, the aqueous phase was diluted with water (50 mL) and subsequently acidified to pH 6 with HCl 0.25 M and adjusted to pH 6.2 with a phosphate buffer (50 mL). This solution was then extracted with EtOAc. The aqueous phase was acidified to pH 2 with conc. HCl and extracted again with EtOAc. The organic phases were combined, dried over MgSO4 and evaporated to give a white solid compound.
N3-Arg(Pbf)-OH. δ H (400 MHz, CDCl3), 1.45 (s, 6H, CH3 dimethyl-dihydrofuran), 1.61–1.90 (m, 4H, CH2 arginine side chain), 2.07 (s, 3H, CH3 tolyl), 2.45 (s, 3H, CH3 tolyl), 2.51 (s, 3H, CH3 tolyl), 2.94 (s, 2H, CH2 dihydrofuran), 3.23 (s, 2H, CH2 arginine side chain), 3.95 (m, 1H, CH arginine), 6.26–7.39 (m, 3H, N-H guanidinium); δC (100.6 MHz, CDCl3), 12.50, 17.86, 19.31, 28.60, 29.72, (C-H3 dimethyl-dihydrofuran, CnH3 tolyl, C-H3 tolyl, C-H2 arginine, C-H3 tolyl), 43.13 (C-H2 dihydrofuran), 60.48 (C-H2 arginine), 61.88 (C-H arginine), 86.82, (C–N3), 117.94, 125.04 (6 × C–Ar), 156.14 (C-guanidinium), 174.18 (C
O).
N3-Arg(Mtr)-OH. δ H (400 MHz, D2O), 1.57–1.72 (m, 4H, CH2 arginine side chain), 2.08 (s, 3H, CH3 Ar), 2.53 (s, 3H, CH3 Ar), 2.61 (s, 3H, CH3 Ar), 3.19 (s, 2H, CH2 arginine side chain), 3.79 (s, 3H, OCH3), 3.95 (m, 1H, CH arginine), 6.50 (s, 1H, Ar-H), 6.26–6.90 (m, 3H, N-H Guanidinium); δC (100.6 MHz, CDCl3), 11.92, 18.17, 22.70, 24.11, 29.37, 31.93 (3 × C–H3 Ar, 3 × C-H2 arginine), 43.13 (O-CH3), 55.47 (C–H arginine), 86.82, (C–N3), 111.91, (C–Ar–H), 125.18, 138.62 (5 × C–Ar), 156.23 (C-guanidinium), (C
O not observed).
– Coupling of azido arginine
The resin-bound peptide sequence assembled up to the penultimate residue was placed in a syringe for manual synthesis. The resin was swelled in DMF for 30 min. 3 equivalents of N3-Arg(R)-OH, DIC and HOBt were dissolved in DMF and added to the resin. The coupling reaction was monitored by the Kaiser Test. Upon completion of the coupling reaction, the resin was washed with DMF and DCM.
Isolation of the azido peptides. Azido peptides were isolated by treatment of the resin with a mixture of 3% triisopropylsilane, 3% water, 3% thioanisole and 3% phenol per ml of TFA for 3.5 h.
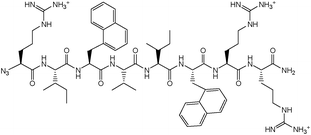
N3-Arg-Ile-2-Nal-Val-Ile-2-Nal-Arg-Arg-NH2.
LRMS m/z: 1231.9 (100%, M+H+).
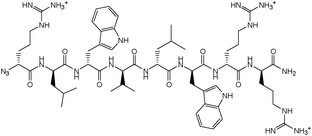
N3-Arg-Leu-Trp-Val-Leu-Trp-Arg-Arg-NH2 (7).
LRMS m/z: 1209.80 (100%, M+H+).
Solid phase synthesis of the triazole peptide. The resin of the azido peptide (0.05 mmol) was swelled during one hour in DMF. Propargylamine (0.15 mmol, 10 μL), CuI (9.5 mg, 0.05 mmol) and diisopropylethylamine (0.3 mmol, 52.1 μL) were then added. A mixture of DMF/MeOH (4/1) was then added and the mixture vortexed during 6 h. The coupling reaction was repeated in a mixture of THF/MeOH (4/1) as the solvent. The resin was then washed successively with DMF, MeOH and DCM. The peptide was cleaved and purified (92%) as described in paragraph 4.1. LRMS m/z: 1265.0 (100%, M+H+).
Hybrid solution-solid phase synthesis of a triazole peptide. – N-Fmoc-propargylamine
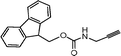 O).
O).
– Fmoc- D-Arg-triazole-ε2-amino acid
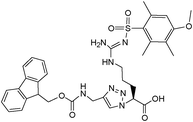
δ H (400 MHz, MeOD), 0.78–0.93, (m, 2H, CH2 arginine), 1.18–1.40 (m, 2H, CH2 arginine), 2.00(s, 3H, CH3 Ar), 2.53 (s, 3H, CH3 Ar), 2.60 (s, 3H, CH3 Ar), 3.12 (s, 2H, CH2 arginine), 3.74 (s, 3H, OCH3), 4.12 (t, 1H, J = 6.8 Hz, CH Fmoc), 4.30 (d, J = 7.2 Hz 2H, CH2 Fmoc), 4.33 (s, 2H, CH2 C6), 5.03 (m, 1H, CH C2), 4.43 (1H, CH arginine), 6.50 (s, 1H, Ar–H Mtr), 7.21–7.78 (m, 8H, Ar–H Fmoc), 7.82 (s, 1H, triazole); LRMS m/z: found 690.1 (M+H+).
:1). 160 μL of the first two solutions and 240 μL of the last one were taken and added along with 600 μL of tert-butanol. The mixture was stirred at 30 °C for 4 to 12 h. The product was precipitated by addition of diethyl ether and isolated by centrifugation. The residue dissolved in water for HPLC purification, performed as described above, except 0.08% formic acid was added to the buffers in place of 0.1% TFA and dual wavelength UV detection (214 and 260 nm) was performed. Collected fractions containing the conjugate were lyophilised (5.9 mg, 3.5 μmol, 29%; purity 91%). NMR analysis was recorded on a BRUCKER AC 600 and LRMS analysis was performed at 35 eV. LRMS m/z found 822.4 (M+2H)2+.
– O-prop-2-ynyl-hydroxylamine hydrochloride
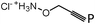
– (6R, 7R)- 7-(2-thiophen-2-yl-acetylamino)-3-formyl-3-cephem-4-carboxylic acid benzhydryl ester
The reaction was carried out in a solid phase reaction vessel. Compound 4 (100 mg, 0.192 mmol) was dissolved in 3 mL of dry DCM; 3 equivalent of IBX resin (0.576 mmol) were then added and the mixture was stirred overnight. The resin was removed by filtration and subsequently washed with 3 × 5 mL of DCM. The DCM fractions were combined and evaporated, yielding quantitatively the product which can be used directly without purification. The resin can be reused after oxidation. δH (400 MHz, CDCl3), 3.26 (d, J = 18.8 Hz, 1H, H-2A), 3.86 (s, 2H, CH2 thienyl), 3.97 (d, J = 18.8 Hz, IH, H-2B), 5.03 (d, J = 5.6 Hz, 1H, H-6), 5.99 (m, 1H, H-7), 6.37 (d, J = 9.2 Hz, 1H, N–H), 6.64 (s, 1H, CH thienyl), 6.97–7.01 (m, 2H, 2 × CH thienyl), 7.06 (s, 1H, CH(Ar)2), 7.26–7.37 (m, 10H, Ar–H), 9.63 (s, 1H, H-3′); δC (100.6 MHz, CDCl3) 22.29, 37.05, 58.91, 59.84 (C-2, CH2 thienyl, C-6, C-7), 80.86 (CH(Ar)2), 123.87, 126.34, 127.09, 127.30, 127.60, 127.71, 127.92, 128.10, 128.31, 128.51, 128.64, 128.69, 128.73, 130.10, 132.47, 134.35, 138.09, 138.41, 138.44 (C-3, C-4, 4 × (C) thienyl, 8 × (C-Ar)) 159.63, 165.01, 169.98 (3 × CO) , 187.76 (C-formyl).
– (6R, 7R)-7-(2-thiophen-2-yl-acetylamino)-3-(prop-2-ynyloxyimino-methyl)-3-cephem-4-carboxylic acid benzhydryl ester
6R, 7R)- 7-(2-thiophen-2-yl-acetylamino)-3-formyl-3-cephem-4-carboxylic acid benzhydryl ester (90 mg, 0.188 mmol) was dissolved in 6 mL of dry acetonitrile. The solution was chilled to 0 °C and O-prop-2-ynyl-hydroxylamine hydrochloride (18 mg, 0.17 mmol) was then added. The mixture was stirred overnight and the solvent was then evaporated to recover the oxime product (107.6 mg, 0.19 mmol, quantitative). δH (400 MHz, CDCl3), 2.47 (t, J = 2.4 Hz, 1H, CH alkyne), 3.49 (d, J = 18.4 Hz, 1H, H-2 A), 3.86 (s, 2H, CH2 thienyl), 4.05 (d, J = 18.4 Hz, 1H, H-2 B), 4.67 (d , J = 2.4 Hz, 2H, CH2 propargyl, 5.02 (d, J = 4.8 Hz,1H, H-6), 5.91 (m, 1H, H-7), 6.33 (d, J = 9.2 Hz, 1H, N–H), 6.97 (s, 1H, CH(Ar)2), 7.01 (m, 2H, CH cephalothin), 7.26–7.41 (m, 11H, CH cephalothin, 10 × Ar–H), 8.34 (s, 1H, H-3′); δC (100.6 MHz, CDCl3) 22.52, 36.06, 56.91, 58.61, 61.18 (C-2, CH2 thienyl, C-6, C-7, CH2 propargyl), 73.93, 78.99, (C-alkyne, C-H alkyne), 121.80, 125.18, 126.03, 126.48, 126.61, 126.71, 126.98, 127.21, 127.29, 127.46, 127.62, 133.49, 137.70, 137.98, (C-3, C-4, 10 × (C–Ar), 4 × (C) thienyl), 146.29 (C-3′), 159.18, 163.65, 168.92 (3 × CO).
– Control 11
The azide-alkyne cyclo-addition reaction was performed by using one molar equivalent of the fully deprotected azido-peptide described in 2b (10.4 mg, 8.6 μmol) and 2 molar equivalents of compound (6R, 7R)- 7-(2-thiophen-2-yl-acetylamino)-3-(prop-2-ynyloxyimino-methyl)-3-cephem-4-carboxylic acid benzhydryl ester (9.8 mg, 17 μmol). Three different aqueous solutions were prepared containing respectively 0.4 equivalent of CuSO4·5H2O (20 mg/2.5 mL), 0.8 equivalents of sodium ascorbate (31.6 mg/2.5 mL) and 7 equivalents of lutidine (170 μL/2.5 mL) in a mixture of water. 107 μL of each solution were withdrawn and added along with 321 μL of tert-butanol. The mixture was stirred at 30 °C for 4 to 12 h and then evaporated. The residue was dissolved in water for HPLC purification, performed as described above, with 0.08% formic acid added to the buffers in place of 0.1% TFA. Collected fractions containing the control, deprotected, peptide (deprotection of the benzhydryl ester occurs during HPLC purification) were lyophilised (0.168 mg, 0.1 μmol, 1.2%; purity 75.23%). LRMS m/z: found 1615.1 (M+H+).
4.4. Activation assays
These assays were performed at pH 7.25 in a 10 mM PBS buffer, by monitoring with a UV-spectrophotometer the disappearance of the cephalothin's β-lactam bond at 260 nm. The chemical hydrolysis assay was carried out in a 0.41 M NaOH solution using a 0.140 mM solution of 8. β-Lactamase-mediated reactivation assays were performed with a purified P99 enzyme from Enterobacter cloacae (0.8 mg, 0.32 μM) using a 13 mM solution of 8.4.5. Susceptibility assays
For determination of minimum inhibitory concentration (MIC) of peptides, the broth microdilution method was used according to the guidelines of the Clinical Laboratory Standards Institute (CLSI) with modifications for testing of cationic peptides as described by Wu and Hancock.26 Briefly bacterial strains from the American Type Culture Collection (ATCC) or the National Collection of Type Cultures (NCTC) or isogenic strains (this study) were grown overnight on Mueller Hinton (MH) agar at 37 °C and single colonies were resuspended in MH broth to the density of a 1 McFarland standard (5 × 107 cells approx) which was further diluted 1/100 in MH broth as the inoculum. Peptides were resuspended in a solution containing 0.2% (w/v) bovine serum albumin and 0.01% (v/v) acetic acid and serial doubling dilutions were prepared at ten times the required concentration in Eppendorf tubes (dilutions tested were from 160 to 0.3125 μg ml−1 final peptide concentration per well). Assays were prepared in 96 well polypropylene plates (Costar Corp., Cambridge MA) and contained 10 μl of each dilution of peptide and 90 μl of inoculum. Sterility controls for each peptide dilution and for MH medium were included. Plates were incubated overnight (18 h) and MIC was recorded as the lowest concentration of peptide at which no growth was observed with reference to wells containing no peptide.4.6. Generation of isogenic strains ± blaCTX-M-15
Plasmid pET-30a(+) (Novagen) was double-restricted with Eco R I/Hind III in a reaction containing 10 U of each restriction enzyme and 1 U of thermosensitive alkaline phosphatase (TSAP) (all Promega) for 3 h at 37 °C. Linearized pET30a plasmid was separated by agarose gel electrophoresis and purified using Qiagen's QIAquick® Gel Extraction Kit. The blaCTX-M-15 insert (924 bp) was amplified from genomic DNA isolated from a CTX-M-producing clinical E. coli isolate using PCR primers; CTX-MEco-FW, 5′-GG![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) ACTATTCATGTTGTTGTTATT-3′27 and CTX-M-R, 5′-CCCTTACAAACCGTCGGTGACGAT-3′.28 The forward primer contained a 5′ EcoR I restriction site (underlined) and included a homologous promoter sequence 29-42 bp upstream of the start codon.27 The reverse primer included a 3′ Hind III restriction site (underlined).28 Insert DNA was purified, double-digested with Eco R I/Hind III restriction enzymes (Promega) and re-purified. A Clonables™ Ligation/Transformation Kit (Novagen) was used to ligate the blaCTX-M-15 insert into the linearized pET-30a(+) vector. NovaBlue Singles™ Competent Cells were transformed with the ligation mixture by heat shock. Positive transformants were selected by growth on Luria-Bertani (LB) agar containing 30 μg ml−1 kanamycin (Kam) and identification of transformants containing the blaCTX-M-15 insert was confirmed by colony PCR screening using T7 vector specific primers which annealed to the T7 promoter (T7F) and T7 terminator (T7R) regions on the pET-30a(+) vector amplifying a 1150 bp target sequence that incorporated the blaCTX-M-15 insert. The recombinant vector pET-CTX-M-15 was purified from a 10 ml overnight LB Kam 30 μg ml−1 broth culture.
ACTATTCATGTTGTTGTTATT-3′27 and CTX-M-R, 5′-CCCTTACAAACCGTCGGTGACGAT-3′.28 The forward primer contained a 5′ EcoR I restriction site (underlined) and included a homologous promoter sequence 29-42 bp upstream of the start codon.27 The reverse primer included a 3′ Hind III restriction site (underlined).28 Insert DNA was purified, double-digested with Eco R I/Hind III restriction enzymes (Promega) and re-purified. A Clonables™ Ligation/Transformation Kit (Novagen) was used to ligate the blaCTX-M-15 insert into the linearized pET-30a(+) vector. NovaBlue Singles™ Competent Cells were transformed with the ligation mixture by heat shock. Positive transformants were selected by growth on Luria-Bertani (LB) agar containing 30 μg ml−1 kanamycin (Kam) and identification of transformants containing the blaCTX-M-15 insert was confirmed by colony PCR screening using T7 vector specific primers which annealed to the T7 promoter (T7F) and T7 terminator (T7R) regions on the pET-30a(+) vector amplifying a 1150 bp target sequence that incorporated the blaCTX-M-15 insert. The recombinant vector pET-CTX-M-15 was purified from a 10 ml overnight LB Kam 30 μg ml−1 broth culture.
The expression host E. coli BL21(DE3) was transformed with recombinant plasmid pET-CTX-M-15 or the empty vector pET30a(+) and positive transformants were selected on Mueller Hinton (MH) Kam 30 μg mL−1 agar. Transformation screening was performed by colony PCR using the T7 primers as previously described. Cloning success was confirmed by sequence analysis of blaCTX-M-15 amplified from pET-CTX-M-15 using CLC DNA Workbench 6 software (CLC bio, Denmark) by comparison to the blaCTX-M-15 sequence from Genbank (Accession no: ACX54236.1). Functional CTX-M-15 enzyme was confirmed in the BL21 cells containing pET-CTX-M-15 by ESBL disk diffusion phenotypic confirmatory tests using MASTDISCS™ ID ceftazidime and cefotaxime ESBL ID Disc Sets (Mast Diagnostics, Merseyside, UK) and by ESBL broth microdilution phenotypic confirmatory tests. Both tests were performed and interpreted according to CLSI guidelines.29 Lack of expression of CTX-M-15 in BL21 cells containing pET30a(+) was confirmed using the same methods.
Acknowledgements
This publication has emanated from research conducted with the financial support of Science Foundation Ireland (SFI 05/RFP/CHE0063 and 06/RFP/CHO024/EC07), Enterprise Ireland (EI PC/2005/164) and the Health Research Board (HRB PHD/2007/11).References
- B. B. Finlay and R. E. W. Hancock, Nat. Rev. Microbiol., 2004, 2, 497 CrossRef CAS.
- M. Zasloff, Nature, 2002, 415, 389 CrossRef CAS.
- (a) R. E. W. Hancock and H.-G. Sahl, Nat. Biotechnol., 2006, 24, 1551 CrossRef CAS; (b) L. Zhang and T. J. Falla, Expert Opin. Pharmacother., 2006, 7, 653 Search PubMed; (c) M. G. Scott, E. Dullaghan, N. Mookherjee, N. Glavas, M. Waldbrook, A. Thompson, A. Wang, K. Lee, S. Doria, P. Hamill, J. J. Yu, Y. Li, O. Donini, M. M. Guarna, B. B. Finlay, J. R. North and R. E. W. Hancock, Nat. Biotechnol., 2007, 25, 465 CrossRef CAS; (d) J. S. Mader and D. W. Hoskin, Expert Opin. Invest. Drugs, 2006, 15, 933 Search PubMed; (e) P. H. Mygind, R. L. Fischer, K. M. Schnorr, M. T. Hansen, C. P. Sönksen, S. Ludvigsen, D. Raventós, S. Buskov, B. Christensen, L. De Maria, O. Taboureau, D. Yaver, S. G. Elvig-Jørgensen, M. V. Sørensen, B. E. Christensen, S. Kjærulff, N. Frimodt-Moller, R. I. Lehrer, M. Zasloff and H.-H. Kristensen, Nature, 2005, 437, 975 CrossRef CAS.
- R. E. W. Hancock, Lancet Infect. Dis., 2001, 1, 156 CrossRef CAS.
- (a) R. A. Rajewski and M. P. McIntosh, in Prodrugs: Challenges and Rewards, ed. V. J. Stellaet al., AAPS Press/Springer, New York, 2007, Part 1, ch. 2.5.1, pp. 429–445 Search PubMed; (b) V. J. Stella, Expert Opin. Ther. Pat., 2004, 14, 277 Search PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255 CrossRef CAS.
- K. Hilpert, M. R. Elliott, R. Volkmer-Engert, P. Henklein, O. Donini, Q. Zhou, D. F. H. Winkler and R. E. W. Hancock, Chem. Biol., 2006, 13, 1101 CrossRef CAS.
- M. B. Strøm, B. E. Haug, M. L. Skar, W. Stensen, T. Stiberg and J. S. Svendsen, J. Med. Chem., 2003, 46, 1567 CrossRef CAS.
- (a) S. Vunnam, P. Juvvadi and R. B. Merrifield, J. Pept. Res., 1997, 49, 59 Search PubMed; (b) R. E. W. Hancock and G. Diamond, Trends Microbiol., 2000, 8, 402 CrossRef CAS; (c) G. Kragol, R. Hoffmann, M. A. Chattergoon, S. Lovas, M. Cudic, P. Bulet, B. A. Condie, K. J. Rosengren, L. J. Montaner and L. Otvos Jr, Eur. J. Biochem., 2002, 269, 4226 Search PubMed.
- (a) S. Mobashery and M. Johnston, J. Biol. Chem., 1986, 261, 7879 CAS; (b) T. P. Smyth, M. E. O'Donnell, M. J. O'Connor and J. O. St Ledger, Tetrahedron, 2000, 56, 5699 CrossRef CAS.
- (a) J. D. D. Pitout and K. B. Laupland, Lancet Infect. Dis., 2008, 8, 159 Search PubMed; (b) H. C. Maltezou, Int. J. Antimicrob. Agents, 2009, 33, 405 Search PubMed .e1.
- L. A. Carpino, H. Shroff, S. A. Triolo, E.-S. M.E. Mansour, H. Wenschuh and F. Albericio, Tetrahedron Lett., 1993, 34, 7829 CrossRef.
- A. Llinas, B. Vilanova, J. Frau, F. Munoz, J. Donoso and M. Page, J. Org. Chem., 1998, 63, 9052 CrossRef CAS.
- K. Hilpert, R. Volkmer-Engert, T. Walter and R. E. W. Hancock, Nat. Biotechnol., 2005, 23, 1008 CrossRef CAS.
- M. H. Van Regenmortel, Biologicals, 2001, 29, 209 CrossRef CAS.
- (a) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565 CrossRef; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (c) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS.
- R. B. Merrifield, Science, 1986, 232, 341.
- T. P. Smyth, M. E. O'Donnell, M. J. O'Connor and J. O. St Ledger, J. Org. Chem., 1998, 63, 7600 CrossRef CAS.
- (a) G. A. Jacoby and L. S. Munoz-Price, N. Engl. J. Med., 2005, 352, 380 Search PubMed; (b) J. D. D. Pitout and K. B. Laupland, Lancet Infect. Dis., 2008, 8, 159 Search PubMed.
- G. W. Stone, Q. Zhang, R. Castillo, V. R. Doppalapudi, A. R. Bueno, J. Y. Lee, Q. Li, M. Sergeeva, G. Khambatta and N. H. Georgopapadakou, Antimicrob. Agents Chemother., 2004, 48, 477 Search PubMed.
- G. H. Talbot, J. Bradley, J. E. Edwards, D. Gilbert, M. Scheld and J. G. Bartlett, Clin. Infect. Dis., 2006, 42, 657 CrossRef.
- S. Petursson and S. G. Waley, Tetrahedron, 1983, 39, 2465 CrossRef.
- Tetrachloroethylchloroformate can be prepared as follows (note that phosgene is formed during this reaction; phosgene is a colorless, volatile liquid and poisonous gas; this reaction must be performed with appropriate phosgene-handling equipment and by trained personnel): chloral (2.5 ml, 25.6 mmol), was cooled to −84 °C under nitrogen. Diphosgene (5 ml, 41.4 mmol) and pyridine (133 μl, 1.64 mmol) were added at −84 °C and the reaction mixture was allowed to warm up gradually to room temperature. The reaction mixture was stirred overnight. Nitrogen was bubbled through the reaction mixture for 30 min to remove the excess phosgene and the reaction mixture was then concentrated on a rotary evaporator to yield the crude product (5 g). Purification was performed by vacuum distillation at 60 °C and 13 mm Hg, yielding the pure product (1.82 g, 6.93 mmol, 27%). δH (400 MHz, CDCl3) 6.70 (s, 1H, CHCl); δC (100.6 MHz, CDCl3) 91.29, 96.24, (CHCl, CCl3) 149.06 (CO).
- J. D. Lundquist and C. Pelletier, Org. Lett., 2001, 3, 781 CrossRef.
- M. Wu and R. E. W. Hancock, J. Biol. Chem., 1999, 274, 29 Search PubMed.
- Â. Novais, R. Cantón, T. M. Coque, A. Moya, F. Baquero and J. C. Galán, Antimicrob. Agents Chemother., 2008, 52, 2377 Search PubMed.
- Y. Nagano, N. Nagano, J.-i Wachino, K. Ishikawa and Y. Arakawa, Antimicrob. Agents Chemother., 2009, 53, 69 Search PubMed.
- CLSI, Performance Standards for Antimicrobial Susceptibility Testing, in 20th Informational Supplement 2010, Clinical and Laboratory Standards Institute, Wayne, PA Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra01351g |
| This journal is © The Royal Society of Chemistry 2012 |