Mutual interactions between flavonoids and enzymatic and transporter elements responsible for flavonoid disposition via phase II metabolic pathways
Wen
Jiang
ab and
Ming
Hu
a
aDepartment of Pharmacological and Pharmaceutical Sciences, College of Pharmacy, University of Houston, 1441 Moursund Street, Houston, TX 77030, USA. E-mail: mhu@uh.edu; Tel: 001-713-795-8320
bPharmaceutics Graduate Program, College of Pharmacy, University of Houston, 1441 Moursund Street, Houston, TX 77204, USA
First published on 13th June 2012
Abstract
Flavonoids, existing mainly as glycosides in nature, have multiple “claimed” beneficial effects in humans. Flavonoids are extensively metabolized in enterocytes and hepatocytes by phase II enzymes such as UGTs and SULTs to form glucuronides and sulfates, respectively. These glucuronides and sulfates are subsequently excreted via ABC transporters (e.g., MRP2 or BCRP). Therefore, it is the interplay between phase II enzymes and efflux transporters that affects the disposition of flavonoids and leads to the low bioavailability of flavonoid aglycones. Flavonoids can also serve as chemical regulators that affect the activity or expression levels of phase II enzymes including UGTs, SULTs and GSTs, and transporters including P-gp, MRP2, BCRP, OATP and OAT. In general, flavonoids may exert the inhibitory or inductive effects on the phase II enzymes and transporters via multiple mechanisms that may involve different nuclear receptors. Since flavonoids may affect the metabolic pathways shared by many important clinical drugs, drug–flavonoid interaction is becoming an increasingly important concern. This review article focuses on the disposition of flavonoids and effects of flavonoids on relevant enzymes (e.g. UGTs and SULTs) and transporters (e.g. MRP2 and BCRP) involved in the interplay between phase II enzymes and efflux transporters. The effects of flavonoids on other metabolic enzymes (e.g. GSTs) or transporters (e.g. P-gp, OATP and OAT) are also addressed but that is not the emphasis of this review.
 Wen Jiang | Wen Jiang graduated from Nanjing University of Technology with a Masters degree in Biochemical Engineering in 2003 (P.R.China). She received her PhD degree in Pharmaceutics from the Department of Pharmacological and Pharmaceutical Sciences at the University of Houston in 2011 (USA), under primary direction of Dr Ming Hu. Her main research work is focused on the mechanisms of interplay between UGTs and efflux transporters in flavonoid disposition. |
 Ming Hu | Ming Hu received his PhD in Pharmaceutics from College of Pharmacy at the University of Michigan. Currently, he is a full professor in the Department of Pharmacological and Pharmaceutical Sciences of University of Houston (USA). His primary research interests include bioavailability of drugs, nutrients and micronutrients with emphasis on mechanisms of absorption and metabolism of flavonoids and phenolic drugs. He published and contributed numerous pioneer papers and book chapters. He has served in editorial boards from multiple journals. Most of his current research are funded by the National Institutes of Health of USA. |
1. Introduction
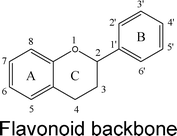
Flavonoids are a class of phenolic and polyphenolic compounds produced as secondary plant metabolites. Flavonoids are widely distributed in nature. The typical sources of flavonoids include vegetables, fruits, beans, grains and herbs. Therefore, an average person could have a significant intake of flavonoids from their daily diet. Published studies indicate that the typical daily intake of flavonoids is in the range of 20–190 mg day−1 in typical Western populations.1,2,3In nature, flavonoids are usually presented as glycosides and to a less extent as aglycones as well as their methylated derivatives. For aglycones, they share the backbone consisting of three carbon rings: a 1,4-benzopyran ring (A), in which the 2 position is linked to a phenyl ring (B) as a substituent.4 For glycosides, the sugars are normally attached to the hydroxyl groups at position 3 or 7 of the A ring. The common sugars or carbohydrates linked to aglycones are D-glucose, glucorhamnose, galactose, L-rhamnose, or arabinose.4 For methylated derivatives, the methylation usually occurs at position 5′ and 3′ of the B ring or at position 7 of the A ring.5 According to their chemical structures, flavonoids (aglycones) are classified into several subfamilies: flavones, flavonols, flavanone, flavanonol, flavanol, isoflavones, anthocyanidin and chalcones (Table 1).6,7,8,9
| Subclass | Flavonoid | Substitution pattern |
|---|---|---|
| a Silybin: (2R,3R)-3,5,7-trihydroxy-2-[(2R,3R)-3-(4-hydroxy-3-methoxyphenyl)-2-(hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxin-6-yl]chroman-4-one. b Ellagic acid: 2,3,7,8-tetrahydroxy-chromeno[5,4,3-cde]chromene-5,10-dione. c Cyanidin: 2-(3,4-dihydroxyphenyl) chromenylium-3,5,7-triol. d Phloretin: 3-(4-hydroxyphenyl)-1-(2,4,6-trihydroxyphenyl)propan-1-one. | ||
Flavones  |
Chrysin | 5,7-OH |
| Apigein | 5,7,4′-OH | |
| Luteolin | 5,7,3′,4′-OH | |
| Eupatilin | 5,7-OH, 6,4′,5′-OMe | |
| Baicalein | 5,6,7-OH | |
| Nobiletin | 5,6,7,8,4′5′-OMe | |
| Tangeritin | 5,6,7,8,4′-OMe | |
| Tricin | 5,7,4′-OH,3′,5′-OMe | |
| Wogonin | 5,7-OH,8-OMe | |
Flavonol  |
Isorhamnetin | 3,5,7,4′-OH,3′-OMe |
| Kaempferol | 3,5,7,4′-OH | |
| Myricetin | 3,5,7,3′,4′,5′-OH | |
| Quercetin | 3,5,7,3′,4′-OH | |
| Morin | 3,5,7,2′,4′-OH | |
| Galangin | 3,5,7-OH | |
| Fisetin | 3,7,4′,5′-OH | |
Flavanone  |
Naringenin | 5,7,4′-OH |
| Hesperetin | 5,7,3′-OH,4′-OMe | |
| Eriodictyol | 5,7,3′,4′-OMe | |
Flavanonol 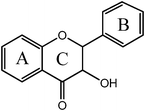 |
Silybina | 3,5,7,-OH |
Flavanol 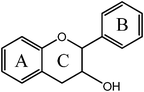 |
(−)-Epicatechin (EC) | 3,5,7,3′,4′-OH (2-S, 3-R) |
| (+)-Catechin | 3,5,7,3′,4′-OH (2-S, 3-S) | |
| Epigallocatechin gallate (EGCG) | 3,5,7,3′,4′,5′,-OH, 3-gallate | |
| Ellagic acidb | 2,3,7,8-OH | |
Isoflavones 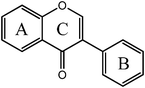 |
Daidzein | 7,4′-OH |
| Genistein | 5,7,4′-OH | |
| Glycitein | 6,4′-OH,7-OMe | |
| Formononetin | 7-OH,4′-OMe | |
| Biochanin A | 5,7-OH,4′-OMe | |
| Prunetin | 5,4′-OH,7-OMe | |
Anthocyanidin 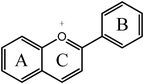 |
Cyanidinc | 3,5,7,3′,4′-OH |
Chalcones  |
Phloretind | 2,4,6, 4′-OH |
| Isoform | Expressiona45,47,48 | Flavonoids as substrates | Flavonoids as modulators | Ref. | |
|---|---|---|---|---|---|
| Inducer or activator | Inhibitor | ||||
| a Expression was mostly referred to expression in those organs, which leads to metabolism and excretion. | |||||
| UGT1A1 | Liver, small intestine, colon, stomach | Quercetin, fisetin, naringenin, luteolin, genistien, daidzein, eupatilin, glycitein, formononetin, biochanin A, prunetin | Chrysin, apigenin, quercetin | Chrysin | 44,54,78–80,85,87 |
| UGT1A3 | Liver, small intestine, colon | Isorhamnetin, sylibin, kaempferol, daidzein, morin, avicularin, eupatilin quercetin xylopyranoside quecetin-3′,4′–OCHO– | — | — | 51,54 |
| UGT1A6 | Liver, small intestine, colon, stomach | Luteolin and chrysin | Quercetin | Silymarin | 52,54,32,87 |
| UGT1A7 | Colon Stomach Esophagus | Eupatilin | — | — | 54 |
| UGT1A8 | Small intestine Colon Esophagus | Naringenin, genistein, daidzein, eupatilin, glycitein, formononetin, biochanin A, prunetin, apigenin, chrysin, 7-hydroxyflavone | — | — | 44,50,54 |
| UGT1A9 | Liver, colon, esophagus, (kidney) | Luteolin, genistein, glycitein, formononetin, biochanin A, prunetin, daidzein,morin, avicularin, quercetin, eupatilin, quercetin xylopyranoside, quecetin-3′,4′–OCHO– | — | Silymarin | 44,51,54,84 |
| UGT1A10 | Small intestine, colon, stomach, esophagus | Naringenin, genistein, apigenin, chrysin, 7-hydroxyflavone, eupatilin | — | — | 44,49,54 |
| UGT2B7 | Liver, small intestine, colon, esophagus | Luteoin and quercetin | — | — | 52 |
| UGT2B15 | Liver, small intestine, colon, stomach, esophagus | Luteoin and quercetin | — | — | 52 |
| UGT2B17 | Liver, small intestine, colon, stomach | Galangin, chrysin, naringin, 7-hydroxyflavone | — | — | 55 |
| Unspecified isoforms | — | Biochanin A, daidzein, formononetin, genistein, prunetin, apigenin, galangin, kaempferol, naringenin, quercetin, chrysin, nobiletin, silymarin, tangeritin | Chrysin, nobiletin, silymarin, baicalein, wogonin | 77,83,80,88,89–91 | |
| Isoform | Expressiona58–61 | Flavonoids as substrates | Flavonoids as modulators | Ref | |
|---|---|---|---|---|---|
| Inducer or activator | Inhibitor | ||||
| a Expression was mostly referred to expression in those organs, which leads to metabolism and excretion. | |||||
| SULT1A1 | Liver, small intestine, colon, stomach | EC, galangin, hesperetin, eriodictyol, (+)catechin | Genistein, biochanin A | Apigenin, chrysin, quercetin, myricetin kaempferol, genistein, daidzein, hesperetin, eriodictyol, catechin EC, luteolin 3,4′-dihydroxyflavone, 3′,4′,7-trihydroxyisoflavone | 62,63,40,67,92–96,99,100 |
| SULT1A3 | Liver, small intestine, colon | Galangin, hesperetin, eriodictyol, (+)catechin, EC Similar to SULT1A1 | — | Baicalein, hesperetin, eriodictyol | 62,63,40,66,95,97 |
| SULT1E1 | Liver, small intestine, colon, stomach | EC, galangin Similar to SULT1A1 | Biochanin A | Tricin, galangin, genistein, equol, daidzein, quercetin | 40,95,97,98,100 |
| Unspecified isoform | — | Genistein, apigenin, baicalein | — | — | 23,64,65 |
| SULT2A1 | Liver, small intestine, colon | — | Genistein, biochanin A | Apigenin, myricetin, baicalein, galangin, 7-hydroxyflavone | 96,99,100 |
| Transporters | Flavonoids as substrates | Flavonoids as modulators | Ref. | ||
|---|---|---|---|---|---|
| Inducer or activator | Inhibitor | No effect | |||
| P-gp | Flavonoid aglycone | Catechin, epicatechin, grapefruit juice, quercetin and kaempherol (low concentration) | EGCG, quercetin kaempherol (high concentraion), chrysin, flavones, hesperetin, naringenin, genistein | Grapefruit juice, narigin, hesperidin, rutin | 109–121 |
| MRP2 | Flavonoid glucuronides and sulfates | Chrysin | Genistein, kaempferol, flavopiridol, chrysin, quercetin, biochanin A, catechin, EGCG, quercetin-7-O-glucuronide | Genistin | 125–131 |
| BCRP | Flavonoid glucuronides and sulfates | Quercetin, chrysin and flavone | Chrysin, biochanin A, apigenin, genistein, fisetin, kaempferol, hesperetin, naringenin, quercetin, luteolin-4-glucoside, daidzein-7-glucuronide, daidzein-4-sulfate, daidzin, ononin, genistin, sissotrin, glycitin, coumestrin | Naringin and phloridzin | 139–147 |
| OATP | — | Rutin | Naringin, naringenin, quercetin, hesperidin, biochanin A, genistein, EGCG | Genistin and quercetin | 149,151,153 |
| OAT | Ellagic acid quercetin-3-O-glucuronide, quercetin-3′-O-glucuronide, quercetin-3′-O-sulfate | — | Ellagic acid, naringenin, morin, silybin, quercetin, quercetin-3-O-glucuronide, quercetin-3′-O-glucuronide, quercetin-3′-O-sulfate | — | 156,157,148 |
There is growing scientific and public interest in flavonoids because of their potential uses for improving human health. Multiple investigations showed that higher consumption of a flavonoid-rich diet was associated with a lower incidence and mortality rates of various degenerative diseases such as cancer, cardiovascular disease and immune dysfunction.1,10,11,12 Extensive research has uncovered that flavonoids have a myriad of biological activities (e.g. anti-cancer, anti-inflammation, antioxidant, anti-microbial, cardiotonic and lipid lowering activities), which could affect a plethora of enzyme systems and signaling cascades involved in the diseases.4 Therefore, the beneficial impacts of flavonoids are likely to be derived from their pleiotropic biological activities.
To exert these biological activities, flavonoids must be absorbed and remain as aglycones in the body, since phase II metabolites of flavonoids are rarely reported as the active species.13 It is believed that flavonoid aglycones are able to passively diffuse across the gut wall, though evidence suggests that the absorption of intact glycosides may also occur, albeit at a significantly slower rate.14,15 For most flavonoid glycosides, they must be first hydrolyzed to aglycones either by microorganisms residing in the intestine or by brush-border enzymes (e.g. lactase phloridzin hydrolase, LPH) before their absorption.16,17,18,19 Because the vast majority of flavonoids are present as glycosides in nature, absorption of flavonoids present in most fruits, vegetables, and herbs are slow and often incomplete. The exception to this rule is certain flavonoid mono-glucosides, which are rapidly hydrolyzed by intestinal LPH, releasing aglycones that are rapidly absorbed.20,21,22 On the other hand, for flavonoid aglycones, which are normally given at a dose that is intended to be pharmacologically active, the dose could be quite high (e.g., 1000 mg), which could result in higher concentration in the intestinal lumen. In general, the absorption of flavonoids (aglycones) is a rapid process. After entering the enterocytes, flavonoids are subjected to extensive metabolism (especially phase II) by intestine conjugating enzymes.23 For flavonoids that have passed through the intestinal epithelium intact, they remain to be subject to the equally rapid metabolism by liver conjugating enzymes.24
After absorption, phase I metabolism (e.g. cytochrome P450) other than hydrolysis of glycosides makes only a minor contribution to flavonoid clearance. In contrast, phase II metabolism plays a significant role in flavonoid clearance. Most of the flavonoids undergo glucuronidation by uridine-5′-diphosphate glucuronosyltransferases (UGTs) and/or sulfation (or sulfonation) by sulfotransferases (SULTs) in either enterocytes or hepatocytes, which convert them to glucuronides and sulfates, respectively.25,26 These glucuronides and sulfates of flavonoids could be subsequently excreted into the lumen or bile by efflux transporters such as multi-drug resistance protein 2 (MRP2) and breast cancer resistance protein (BCRP).26,27 Interestingly, the excreted glucuronides and sulfates in the intestine can be hydrolyzed by microflora β-glucuronidases and sulfatases back to aglycones, which can again undergo absorption and metabolism. Therefore, it is not surprising to speculate that the low bioavailability of flavonoids (usually, the oral bioavailability of flavonoid aglycones with a range of 2–20%) is due to the presence of a potent disposition network that mainly consists of phase II conjugating enzymes and efflux transporters of phase II enzyme conjugates.28,29,30,31 In this paper, the bioavailability of flavonoids refers to intact aglycones in the systematic circulation, according to definition of the US Food and Drug Administration. Conjugates of flavonoids in the blood are not considered as bioavailable according to this definition.
Flavonoids are not only substrates of drug metabolizing enzymes (DMEs) and active efflux transporters; they are also regulators for some of those DMEs and transporters. Substantial amounts of evidence demonstrated that flavonoids could inhibit the activities of phase II enzymes (e.g. UGTs, SULTs and glutathione-S-transferases (GSTs)) and active transporters such as P-gp (P-glycoprotein), BCRP, MRP2 or OATP (organic anion-transporting peptide) by a variety of mechanisms (e.g., competitive inhibition).32,33,34,35 In addition, flavonoids can also modify the expression of phase II enzymes and transporters at both the mRNA and the protein level. Thus, the enzymes and transporters involved in the flavonoid disposition could be affected by flavonoids. Due to the fact that many clinical drugs share the same metabolic pathways with flavonoids, potential drug–flavonoid interactions are expected. Specifically, when large amounts of flavonoids, taken up from dietary or supplementary sources, are administered with prescription or OTC (over the counter drugs) for a prolonged period of time, the pharmacokinetics of therapeutic drugs can be altered. As a result, it is meaningful to investigate the effects of flavonoids on functions and expression of the DMEs and transporters.
Based on the above considerations, we will present a current overview of flavonoid disposition and the effects of flavonoids on phase II enzymes and transporters, most of which are involved in their disposition as well. The paper reviews the literature mainly from the period 1990 to 2011. The effects of DMEs and transporters on flavonoid disposition will be addressed in detail, the effects of flavonoids on the activity or expression of DMEs and transporters will be discussed, and lastly the potential impact of the interactions between flavonoids and DMEs as well as transporters on the approved drugs will be discussed. The focus of this review is on the in vitro or in vivo observations from human enzymes and transporters, though animal studies were also summarized.
Unless otherwise specified, flavonoids refer to aglycones. Since phase I enzymes lead to minor metabolism of flavonoids and many other reviews have described their modulation by flavonoids in detail,32,35,36,37 the effects of flavonoids on phase I enzymes were not included in this review. Though phase II enzymes like GSTs and transporters like P-gp, OATP and OAT, are not mainly involved in flavonoid disposition on the basis of current knowledge, their modulation by flavonoids are still covered in this review, considering that many clinical drugs and endogenous compounds are their substrates. We believe that the information discussed in this review can be used as an effective and updated research reference on flavonoid study, especially for flavonoid disposition and drug–flavonoid interactions. Hopefully, the efforts made on flavonoid disposition will eventually lead to an effective way to improve flavonoid bioavailability, and allow flavonoids to exert more therapeutic or disease-prevention effects on humans.
2. Disposition of flavonoids
2.1. Phase I metabolism of flavonoids
Flavonoids, like other drugs or xenobiotics, can be metabolized by cytochrome P450 (CYP450) enzymes. It was reported that CYP2C9 was the most efficient isoform for the oxidation of galangin followed by CYP1A3 and CYP1A1.38 For kaempferide, CYP1A2 was predominantly responsible for its oxidation followed by CYP2C9 and CYP1A1.39Compared with phase II metabolic pathways, however, CYP450-mediated metabolism usually contributes little to the overall metabolism of flavonoids. In addition, CYP-mediated metabolism of flavonoids has never been shown to be important in vivo or in intact cells. For example, apigenin was metabolized by rat liver microsomes to form three monohydroxylated derivatives, one of which was luteolin. Further investigation indicated the involvement of CYP1A1, CYP3B and CYP2E1 in apigenin hydroxylation in vitro.24 However, when apigenin was perfused through an isolated rat liver, none of the phase I metabolites could be recovered in the effluent perfusate, suggesting that CYP-mediated metabolism has little impact on in vivo flavonoid clearance, when the phase II metabolic pathway was functioning.24 Therefore, metabolism via CYP is not a major clearance mechanism for flavonoids and is not the main reason for their poor bioavailability in vivo.
2.2. Phase II metabolism of flavonoids
In contrast to minor contributions of phase I metabolism, phase II metabolism is the predominant pathway for the flavonoid disposition both in vitro and in vivo. There are two main types of phase II metabolites of flavonoids present in vivo: glucuronides and sulfates, which are produced by UGTs and SULTs, respectively. In a study of galangin metabolism, glucuronides are found to be the primary metabolites followed by sulfates, and then oxidative metabolites.40 These results supported the hypothesis that the efficiency of glucuronidation of flavonoids was very high, followed by sulfation, with only a very minor contribution by CYP-mediated oxidation. This metabolism rank (glucuronidation > sulfation > oxidation) could be extended to most flavonoids known in the literature.Both enterohepatic and enteric recycling plays a critical role in flavonoid disposition. Therefore, UGT and SULT isoforms expressed in the liver or intestine are functionally more important than other isoforms in flavonoid metabolism. It is expected that different UGT and SULT isoforms can convert the same flavonoid at different rates, and conjugations at different positions will produce regiospecific glucuronides and sulfates at different ratios.41–43 On the other hand, flavonoids with different structural features are targets of different UGT or SULT isoforms.41–44 Lastly, the conditions of glucuronidation or sulfation reactions can also affect the structural preference of UGT or SULT isoforms.
The human UGT superfamily consists of UGT1, UGT2, UGT3 and UGT8 families. The UGT1 family currently contains only the UGT1A subfamily, while the UGT2 family can be classified into UGT2A (including UGT2A1, 2A2 and 2A3) and UGT2B subfamilies. The UGT3 family is currently composed of the UGT3A subfamily (including UGT3A1 and 3A2). The only known member of the UGT8 family is UGT8A1.47
Among all the UGT subfamilies, the UGT1A and UGT2B subfamilies, which are classified according to their primary amino acid sequences, are predominantly responsible for metabolism of flavonoids and have more clinical relevance than other subfamilies with respect to drug metabolism. As a result of different exon splicing the same gene, transcription of the UGT1A gene cluster can produce the following isoforms: UGT1A1, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9 and UGT1A10.47 Unlike isoforms from the UGT1A subfamily, each isoform (UGT2B4, UGT2B7, UGT2B10, UGT2B11, UGT2B15 and UGT2B17) from the UGT2B subfamily is encoded by an individual gene.47 UGTs are widely distributed in the human body. The liver, intestines, kidney and stomach are enriched with various and sometimes unique UGT isoforms.48
Flavonoids are extensively metabolized by variations of UGT isoforms (Table 2). It was reported that UGT1A1, UGT1A8, UGT1A9 and UGT1A10 mainly contribute to the glucuronidation of isoflavones including daidzein, genistein, glycitein, formononetin, biochanin A and prunetin in a concentration-dependent manner.44 Under different concentrations, the glucuronidation pattern of each isoflavone by UGT isoforms was sometimes changed. For example, genistein was mostly metabolized by UGT1A9 at 2.5 μM, whereas it was catalyzed mainly by UGT1A8 at 10 and 35 μM.44 In fact, isoflavones were the best substrates among all the 42 flavonoids tested on the basis of the structure–function relationship of UGT1A10, suggesting that the hydroxyl group at C6 or C7 other than C5 of the A-ring or on C4′ of the B ring was preferred.49 UGT1A8, which had a 94% similarity to the primary amino acid sequence of UGT1A10, not only had maximal overlapping substrates with UGT1A10, but also displayed a higher glucuronidation rate toward flavones (7-hydroxyflavone, chrysin and apigenin), a flavanone (naringenin) and an isoflavone (genistein) than UGT1A10.50 In another study, UGT1A9 and UGT1A3 were able to catalyze both flavonoid aglycones and glycoside, though their substrate preference was clearly aglycones.51 The newly reported substrates of UGT1A3 included isorhamnetin, silybin, kaempferol, daidzein, morin, quercetin-3′, 4′-OCHO-, quercetin xylopyranoside, and avicularin, most of which were also the substrates of UGT1A9 except isorhamnetin, silybin and kaempferol.51 With respect to shared substrates, UGT1A9 always had a higher catalytic efficiency or intrinsic clearance (Vmax/Km) than UGT1A3.51 Other than the above UGT isoforms, UGT1A6 and UGT1A7 are also involved in glucuronidation of flavonoids. UGT1A6 was the predominant isoform responsible for glucuronidation of luteolin at the 7 position.52 It was also found that chrysin was metabolized by UGT1A6, an isoform highly expressed in Caco-2 cells.53 In another study, UGT1A7 was found to be involved in glucuronidation of eupatilin, which was also the substrate of UGT1A1, UGT1A3, UGT1A8, UGT1A9 and UGT1A10.54
In addition to UGT1A isoforms, UGT2B isoforms can also metabolize flavonoids, though they may not be as efficient as UGT1A isoforms. Evidence showed that galangin, chrysin, 7-hydroxyflavone and naringin were substrates for UGT2B17, among which galangin had the highest conversion value, whereas apigenin, baicalein, fisetin, quercetin, genistein and biochanin A were not conjugated by UGT2B17.55 Another example of flavonoids as UGT2B substrates was from the study of the regioselectivity of the phase II metabolism of luteolin and quercetin. It was demonstrated that luteoin and quercetin were substrates of UGT2B15 and UGT2B7 with glucuronidation primarily occurring at position 7 and 3′, respectively.52 Between these two isoforms, UGT2B7 had a relatively higher conversion than UGT2B15. Generally, as substrates of UGT2B7, flavonol has a higher glucuronidation activity than flavones and isoflavones.56
After glucuronidation mediated by various UGT isoforms, flavonoids are converted to corresponding glucuronides. These glucuronides are more hydrophilic and can be easily excreted via urine or feces. Usually, the biological activity of glucuronides is less than corresponding flavonoids with a few exceptions (e.g. quercetin-7-O-glucuronide).57
Compared with the glucuronidation of flavonoids, research on the sulfation of flavonoids, which is regarded as a metabolic pathway that can compete with glucuronidation, is relatively scant. Previously, sulfation primarily catalyzed by SULT1A1 and SULT1A3, was the major pathway in the metabolism of (−)-epicatechin (EC) in human liver and intestine without detectable glucuronidation.62 This sulfation result was consistent with another report showing that SULT1A1 and SULT1A3 metabolized EC in addition to hesperetin, eriodictyol, (+)-catechin.63 On the other hand, rapid sulfation and glucuronidation of EC in rats implied a large species difference in metabolism compared to humans.62 As mentioned previously, sulfation of galangin was one of metabolic pathways contributing to metabolism of galangin. Three SULT isoforms (SULT1A1, SULT1A3 and SULT1E1) were involved in sulfation of galangin, among which SULT1A1 displayed the most sulfation efficiency. On the contrary to the above three SULT isoforms, SULT2A1 had no activity against galangin.40 In Caco-2 cells, it is common to observe that flavonoids (e.g. genistein, apigenin and baicalein) undergo both glucuronidation and sulfation.23,64,65 Sulfation might be attributed to SULT isoforms being expressed in Caco-2 cells such as SULT1A1, SULT1A2 and SULT1A3.66 Recently, Meng et al. showed the SULT1A3 exclusively metabolized flavonoids at the 7-OH position.41
2.3. Efflux of flavonoids and their phase II metabolites
In addition to extensive metabolism of flavonoids, efflux of flavonoids and their metabolites is another factor that leads to low bioavailability of flavonoids. ATP-binding cassette (ABC) transporters are ubiquitous integral membrane proteins that outwardly transport various ligands actively across the membrane with hydrolysis of ATP.67 Accumulating evidence showed that members of ABC such as P-gp, MRP2 and BCRP were involved in transportation of flavonoids or their phase II metabolites out of cells.68–71 Experiments on Caco-2 cells demonstrated that both quercetin and naringenin were substrates of MRP2 while only naringenin was substrate of P-gp.68 Moreover, glucuronides of quercetin (quercetin-7-O-β-D-glucuronide, -3-O-β-D-glucuronide, -3′-O-β-D-glucuronide and -4′-O-β-D-glucuronide) were also substrates of MRP2.69 Kaempferol, a major constituent of Ginkgo biloba extract, was found to be a substrate of BCRP by An and co-workers.71 In fact, glucuronides and sulfates of flavonoids are more likely to be substrates of MRP2, MRP3 and BCRP.26,27,69–70,722.4. Interplay between phase II enzymes and efflux transporters in disposition of flavonoids
As an extension to the idea of interplay between CYP3A and P-gp, interplay between phase II enzymes and BCRP/MRP describes a sequential process in which substrates are metabolized by phase II enzymes (e.g., UGTs) and the produced hydrophilic phase II metabolites are transported out of cells by the efflux transporters (e.g., BCRP or MRP). This kind of interplay was commonly identified in the study of flavonoid disposition in both animal and cell models. This is because flavonoids as substrates can passively diffuse into cells and subsequently be metabolized to glucuronides and sulfates via glucuronidation and sulfation, respectively. The glucuronides and sulfates, which are too hydrophilic to diffuse across the cell membrane, can only exit cells via the action of the efflux transporters [Fig. 1]. The engaged efflux transporters include BCRP, MRP2 and MRP3.26,27,69,70,72 Previously, the efflux step of flavonoid conjugates (phase II metabolites) was regarded most likely as the rate-limiting step for the interplay due to the discrepancy of results between in vitro glucuronidation assay and the Caco-2 cell model or rat perfusion model. For example, glucuronidation of biochanin A was found to be faster than formononetin in rat intestinal and liver microsomes; however, excretion of formononetin conjugates was more efficient than biochanin A in the rat intestine perfusion model and Caco-2 cell model.73 Recently, the idea that the rate-limiting step of efflux is challenged when it was applied to a simple model of one phase II enzyme (UGT1A9) and one dominant efflux transporter (BCRP). In this simple model, we showed clearly that the efflux transporter was simply a gate-keeper and kinetic interplay between UGT1A9 and BCRP ultimately determined how much metabolite was formed and then excreted.74 Efforts are underway to understand more detail about the interplay between phase II enzymes and efflux transporters.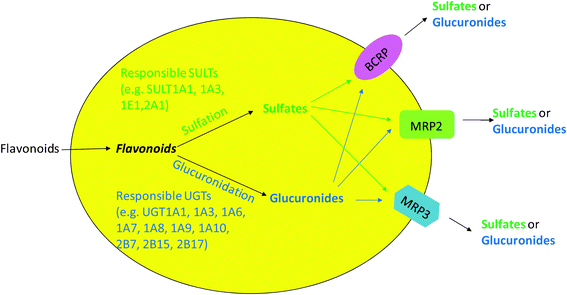 | ||
| Fig. 1 Cellular interplay between phase II enzymes and efflux transporters in flavonoid disposition. | ||
3. Effects of flavonoids on phase II enzymes
Flavonoids are likely to be metabolized by phase II enzymes as described previously. Reciprocally, flavonoids can also exert some effect on phase II enzymes [Fig. 2]. As an activator or inducer, a flavonoid may induce or activate the target enzymes via multiple mechanisms, which possibly require the involvement of various nuclear receptors. For example, some flavonoids could induce phase II enzymes by disrupting the interaction between Nrf2 and Keap1 and by making Nrf2 relocate to the nucleus and bind to the antioxidant/electrophile response element (ARE).35,75 In addition, flavonoids may interact with constitutive androstane receptor (CAR) and pregnane X receptor (PXR), which were reported to regulate phase II enzymes and transporters.76 On the other hand, flavonoids can act as inhibitors of phase II enzymes, more likely through simple mechanisms such as competitive inhibition. In the following parts, we will discuss the research developments on how flavonoids affect the phase II enzymes, including UGTs, SULTs and GSTs.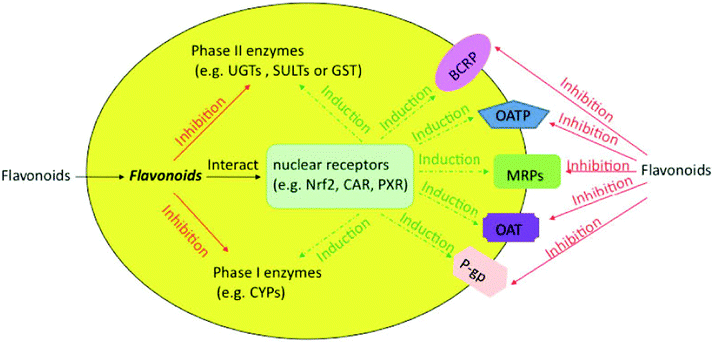 | ||
| Fig. 2 Effects of flavonoids on phase I and II enzymes and transporters in cells. The solid line represents the inhibition, whereas the dotted line represents induction via various nuclear receptors. | ||
3.1. Effects of flavonoids on UGTs (Table 2)
Early study on the prostate cancer cell line LNCaP showed that certain glucuronidation activities (i.e., glucuronidation of testosterone via UGT2Bs) were increased after the cells were exposed to the flavonoids including biochanin A, daidzein, formononetin, genistein, prunetin, apigenin, galangin, kaempferol, naringenin and quercetin.77 Among the tested flavonoids, biochanin A was the most potent inducer of UGTs, increasing the glucuronidation activity by 10 fold.77 At the same time, due to less availability of testosterone resulting from glucuronidation, the production and release of prostate specific antigen (PSA) as a prostate tumor marker were significantly decreased in the presence of biochanin A.77 It suggested that flavonoids could play an important role in modulation of sex hormones in humans and provide us with a possible means to prevent and manage prostate cancers. In addition, chrysin was reported to be able to induce UGT1A1 specifically, both in Caco-2 and HepG2 cells.78–80 However, UGT1A6, UGT1A9 and UGT2B7 were not affected by the chrysin treatment.80 Further study from the same group showed that induction of UGT1A1 by chrysin was not dependent on an aryl hydrocarbon receptor (AhR) since galangin and isorhamnetin, both of which are inducers of CYP1A1 via AhR, did not display any induction of UGT1A1.81 More recently, UGT1A1 was believed to be under the regulation of Nrf2.82 In addition to be an inducer of UGT1A1, chrysin was also an inhibitor for the glucuronidation of estradiol at substrate concentrations above 25 μM.83 Similar inhibitory effects on glucuronidation of estradiol were also observed when nobiletin and silymarin were treated at substrate concentrations over 25 μM.83 On the contrary, the same three flavonoids had little effect or possibly even slight stimulation effects on estradiol glucuronidation at the lower concentrations (5 and 10 μM). As for tangeritin and quercetin, both exerted inhibitory effects on glucuronidation of estrodiol. Tangeritin was the most potent inhibitor, while quercetin was a relatively weak inhibitor.83 In addition, naringenin could inhibit glucuronidation of estradiol to a similar extent at all tested naringenin concentrations (5–100 μM), suggesting that it did not act as a competitive inhibitor.83 Furthermore, the milk thistle extract silymarin and its major ingredient silybin could inhibit UGT1A6 and UGT1A9 in human hepatocytes at a concentration of 100 μM.32,84 As for apigenin, it was found to be able to induce UGT1A1 transcription 4 fold in undifferentiated Caco-2 cells.85 The combination of apigenin and sulforaphane could further lead to significant induction of UGT1A1 mRNA (up to 12 fold).85 Considering that SN50 (a NF-kB translocation inhibitor) could enhance the induction of UGT1A1 by apigenin, it was believed that UGT1A1 induction by apigenin might be associated with NF-kB translocation.85 In addition to apigenin, quercetin was also shown to markedly increase the glucuronidation activity in Caco-2 cells.80 The significant increase in glucuronidation activity might result from increasing expression levels of UGT1A6 (at 50 μM of quercetin) and UGT1A1.86,87Regulation studies done on the cell culture could be replicated in vivo, although the results were not always consistent. For example, feeding rats with a diet containing 1% quercetin significantly increased the activity of UGTs in liver and to a lesser extent in intestine.88 Similar results of increasing hepatic UGT activity were obtained after flavone, flavanone and tangeritin were fed to rats at a dietary concentration of 0.3% (w/w) or soy isoflavones (0.81 mg kg−1 diet).89,90 In contrast, hepatic UGT activity was inhibited after exposing mice to a liquid diet containing 5 mM baicalein or wogonin for 1 week.91 In one study, where a human jejunum was perfused with an onion and broccoli extract containing 60 μM quercetin for 2 h, mRNA of UGT1A1 in shed enterocytes was increased.87 The results from humans were consistent with the observation in Caco-2 cells treated with pure quercetin.
3.2. Effects of flavonoids on SULTs (Table 3)
It is not surprising to observe the inhibition of SULT activities at higher flavonoid concentrations (> 10 μM). Indeed, auto-inhibition or substrate-inhibition is a very common phenomenon in SULT-mediated metabolism of flavonoids. For example, metabolism of genistein and daidzein by SULT1A1 followed the substrate-inhibition kinetic profile. When substrate concentrations were over 1 μM, the sulfation activities were substantially decreased.92 In addition to substrate-inhibition, a large body of studies showed that flavones (e.g. apigenin and chrysin), flavonols (e.g. quercetin, myricetin and kaempferol), isoflavones (e.g. genistein and daidzein), flavanone (e.g. hesperetin and eriodictyol) and flavanols (e.g. catechin and epicatechin) were potent inhibitors of SULT1A1.92,93,94,63In an investigation conducted by Harris and co-workers, 37 flavonoids were tested to determine their inhibition potency against SULT1A1.95 The results indicated that flavonoids could inhibit SULT1A1 significantly at submicromolar concentrations. Among all the flavonoids selected, 3,4′-dihydroxyflavone was the most potent inhibitor with an IC50 less than 1 nM, whereas luteolin was regarded as the most potent naturally occurring inhibitor with IC50 of 3 nM.95 In contrast to the above flavones and flavonols, isoflavones such as genistein and daidzein had relatively high IC50, which was in the range of 500–600 nM except for 3′,4′,7-trihydroxyisoflavone, which had an IC50 of 20 nM.95 Therefore, the 3′,4′-dihydroxy motif might be a critical feature for flavonoids to have potent inhibitory effects of SULT1A1 based on the fact that flavonoids with the above feature motif had an IC50 of 100 nM or less against SULT1A1-mediated sulfonation of the standard substrate 4-nitrophenol (3 μM).95,96 Though various flavonoids were shown to have potent inhibition of SULT1A1, few of them displayed powerful inhibitory effects on SULT1A3. Baicalein was found to be the only one having substantial inhibition (IC50 500 nM) at less than 1 μM against 10 μM dopamine as the standard substrate.95,96 Recently, hesperetin and eriodictyol were also reported to be potent inhibitors of SULT1A3. Hesperetin and eriodictyol had IC50 values of 20 μM and 15 μM, respectively, against SULT1A3-mediated sulfonation of its typical substrate dopamine (50 μM).63 In the case of SULT1E1, which was inhibited relatively easily by many simple phenols, tricin (a derivative of flavonoid, 3′,5′-dimethoxy-4′,5,7-trihydroxyflavone) was found to be the most potent inhibitor.97 It could competitively inhibit SULT1E1 with an inhibition constant of approximately 1 nM.97 Another potent inhibitor of SULT1E1 was galangin with IC50 of <1 μM against 10 nM estradiol as standard substrate.95 Similarly, genistein and equol are as potent as galangin, they exhibited inhibitory constants at the active site of 400 and 500 nM and at an allosteric site of 2 and 5 μM, respectively (substrate 10 nM estradiol).95 On the other hand, daidzein was less potent than above flavonoids, and had an IC50 of 5 μM against 10 nM estradiol even though its chemical structure was most close to equol and distantly related to genistein.95 In the same system, formononetin was found to be the least potent inhibitor among all the tested compounds.95 In another published study, quercetin had surprisingly 10 times more inhibitory potency for SULT activity in the intact cultured human mammary epithelial cells (HME) (an IC50 of about 0.1 μM) than in recombinant SULT isoform.98 Last but not least, SULT2A1, another SULT isoform expressed in the intestine, was reported to be inhibited by apigenin with a Ki value of around 2.4 μM.96 Similar to apigenin, myricetin, baicalein, galangin and 7-hydroxyflavone also had Ki values between 2–3 μM.96 Possibly, a 7-hydroxyflavone substituent seemed to be a requirement for some inhibition.96 On the contrary, fisetin, kaempferol, luteolin, hesperetin, morin, quecetin, rutin, catechin, and daidzein did not show any inhibitory effect on SULT2A1 even at a concentration of 25 μM.96
Flavonoids not only inhibit the SULT activity, but also induce the expression of SULTs in various model systems. Recently in both HepG2 and Caco-2 cells, genistein was shown to induce SULT1A1 and SULT2A1 at both mRNA and protein levels in a dose- and time-dependent manner.99 In Sprague–Dawley rats, biochanin A significantly induced expression of rat SULT1A1, SULT2A1 and SULT1E1 in liver and intestine after a 7 day treatment period (doses: 0, 2, 10, and 50 mg kg−1 day−1).100
3.3. Effects of flavonoids on GSTs
Glutathione-S-transferases (GSTs) are an another group of phase II enzymes. However, GSTs are not primarily responsible for flavonoid metabolism. They are able to catalyze the binding of a wide range of electrophiles to the sulfydryl group of glutathione in order to neutralize the reactive oxygen species. As a result, GSTs play an important role in the detoxification of various chemical carcinogens. GSTs are widely distributed in many tissues and present in relatively large amounts in the epithelial tissues of the human gastrointestinal tract.35,101 Volumes of evidence showed that considerable numbers of flavonoids contribute to the regulation of GSTs, either in gene expression or protein activities or both. An example from a cell study in vitro indicated that effects of genistein on expression of GSTs through the ER/ARE signaling pathway were cell-type specific.102 In COS1 I cells expressing ERα and ERβ, genistein (1 μM) repressed the GST Ya ARE-dependent gene expression; however, in C4-12-5 cells, which are ER-negative breast cancer cells derived from the MCF-7 cells, genistein (1 μM) showed modest induction of the GST gene following transfection with ERα and ERβ.102 In addition, evidence from an in vivo study showed that GSTs in the stomach were significantly induced when the mice were fed with citrus limonoids and citrus flavonoids (hesperidin, naringin and crude flavonoids mixture) for one week at dose of 20 mg.103 Moreover, in another study, Swiss Webster mice were exposed to flavone, morin, naringenin, (+)-catechin and quercetin for 20 days at 2.5 g kg−1 diet.104 It was revealed that only groups fed with a flavone showed the gender and isozyme-specific induction of GSTs. In female mice, GST activity was increased 7 fold, while in male mice, GST activity was increased 4 fold. With regard to enzyme specific assay, it was found that the activities of GSTM and GSTP increased to a greater extent in females (8 fold and 4 fold respectively) than in males (6 fold and 2 fold).104 In humans, the modification of GSTs by flavonoids is more complicated than that in mice. In a study involving healthy volunteers, who drank juice containing several flavonoids (e.g., quercetin, quercetrin, myricetrin, kaempferol, hesperidin, eriodictyol and naringenin), GSTP1 protein in leucocytes was first suppressed at weeks 4 and 6. However, at week 8, expression of GSTP1, which was reversibly linked with DNA damage, significantly increased.105 The results were contrary to in vitro experiments. In vitro, when leucocytes were treated with polyphenol mixtures containing flavonoids, no change of GST gene expression or activity was observed, though up-regulation of certain CYP450 and SULTs isoforms were observed.1054. Effects of flavonoids on transporters
Currently, the interaction between flavonoids and ABC transporters (Table 4) is drawing more and more attention of scientific researchers [Fig. 2]. In addition to ABC transporters, another class of transporters called solute carrier family (SLC), is also involved in interaction with flavonoids [Fig. 2]. In contrast to ABC transporters, which are mainly responsible for efflux, members of the SLC primarily manage cellular uptake. Flavonoids and their metabolites might act as substrates or inhibitors of both ABC and SLC transporters. As an inducer of transporters either at the mRNA or at the protein level, long exposure to flavonoids is necessary. Various mechanisms underpinning the interaction between ABC transporters and flavonoids have been postulated. Some flavonoids can affect the ATPase activity by inhibition or stimulation; other flavonoids, which are substrates of transporters, can compete for the binding site of transporters and result in competitive inhibition; and still a more selected group of flavonoids can modulate the transporters through post translational modifications (e.g., phosphorylation).4.1. Effects of flavonoids on P-gp
P-glycoprotein (P-gp), encoded by the ABCB1 gene with a molecular weight of 170 KD, is the first human ABC transporter that was cloned and characterized through its ability to confer a multi-drug resistant phenotype to cancer cells.106 As an efflux transporter expressed in many tissues and apical membrane of epithelial cells, P-gp is mainly responsible for outward transportation (i.e., efflux) of hydrophobic, cationic or neutral compounds with a planar structure with some notable exception (e.g., methotrexate and phenytoin, both with negative charges).33,107P-gp consists of two homologous segments, each of which contains six transmembrane domains (TMDs) and a nucleotide binding domain (NBDs) with ATPase activity.108There were several controversial opinions with regard to effects of flavonoids on P-gp. The first controversy was about the effects of green tea. One study supported the notion that P-gp activity was inhibited by green tea polyphenols,109 whereas the other suggested that P-gp function was elevated by catechin in green tea.110 Experiments from the first group indicated that green tea polyphenols at a concentration of 30 μg ml−1 could inhibit the photolabeling of P-gp by 75% and increase the accumulation of rhodamine-123 3 fold in the multidrug-resistant cell line CHRC5.109 Moreover, EGCG as a representative of green tea polyphenols exhibited an inhibitory effect on P-gp activity not only in CHRC5 cells but also in Caco-2 cells.109 In the second study, although some catechins like EGCG displayed inhibitory effects on P-gp, others like (−)-epicatechin were shown to facilitate the P-gp-mediated transport of the fluorescent markers LDS-751 via a heterotropic allosteric mechanism.110 Currently, there is no explanation for these controversial findings.
Another controversy deals with the grapefruit juice effect.111 Some studies show that there were some effects whereas other showed none. Earlier studies from Dr Lown’s group indicated that twice daily consumption of grapefruit juice (8 oz) for 6 days resulted in 62% reduction of CYP3A4 levels without any change of P-gp level in 10 healthy volunteers.112 This result was consistent with the conclusion that naringin and 6′,7′-dihydroxybergamottin, both of which were components of grapefruit juice, improved saquinavir transport in Caco-2 cells by inhibition and down-regulation of the CYP3A4 rather than by modulation of P-gp.113 On the contrary, some papers showed that grapefruit juice had either inhibitory or activating effect on P-gp. Dr Benet’s group found that grapefruit juice stimulated the efflux of a couple of P-gp substrates across the MDCK-MDR1 cells.114 In the study of vincristine transport across MBEC4 cells (cultured mouse brain capillary endothelial cells), quercetin and kaempherol were found to decrease the uptake of vincristine at low concentration (10 μM), but increased the uptake of vincristine at high concentration (50 μM).115 This biphasic in vitro results were further confirmed by the study on ddY mouse in vivo with co-administration of quercetin (0.1 mg kg−1) and (1.0 mg kg−1).115 The reasons were due to changes in P-gp functionality. At a low concentration, P-gp activity was stimulated as a result of enhancing its phosphorylation; while at a high concentration, P-gp function was hindered.115 In addition to quercetin and kaempherol, other flavonoid components, including chrysin, flavone, hesperetin, naringenin, were found to increase the vincristine uptake in MBEC4 cells by inhibiting P-gp at the 10–50 μM,. However, their corresponding glycosides such as hesperidin, naringin and rutin did not affect P-gp.115 Furthermore, P-gp was reported to be inhibited by kaempferol and naringenin in the human immortalized tubular cell line HK-2 cells by down-regulating the protein level and mRNA expressions of P-gp.116
Apart from above controversies, the majority of findings supported the inhibitory role of flavonoids against P-gp. It was reported that genistein (200 μM) could elicit an elevation in intracellular accumulation of rhodamine 123 and daunorubicin in P-gp-expressing cell lines. In addition, genistein could also decrease photoaffinity labeling of P-gp by [3H] azidopine.117 Other related studies also pointed out that flavonoids had an inhibitory effect on P-gp in MDCKII-MDR1 cells, Caco-2 cells and K562 cells, based on their ability to the increase cellular uptake of various testing substrates.118–120
On the basis of accumulating interaction data, the structure–function relationship between flavonoids and P-gp was established. More and more flavonoid derivatives were synthesized to modulate P-gp functions.121 Recently, a 3D linear solvation energy model was developed to quantify the affinity of flavonoid derivatives toward P-gp. According to this 3D model, two major physicochemical parameters (shape parameters and hydrophobicity), were identified to be primarily responsible for the affinity of flavonoid derivatives towards P-gp. At the same time, the hydrogen-bonding capacity was identified as a minor modulator.122
4.2. Effect of flavonoids on MRP2
MRP2 is an efflux transporter with molecular weight of 190 KD, which belongs to the ABCC (also called MRP) subfamily of ABC transporters. MRP2 has two nucleotide binding domains (NBDs) and seventeen transmembrane domains (TMDs).123 As a mechanism leading to the multidrug resistance, MRP2 is highly expressed in carcinoma cells or tumor tissues as well as normal tissues such as liver, kidney and intestine.124 The main function of MRP2, which is located to apical membrane of polarized cells, is to actively transport endogenous (e.g. bilirubin and its polar conjugates) and xenobiotic (e.g. drugs) substances out of the cells.124Early investigation showed that genistein inhibited the efflux of daunorubicin from small-cell lung cancer GL4/ADR cells, in which MRP was overexpressed, in a competitive manner.125 The same result of inhibition of daunorubicin by genistein competitively was also observed in plasma membrane vesicles from marine MRP-transfected NIH3T3 cells.126 Follow-up study confirmed that genistein but not genistin, together with kaempferol and flavopiridol (synthetic flavonoid derivative) could affect MRP-mediated transport of anticancer drugs by a direct interaction with MRP.127 Moreover, chrysin was reported to inhibit the accumulation of the MRP2 substrate CMFDA in Caco-2 cells in a dose-dependent manner.128 The maximal accumulation, which could also be achieved by specific MRP inhibitor-MK571, was observed in the presence of 250 μM chrysin. Interestingly, chrysin was also found to increase the expression of MRP2 5 fold in Caco-2 cells after long-term treatment.128 Over a time period of 48 h, the inhibition of transporter function overtook the enhanced expression of MRP2 by chrysin, resulting in an increased accumulation of topotecan.128 Therefore, chrysin seemed to play a dual role in regulating MRP2 in Caco-2 cells. In fact, the dual role of chrysin was also extended to other efflux transporters—P-gp and BCRP. Evidence from other investigators confirmed the inhibition of MRP2 by chrysin in Caco-2 cells.129 It was found that chrysin, genistein, quercetin and biochanin A (all at 50 μM) could increase transportation of ochratoxin A from the apical side to basolateral side. Enhanced ochratoxin A uptake by the above flavonoids was also observed in Caco-2 cells. Moreover, quercetin displayed concentration-dependent inhibition on ochratoxin A absorption.129 A later study further identified that major phase II metabolites of quercetin inhibited MRP2 function with a similar potency to quercetin itself.130 The results by using Sf9 inside-out vesicles revealed that glucuronides, especially 7-O-glucuronosyl-quercetin significantly increased the potential of quercetin to inhibit MRP2-mediated calcein transport. On the contrary, methylation at the 4′ position of quercetin resulted in a reduction of the potential to inhibit MRP2.130
Quantitative structure–function relationship has been built according to flavonoid-mediated inhibition of MRP2 by using MDCKII-MRP2 cells.131 A total 29 flavonoids with various structures were used in the study. It was revealed that robinetin and myricetin, both of which had an IC50 much lower than 50 μM (15 and 22 μM), respectively, were the two best inhibitors of MRP2 among all the tested flavonoids.131 Furthermore, the kinetic mechanism for robinetin to inhibit MRP2-mediated transportation of calcein was competitive. As a result, a flavonol B-ring pyrogallol group appeared to be an essential structure for inhibition of MRP2-mediated calcein efflux.131
4.3. Effect of flavonoids on BCRP
Breast cancer resistance protein (BCRP), which is encoded by the ABCG2 gene, consists of 655 amino acids with molecular weight of 72 kD.132 Unlike many other members from the ABC transporter family, BCRP is a “half ABC transporter” because it only contains one NBD and six TMDs.133 To function properly, BCRP must form homodimers or homooligomers.134 In addition to its physiological function, BCRP has a role in limiting oral bioavailability of drugs and drug transport across the blood–brain barrier, blood–testis barrier and the maternal–fetal barrier of selected substrates.135–137 Though BCRP was identified originally from cancer cells, it is broadly distributed in humans including kidney, liver and intestine.138 The substrates of BCRP include various compounds from endogenous to exogenous, which are generally organic anion or neutral substances, often time similar to the substrates of MRP2.Currently, the inhibition of BCRP by flavonoids was most extensively investigated among all the ABC transporters. A substantial amount of work has been done to study the interactions between flavonoids and BCRP. It was Zhang and coworkers who first identified chrysin and biochanin A as the most potent inhibitors of BCRP among all the 20 naturally occurring flavonoids.139 In fact, it was found that most tested flavonoids (e.g. apigenin, genistein, fisetin, kaempferol, hesteretin, naringenin and quercetin) could increase mitoxantrone accumulation in BCRP-overexpressing cells (MCF-7-MX100 and NCI-H460-MX20) at concentration of 50 μM, but none of the flavonoid glycosides (e.g.naringin and phloridzin) were effective.139 Specifically, the concentration-dependent enhancement of accumulation of mitoxantrone were observed for apigenin, biochanin A, chrysin, genistein and kaempferol.139 Furthermore, several combinations of multiple flavonoids showed an additive (inhibitory) effect on BCRP.140 For example, apigenin (A), biochanin A (B) and chrysin (C) actively inhibited BCRP-mediated transport in combinations of AB, BC and ABC with equal molar concentration of each individual constituent.140 The combinations were not limited to 3 flavonoids. The additive inhibition of BCRP could be obtained when 5 or 8 flavonoids were combined together.140 Recently, the pharmacokinetic and tissue distributions of mitoxantrone with or without co-administration of 5,7-dimethoxyflavone (5,7-DMF) or multiple flavonoids with low EC50 (7,8-benzoflavone, 5,6,7-trimethoxyflavone and 8-methylflavone) were studied in mice to evaluate the potentially additive or synergistic effect of multiple flavonoids on BCRP inhibition.141 It revealed that there was no significant change of pharmacokinetic parameters with or without flavonoids. However, significant changes in mitoxantrone distribution were observed in several tissues with co-administration of flavonoids. In the presence of 5,7-DMF, the AUC values of mitoxantrone were significantly increased in liver (94.5%), kidney (61.9%) and lung (18.4%); on the other hand, co-administration of multiple flavonoids resulted in increasing AUC values in heart (30.5%), liver (95.9%), kidney (63.3%), lung (30.7%) and muscle (33.8%).141 Therefore, inhibition of BCRP by either individual flavonoid or multiple flavonoids can occur both in vitro and in vivo. However, conclusions from in vitro studies were not always consistent with in vivo studies. Discrepancy was observed when biochanin A was used as inhibitor to treat MDCKII-BCRP or MDCKII-Bcrp cells or given to mice.142In vitro, accumulation of cellular mitoxantrone was enhanced with biochanin A at concentration of 2.5 or 25 μM in MDCK cells overexpressing human BCRP or murine Bcrp. In contrast to in vitro data, biochanin A administrated intravenously at 10 mg kg−1 had no impact on the plasma concentration of mitoxantrone and AUC values in most tissues (brain, heart, liver and lung).142 Thus, when extrapolating results of in vitro inhibition on BCRP by flavonoids to in vivo, we should pay particular attention to the fact the low bioavailability may severely limit their in vivo levels necessary to achieve observed inhibition effects (in vitro).
Unlike flavonoid aglycones, most flavonoid glycosides were reported to have little or no effect on BCRP-mediated drug resistance. Only a few flavonoid glycosides were shown to affect BCRP-mediated drug resistance. Naringenin-7-glucoside and luteolin-4-glucoside displayed moderate effects to reverse the resistance to SN-38 and mitoxantrone in BCRP-transduced K562 cells.143 However, the inhibition of BCRP by naringin might be controversial. Recently, experiments indicated that 6 isoflavonoid glucosides (daidzin, ononin, genistin, sissotrin, glycitin and coumestrin) had 10 to 100 fold lower inhibitory potency (against BCRP) than their corresponding aglycones.144 Interestingly, it was also found that the inhibitory potency of daidzein-7-glucuronide was 100 fold lower than daidzein while daidzein-4-sulfate had a inhibitory potency comparable to daidzein.144 It is very clear that there is a strong structure–inhibition relationship between BCRP and flavonoids. It appeared that flavonoids with following structural features might inhibit BCRP effectively: a hydroxyl group at position 5, double bond between position 2 and 3, and a methoxyl moiety at position 3 or 6.145 With regard to the inhibition mechanisms, interference with substrate-binding sites of BCRP might account for flavonoid inhibition of BCRP since quercetin and daidzein were able to stimulate vanadate-inhibitable ATPase activity in membranes prepared from bacteria expressing BCRP.146 In contrast to inhibition, expression of BCRP in Caco-2 cells can also be induced by flavonoids.147 Evidence indicated that quercetin (25 μM) could strongly induce the protein expression of BCRP in Caco-2 cells. At mRNA level, quercetin, chrysin and flavone had a pronounced induction of BCRP, while genistein showed no effect. The induction of BCRP was also believed to be via regulation of AhR (aryl hydrocarbon receptor).147
4.4. Effects on OATP and OAT
Compared to abundant studies related to interactions of flavonoids with ABC transporters, little is known about the interactions of flavonoids with transporters from the solute carrier family (SLC). Organic anion-transporting peptide (OATP, also called SLC21A) and organic anion transporter (OAT, also called SLC22A) are two subfamilies, which are reported to have an interaction with flavonoids or flavonoid conjugates.148–151Members of the OATP family mediate sodium-independent uptake of a broad spectrum of amphipathic organic anions, organic cations and conjugates. OATP2B1 and OATP1A2 are expressed in the intestine while OATP1B1 and OATP1B3 are expressed in liver.152,153 According to published papers, OATP1B1, OATP1B3 and OATP2B1 are localized on the basolateral membrane, while OATP1A2 is localized on the apical membrane.153 A large volume of evidence showed that various juices, which contained plenty of flavonoids, could inhibit the function of OATP. It was demonstrated that several citrus juice constituents (10 μM), including naringin, naringenin, quercetin, bergamottin and 6,7-dihydroxybergamottin, tangeretin and nobiletin, significantly inhibited the OATP2B1-mediated uptake of estrone-3-sulfate by about 20-60% in OATP2B1-expressing HEK293 cells.154 Another study also found that naringin (grapefruit) and hesperidin (orange) inhibit the OATP1A2-mediated transport of fexofenadine in vitro and in vivo.151 Further analysis of human studies showed that consumption of grapefruit juice concomitantly or 2 h before fexofenadine administration was linked with reduced oral fexofenadine plasma concentration without affecting protein expressions of OATP1A2 and P-gp in intestine.152 In OATP1B1-expressing HeLa cells, 20 naturally occurring flavonoids were tested for their impacts on uptake of [3H]dehydroepiandrosterone sulfate (DHEAS).149 Most flavonoids (e.g. biochanin A, genistein, and epigallocatechin-3-gallate) significantly inhibited DHEAS uptake in a concentration-dependent manner.149 Not surprisingly, flavonoid glycosides showed comparable, opposite or no effect on uptake of DHEAS in contrast to their corresponding flavonoid aglycones. Genistin could not inhibit the uptake but genistein could; rutin, on the other hand, showed activation of uptake, whereas quercetin had no effect on uptake.149 As the most potent inhibitor among 20 flavonoids, biochanin A was selected for in vitro kinetic study. The results suggested that biochanin A inhibited uptake of DHEAS in a noncompetitive way as it was not the substrate for OATP1B1.149
Members of OAT play an important role in the sodium-dependent renal uptake of organic anions. OAT1 and OAT3, which are highly expressed on the basolateral membrane of proximal tubules, are two relatively well-studied transporters within the OAT family.155 Their substrates are highly variable from endogenous metabolites to drugs and toxicants. At present, only a few studies have focused on the interaction of OAT with flavonoids. In one study, ellagic acid was identified to be a substrate and potent inhibitor of OAT1.156 In another study, naringenin, morin, silybin and quercetin were reported to inhibit both OAT1 and OAT3.157 Most recently, quercetin phase II conjugates (quercetin-3-O-glucuronide, 3′-O-glucuronide and 3′-O-sulfate) were found to potently inhibit OAT3-mediated transport of 5-carboxyfluorescein. In addition, quercetin-3′-O-sulfate could also strongly inhibited transport of p-aminohippuric acid, which is a substrate of OAT1.148
5. Clinical significance of flavonoid interaction with disposition machinery
Flavonoids can exert various biological and pharmacological effects that benefit humans. This can be achieved since humans can easily ingest large amounts of flavonoids from their daily dietary intake because of their ubiquitous distribution in vegetables, fruits, herbal supplements and beverages deriving from plants. A couple of clinical studies indicated that oral or intravenous administration of flavonoids, either as individual components (e.g. quercetin alone) or mixtures, could inhibit lymphocyte tyrosine kinase activity (resulting in antitumor effects) or prevent bone loss in late postmenopausal women (without a detectable hyperplasia effect on the endometrium as the side effects of many drugs do).158,159 These and other clinical study results suggest that some flavonoids can reach the effective concentrations in vivo at which their pharmacological activity is exhibited. Although there is extensive phase II metabolism mediated mainly by UGTs and SULTs, when multiple flavonoids are taken together, the bioavailability of one flavonoid might be improved. In an animal study, co-administration of quercetin and EGCG together with biochanin A significantly enhanced the bioavailability of biochanin A due to decrease of clearance by extending enterohepatic cycling.160 Therefore, we should pay more attention to flavonoid–flavonoid interactions in pharmacokinetic and efficacy studies in vivo.Drug–flavonoid interaction is another important issue, especially from a clinical point of view. In one instance, patients with long-term exposure to foods containing considerable amounts of flavonoids may experience changes in their physiological status including expression of drug metabolizing enzymes and transporters. On the other hand, the immediate inhibitory effects on drug metabolizing enzymes and transporters can also be observed when flavonoid-enriched edibles are consumed with medication. As a result, the pharmacokinetics of the medication is changed. For example, an aqueous solution of naringin that contained the same amount of the flavanone as 300 ml grapefruit juice decreased the bioavailability of co-administered fexofenadine.151 In another study, an intake of daidzein (200 mg twice daily) for ten consecutive days increased the bioavailability, maximal plasma concentration and elimination half-life of the bronchodilator theophylline in humans.161 Since flavonoids can interact with multiple drug metabolizing enzymes and transporters, which are involved in absorption, distribution, metabolism and excretion of drugs, drug–flavonoid interaction has become and will remain an important area for clinical research and studies.
6. Conclusion
In conclusion, the mutual interactions between flavonoids and drug-metabolizing enzymes and transporters are very complex and multifaceted. Flavonoids can be substrates of and/or chemical modulators of certain enzymes and transporters that will impact the disposition processes of various compounds including flavonoids themselves. The extensive metabolism of flavonoids by phase II enzymes and subsequent excretion of conjugates by the associated efflux transporters, especially those from the ABC transporter superfamily, explain why flavonoid aglycones are present at very low levels in the systemic circulation. Recent research further suggested that it was the interplay between phase II enzymes (e.g. UGTs and SULTs isoforms) and efflux transporters (e.g. MRP2 and BCRP) that drives the metabolic disposition and clearance processes.On the other hand, flavonoids can also modify expression or activity of those enzymes and transporters relevant for the interplay. For most flavonoids, these mean that they could inhibit the activity of phase II enzymes or transporters acutely; but induce the expression or activate the function of phase II enzymes or efflux transporters on a chronic basis. These interactions are often mediated via the action of various chemosensing mechanisms involving partners such as nucleic receptors and their co-regulators.
Therefore, mutual interactions between flavonoids and enzymes as well as efflux transporters are critical factors that should be taken into considerations when flavonoids are co-administered with medication. This is because pharmacokinetics of the medication can be altered. A better understanding of mutual interactions between disposition of flavonoids and elements responsible for their disposition will greatly help us to identify potential drug–flavonoid interactions and flavonoid–flavonoid interactions. In addition, the mutual interaction study can accelerate the process of building structure–metabolism or structure–activity relationships, allowing us to identify critical structural features that are not subjected to metabolism but may retain essential biological activity.
| ABC | ATP-binding cassette |
| UGT | Uridine-5′-diphosphate glucuronosyltransferases |
| SULT | Sulfotransferases |
| CYP450 | Cytochrome P450 |
| P-gp | P-glycoprotein |
| MRP | Multi-drug resistance protein |
| BCRP | Breast cancer resistance protein |
| GST | Glutathione-S-transferases |
| OATP | Organic anion-transporting peptide |
| OAT | Organic anion transporter |
| SLC | Solute carrier |
| DME | Drug metabolizing enzymes |
References
- O. K. Chun, S. J. Chung and W. O. Song, J Nutr, 2007, 137, 1244 CAS.
- J. H. de Vries, P. L. Janssen, P. C. Hollman, W. A. van Staveren and M. B. Katan, Cancer Lett., 1997, 114, 141 CrossRef CAS.
- M. G. Hertog, P. C. Hollman, M. B. Katan and D. Kromhout, Nutr. Cancer, 1993, 20, 21 CrossRef CAS.
- K. R. Narayana, M. S. Reddy, M. R. Chaluvadi and D. R. Krishna, Indian Journal of Pharmacology, 2001, 33, 2 CAS.
- G. Schroder, E. Wehinger, R. Lukacin, F. Wellmann, W. Seefelder, W. Schwab and J. Schroder, Phytochemistry, 2004, 65, 1085 CrossRef.
- Y. C. Wong, L. Zhang, G. Lin and Z. Zuo, Expert Opin. Drug Metab. Toxicol., 2009, 5, 1399 CrossRef CAS.
- J. B. a. W. C. A. Harborne, Nat. Prod. Rep., 1998, 15, 631 RSC.
- N. C. a. G. R. J. Veitch, Nat. Prod. Rep., 2011, 28, 1626 RSC.
- J. B. Harborne, The flavonoids: advances in research since 1986, Chapman & Hall/CRC, 1994 Search PubMed.
- C. Bosetti, L. Spertini, M. Parpinel, P. Gnagnarella, P. Lagiou, E. Negri, S. Franceschi, M. Montella, J. Peterson, J. Dwyer, A. Giacosa and C. La Vecchia, Cancer Epidemiol., Biomarkers Prev., 2005, 14, 805 CAS.
- D. Grassi, G. Desideri, G. Croce, S. Tiberti, A. Aggio and C. Ferri, Curr. Pharm. Des., 2009, 15, 1072 CrossRef CAS.
- B. N. Ames, M. K. Shigenaga and T. M. Hagen, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 7915 CrossRef CAS.
- T. Koga and M. Meydani, Am J Clin Nutr, 2001, 73, 941 CAS.
- T. Walle, Free Radical Biol. Med., 2004, 36, 829 CrossRef CAS.
- R. A. Walgren, J. T. Lin, R. K. Kinne and T. Walle, J Pharmacol Exp Ther, 2000, 294, 837 CAS.
- T. Walle, Y. Otake, U. K. Walle and F. A. Wilson, J Nutr, 2000, 130, 2658 CAS.
- A. J. Day, M. S. DuPont, S. Ridley, M. Rhodes, M. J. Rhodes, M. R. Morgan and G. Williamson, FEBS Lett., 1998, 436, 71 CrossRef CAS.
- A. J. Day, F. J. Canada, J. C. Diaz, P. A. Kroon, R. McLauchlan, C. B. Faulds, G. W. Plumb, M. R. Morgan and G. Williamson, FEBS Lett., 2000, 468, 166 CrossRef CAS.
- Y. Liu, Y. Liu, Y. Dai, L. Xun and M. Hu, J. Altern. Complementary Med., 2003, 9, 631 CrossRef.
- Y. Liu and M. Hu, Drug Metab. Dispos., 2002, 30, 370 CrossRef CAS.
- A. J. Day, J. M. Gee, M. S. DuPont, I. T. Johnson and G. Williamson, Biochem. Pharmacol., 2003, 65, 1199 CrossRef CAS.
- K. Nemeth, G. W. Plumb, J. G. Berrin, N. Juge, R. Jacob, H. Y. Naim, G. Williamson, D. M. Swallow and P. A. Kroon, Eur. J. Nutr., 2003, 42, 29 CrossRef CAS.
- J. Chen, H. Lin and M. Hu, J. Pharmacol. Exp. Ther., 2003, 304, 1228 CrossRef CAS.
- A. Gradolatto, M. C. Canivenc-Lavier, J. P. Basly, M. H. Siess and C. Teyssier, Drug Metab. Dispos., 2004, 32, 58 CrossRef CAS.
- C. Manach and J. L. Donovan, Free Radical Res., 2004, 38, 771 CrossRef CAS.
- W. Zhu, H. Xu, S. W. Wang and M. Hu, AAPS J., 2010, 12, 525 CrossRef CAS.
- S. R. Miranda, J. K. Lee, K. L. Brouwer, Z. Wen, P. C. Smith and R. L. Hawke, Drug Metab. Dispos., 2008, 36, 2219 CrossRef CAS.
- K. D. Setchell, N. M. Brown, P. Desai, L. Zimmer-Nechemias, B. E. Wolfe, W. T. Brashear, A. S. Kirschner, A. Cassidy and J. E. Heubi, J Nutr, 2001, 131, 1362S CAS.
- A. Crozier, D. Del Rio and M. N. Clifford, Mol. Aspects Med., 2010, 31, 446 CrossRef CAS.
- M. Hu, Mol. Pharmaceutics, 2007, 4, 803 CrossRef CAS.
- A. Crozier, I. B. Jaganath and M. N. Clifford, Nat. Prod. Rep., 2009, 26, 1001 RSC.
- R. Cermak, Expert Opin. Drug Metab. Toxicol., 2008, 4, 17 CrossRef CAS.
- M. E. Morris and S. Zhang, Life Sci., 2006, 78, 2116 CrossRef CAS.
- A. I. Alvarez, R. Real, M. Perez, G. Mendoza, J. G. Prieto and G. Merino, J Pharm Sci, 2010, 99, 598 CAS.
- Y. J. Moon, X. Wang and M. E. Morris, Toxicol. in Vitro, 2006, 20, 187 CrossRef CAS.
- P. Hodek, P. Trefil and M. Stiborova, Chem.-Biol. Interact., 2002, 139, 1 CrossRef CAS.
- R. Z. Harris, G. R. Jang and S. Tsunoda, Clin. Pharmacokinet., 2003, 42, 1071 CrossRef CAS.
- Y. Otake and T. Walle, Drug Metab. Dispos., 2002, 30, 103 CrossRef.
- U. K. Walle and T. Walle, Drug Metab. Dispos., 2007, 35, 1985 CrossRef CAS.
- Y. Otake, F. Hsieh and T. Walle, Drug Metab. Dispos., 2002, 30, 576 CrossRef CAS.
- S. Meng, B. Wu, R. Singh, T. Yin, J. K. Morrow, S. Zhang and M. Hu, Mol. Pharmaceutics, 2012, 9, 862 CrossRef CAS.
- L. Tang, L. Ye, R. Singh, B. Wu, C. Lv, J. Zhao, Z. Liu and M. Hu, Mol. Pharmaceutics, 2010, 7, 664 CrossRef CAS.
- B. Wu, S. Basu, S. Meng, X. Wang and M. Hu, Curr. Drug Metab., 2011, 12, 900 CrossRef CAS.
- L. Tang, R. Singh, Z. Liu and M. Hu, Mol. Pharmaceutics, 2009, 6, 1466 CrossRef CAS.
- R. H. Tukey and C. P. Strassburg, Annu. Rev. Pharmacol., 2000, 40, 581 CrossRef CAS.
- T. B. Joseph, S. W. Wang, X. Liu, K. H. Kulkarni, J. Wang, H. Xu and M. Hu, Mol. Pharmaceutics, 2007, 4, 883 CrossRef CAS.
- P. I. Mackenzie, K. W. Bock, B. Burchell, C. Guillemette, S. Ikushiro, T. Iyanagi, J. O. Miners, I. S. Owens and D. W. Nebert, Pharmacogenet. Genomics, 2005, 15, 677 CrossRef CAS.
- A. Nakamura, M. Nakajima, H. Yamanaka, R. Fujiwara and T. Yokoi, Drug Metab. Dispos., 2008, 36, 1461 CrossRef CAS.
- R. H. Lewinsky, P. A. Smith and P. I. Mackenzie, Xenobiotica, 2005, 35, 117 CrossRef CAS.
- Z. Cheng, A. Radominska-Pandya and T. R. Tephly, Drug Metab Dispos, 1999, 27, 1165 CAS.
- Y. Chen, S. Xie, S. Chen and S. Zeng, Biochem. Pharmacol., 2008, 76, 416 CrossRef CAS.
- M. G. Boersma, H. van der Woude, J. Bogaards, S. Boeren, J. Vervoort, N. H. Cnubben, M. L. van Iersel, P. J. van Bladeren and I. M. Rietjens, Chem. Res. Toxicol., 2002, 15, 662 CrossRef CAS.
- A. Galijatovic, Y. Otake, U. K. Walle and T. Walle, Xenobiotica, 1999, 29, 1241 CrossRef CAS.
- H. S. Lee, H. Y. Ji, E. J. Park and S. Y. Kim, Xenobiotica, 2007, 37, 803 CrossRef CAS.
- D. Turgeon, J. S. Carrier, S. Chouinard and A. Belanger, Drug Metab. Dispos., 2003, 31, 670 CrossRef CAS.
- S. Xie, L. You and S. Zeng, Pharmazie, 2007, 62, 625 CAS.
- K. M. Janisch, G. Williamson, P. Needs and G. W. Plumb, Free Radical Res., 2004, 38, 877 CrossRef CAS.
- N. Gamage, A. Barnett, N. Hempel, R. G. Duggleby, K. F. Windmill, J. L. Martin and M. E. McManus, Toxicol. Sci., 2006, 90, 5 CrossRef CAS.
- K. Nagata and Y. Yamazoe, Annu. Rev. Pharmacol., 2000, 40, 159 CrossRef CAS.
- D. Ung and S. Nagar, Drug Metab. Dispos., 2007, 35, 740 CrossRef CAS.
- S. Nowell and C. N. Falany, Oncogene, 2006, 25, 1673 CrossRef CAS.
- J. B. Vaidyanathan and T. Walle, Drug Metab. Dispos., 2002, 30, 897 CrossRef CAS.
- C. Huang, Y. Chen, T. Zhou and G. Chen, Xenobiotica, 2009, 39, 312 CrossRef CAS.
- M. Hu, J. Chen and H. Lin, J. Pharmacol. Exp. Ther., 2003, 307, 314 CrossRef CAS.
- H. Sun, L. Zhang, E. C. Chow, G. Lin, Z. Zuo and K. S. Pang, J. Pharmacol. Exp. Ther., 2008, 326, 117 CrossRef CAS.
- W. Meinl, B Ebert, H. Glatt and A. Lampen, Drug Metab. Dispos., 2008, 36, 276 CrossRef CAS.
- K. J. Linton, Physiology, 2007, 22, 122 CrossRef CAS.
- M. Nait Chabane, A. Al Ahmad, J. Peluso, C. D. Muller and G. Ubeaud, J. Pharm. Pharmacol., 2009, 61, 1473 CrossRef.
- G. Williamson, I. Aeberli, L. Miguet, Z. Zhang, M. B. Sanchez, V. Crespy, D. Barron, P. Needs, P. A. Kroon, H. Glavinas, P. Krajcsi and M. Grigorov, Drug Metab. Dispos., 2007, 35, 1262 CrossRef CAS.
- W. Brand, P. A. van der Wel, M. J. Rein, D. Barron, G. Williamson, P. J. van Bladeren and I. M. Rietjens, Drug Metab. Dispos., 2008, 36, 1794 CrossRef CAS.
- G. An, J. Gallegos and M. E. Morris, Drug Metab. Dispos., 2011, 39, 426 CrossRef CAS.
- K. van de Wetering, W. Feddema, J. B. Helms, J. F. Brouwers and P. Borst, Gastroenterology, 2009, 137, 1725 CrossRef CAS.
- X. Jia, J. Chen, H. Lin and M. Hu, J. Pharmacol. Exp. Ther., 2004, 310, 1103 CrossRef CAS.
- W. Jiang, B. Xu, B. Wu, R. Yu and M. Hu, Drug Metab Dispos, 2011 Search PubMed.
- Y. Zhang and G. B. Gordon, Mol Cancer Ther, 2004, 3, 885 CAS.
- B. L. Urquhart, R. G. Tirona and R. B. Kim, J. Clin. Pharmacol., 2007, 47, 566 CrossRef CAS.
- X. Y. Sun, C. A. Plouzek, J. P. Henry, T. T. Wang and J. M. Phang, Cancer Res, 1998, 58, 2379 CAS.
- T. Walle, Y. Otake, A. Galijatovic, J. K. Ritter and U. K. Walle, Drug Metab Dispos, 2000, 28, 1077 CAS.
- A. Galijatovic, U. K. Walle and T. Walle, Pharm. Res., 2000, 17, 21 CrossRef CAS.
- A. Galijatovic, Y. Otake, U. K. Walle and T. Walle, Pharm. Res., 2001, 18, 374 CrossRef CAS.
- U. K. Walle and T. Walle, Drug Metab. Dispos., 2002, 30, 564 CrossRef CAS.
- M. F. Yueh and R. H. Tukey, J. Biol. Chem., 2007, 282, 8749 CrossRef CAS.
- J. A. Williams, B. J. Ring, V. E. Cantrell, K. Campanale, D. R. Jones, S. D. Hall and S. A. Wrighton, Drug Metab. Dispos., 2002, 30, 1266 CrossRef CAS.
- R. Venkataramanan, V. Ramachandran, B. J. Komoroski, S. Zhang, P. L. Schiff and S. C. Strom, Drug Metab Dispos, 2000, 28, 1270 CAS.
- V. Svehlikova, S. Wang, J. Jakubikova, G. Williamson, R. Mithen and Y. Bao, Carcinogenesis, 2004, 25, 1629 CrossRef CAS.
- K. W. Bock, T. Eckle, M. Ouzzine and S. Fournel-Gigleux, Biochem. Pharmacol., 2000, 59, 467 CrossRef CAS.
- N. Petri, C. Tannergren, B. Holst, F. A. Mellon, Y. Bao, G. W. Plumb, J. Bacon, K. A. O'Leary, P. A. Kroon, L. Knutson, P. Forsell, T. Eriksson, H. Lennernas and G. Williamson, Drug Metab. Dispos., 2003, 31, 805 CrossRef CAS.
- E. M. van der Logt, H. M. Roelofs, F. M. Nagengast and W. H. Peters, Carcinogenesis, 2003, 24, 1651 CrossRef CAS.
- M. C. Canivenc-Lavier, M. F. Vernevaut, M. Totis, M. H. Siess, J. Magdalou and M. Suschetet, Toxicology, 1996, 114, 19 CrossRef CAS.
- L. C. Appelt and M. M. Reicks, J Nutr, 1999, 129, 1820 CAS.
- Y. F. Ueng, C. C. Shyu, Y. L. Lin, S. S. Park, J. F. Liao and C. F. Chen, Life Sci., 2000, 67, 2189 CrossRef CAS.
- H. Nakano, K. Ogura, E. Takahashi, T. Harada, T. Nishiyama, K. Muro, A. Hiratsuka, S. Kadota and T. Watabe, Drug Metab. Pharmacokinet., 2004, 19, 216 CrossRef CAS.
- E. A. Eaton, U. K. Walle, A. J. Lewis, T. Hudson, A. A. Wilson and T. Walle, Drug Metab Dispos, 1996, 24, 232 CAS.
- T. Walle, E. A. Eaton and U. K. Walle, Biochem. Pharmacol., 1995, 50, 731 CrossRef CAS.
- R. M. Harris, D. M. Wood, L. Bottomley, S. Blagg, K. Owen, P. J. Hughes, R. H. Waring and C. J. Kirk, J. Clin. Endocrinol. Metab., 2004, 89, 1779 CrossRef CAS.
- R. M. Harris and R. H. Waring, Curr. Drug Metab., 2008, 9, 269 CrossRef CAS.
- R. H. Waring, S. Ayers, A. J. Gescher, H. R. Glatt, W. Meinl, P. Jarratt, C. J. Kirk, T. Pettitt, D. Rea and R. M. Harris, J. Steroid Biochem. Mol. Biol., 2008, 108, 213 CrossRef CAS.
- Y. Otake, A. L. Nolan, U. K. Walle and T. Walle, J. Steroid Biochem. Mol. Biol., 2000, 73, 265 CrossRef CAS.
- Y. Chen, C. Huang, T. Zhou and G. Chen, Basic Clin. Pharmacol. Toxicol., 2008, 103, 553 CrossRef CAS.
- Y. Chen, C. Huang, T. Zhou, S. Zhang and G. Chen, J. Biochem. Mol. Toxicol., 2010, 24, 102 CrossRef CAS.
- W. H. Peters, L. Kock, F. M. Nagengast and P. G. Kremers, Gut, 1991, 32, 408 CrossRef CAS.
- P. J. Ansell, C. Espinosa-Nicholas, E. M. Curran, B. M. Judy, B. J. Philips, M. Hannink and D. B. Lubahn, Endocrinology, 2004, 145, 311 CrossRef CAS.
- B. S. Patill, J. S. Brodbelt, E. G. Miller, N. D. Turner, in Potential Health Benefits of Citrus, ed. B. S. Patil, N. D. Turner , E. G. Miller and J. S. Brodbelt, 2006, vol. 936 Search PubMed.
- A. E. Mitchell, S. A. Burns and J. L. Rudolf, Arch. Toxicol., 2007, 81, 777 CrossRef CAS.
- T. Hofmann, U. Liegibel, P. Winterhalter, A. Bub, G. Rechkemmer and B. L. Pool-Zobel, Mol. Nutr. Food Res., 2006, 50, 1191 CAS.
- M. Dean, The human ATP-binding cassette (ABC) transporter superfamily, National center for biotechnology information, 2002 Search PubMed.
- M. M. Gottesman and I. Pastan, Annu. Rev. Biochem., 1993, 62, 385 CrossRef CAS.
- U. A. Germann, I. Pastan and M. M. Gottesman, Seminars in Cell Biology, 1993, 4, 63 CrossRef CAS.
- J. Jodoin, M. Demeule and R. Beliveau, Biochim. Biophys. Acta, Mol. Cell Res., 2002, 1542, 149 CrossRef CAS.
- E. J. Wang, M. Barecki-Roach and W. W. Johnson, Biochem. Biophys. Res. Commun., 2002, 297, 412 CrossRef CAS.
- S. Zhou, L. Y. Lim and B. Chowbay, Drug Metab. Rev., 2004, 36, 57 CrossRef CAS.
- K. S. Lown, D. G. Bailey, R. J. Fontana, S. K. Janardan, C. H. Adair, L. A. Fortlage, M. B. Brown, W. Guo and P. B. Watkins, J. Clin. Invest., 1997, 99, 2545 CrossRef CAS.
- V. A. Eagling, L. Profit and D. J. Back, Br. J. Clin. Pharmacol., 1999, 48, 543 CrossRef CAS.
- A. Soldner, U. Christians, M. Susanto, V. J. Wacher, J. A. Silverman and L. Z. Benet, Pharm. Res., 1999, 16, 478 CrossRef CAS.
- Y. Mitsunaga, H. Takanaga, H. Matsuo, M. Naito, T. Tsuruo, H. Ohtani and Y. Sawada, Eur. J. Pharmacol., 2000, 395, 193 CrossRef CAS.
- N. Romiti, G. Tramonti, A. Donati and E. Chieli, Life Sci., 2004, 76, 293 CrossRef CAS.
- A. F. Castro and G. A. Altenberg, Biochem. Pharmacol., 1997, 53, 89 CrossRef.
- X. L. Liu, H. W. Tee and M. L. Go, Bioorg. Med. Chem., 2008, 16, 171 CrossRef CAS.
- R. Hayeshi, C. Masimirembwa, S. Mukanganyama and A. L. Ungell, Eur. J. Pharm. Sci., 2006, 29, 70 CrossRef CAS.
- T. Ikegawa, H. Ohtani, N. Koyabu, M. Juichi, Y. Iwase, C. Ito, H. Furukawa, M. Naito, T. Tsuruo and Y. Sawada, Cancer Lett., 2002, 177, 89 CrossRef CAS.
- M. Hadjeri, M. Barbier, X. Ronot, A. M. Mariotte, A. Boumendjel and J. Boutonnat, J. Med. Chem., 2003, 46, 2125 CrossRef CAS.
- J. Boccard, F. Bajot, A. Di Pietro, S. Rudaz, A. Boumendjel, E. Nicolle and P. A. Carrupt, Eur. J. Pharm. Sci., 2009, 36, 254 CrossRef CAS.
- E. M. Leslie, R. G. Deeley and S. P. Cole, Toxicology, 2001, 167, 3 CrossRef CAS.
- E. M. Leslie, R. G. Deeley and S. P. Cole, Toxicol. Appl. Pharmacol., 2005, 204, 216 CrossRef CAS.
- C. H. Versantvoort, H. J. Broxterman, J. Lankelma, N. Feller and H. M. Pinedo, Biochem. Pharmacol., 1994, 48, 1129 CrossRef CAS.
- S. Paul, M. G. Belinsky, H. Shen and G. D. Kruh, Biochemistry, 1996, 35, 13647 CrossRef CAS.
- J. H. Hooijberg, H. J. Broxterman, M. Heijn, D. L. Fles, J. Lankelma and H. M. Pinedo, FEBS Lett., 1997, 413, 344 CrossRef CAS.
- M. Schumacher, A. Hautzinger, A. Rossmann, S. Holzhauser, D. Popovic, A. Hertrampf, S. Kuntz, M. Boll and U. Wenzel, Biochem. Pharmacol., 2010, 80, 471 CrossRef CAS.
- T. Sergent, S. Garsou, A. Schaut, S. De Saeger, L. Pussemier, C. Van Peteghem, Y. Larondelle and Y. J. Schneider, Toxicol. Lett., 2005, 159, 60 CrossRef CAS.
- J. J. van Zanden, H. van der Woude, J. Vaessen, M. Usta, H. M. Wortelboer, N. H. Cnubben and I. M. Rietjens, Biochem. Pharmacol., 2007, 74, 345 CrossRef CAS.
- J. J. van Zanden, H. M. Wortelboer, S. Bijlsma, A. Punt, M. Usta, P. J. Bladeren, I. M. Rietjens and N. H. Cnubben, Biochem. Pharmacol., 2005, 69, 699 CrossRef CAS.
- L. A. Doyle, W. Yang, L. V. Abruzzo, T. Krogmann, Y. Gao, A. K. Rishi and D. D. Ross, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15665 CrossRef CAS.
- R. Allikmets, L. M. Schriml, A. Hutchinson, V. Romano-Spica and M. Dean, Cancer Res, 1998, 58, 5337 CAS.
- C. Ozvegy, T. Litman, G. Szakacs, Z. Nagy, S. Bates, A. Varadi and B. Sarkadi, Biochem. Biophys. Res. Commun., 2001, 285, 111 CrossRef CAS.
- K. M. Giacomini, S. M. Huang, D. J. Tweedie, L. Z. Benet, K. L. Brouwer, X. Chu, A. Dahlin, R. Evers, V. Fischer, K. M. Hillgren, K. A. Hoffmaster, T. Ishikawa, D. Keppler, R. B. Kim, C. A. Lee, M. Niemi, J. W. Polli, Y. Sugiyama, P. W. Swaan, J. A. Ware, S. H. Wright, S. W. Yee, M. J. Zamek-Gliszczynski and L. Zhang, Nat. Rev. Drug Discovery, 2010, 9, 215 CrossRef CAS.
- A. E. van Herwaarden and A. H. Schinkel, Trends Pharmacol. Sci., 2006, 27, 10 CrossRef CAS.
- M. L. Vlaming, J. S. Lagas and A. H. Schinkel, Adv. Drug Delivery Rev., 2009, 61, 14 CrossRef CAS.
- R. W. Robey, K. K. To, O. Polgar, M. Dohse, P. Fetsch, M. Dean and S. E. Bates, Adv. Drug Delivery Rev., 2009, 61, 3 CrossRef CAS.
- S. Zhang, X. Yang and M. E. Morris, Mol. Pharmacol., 2004, 65, 1208 CrossRef CAS.
- S. Zhang, X. Yang and M. E. Morris, Pharm. Res., 2004, 21, 1263 CrossRef CAS.
- G. An, F. Wu and M. E. Morris, Pharm. Res., 2011, 28, 1090 CrossRef CAS.
- G. An and M. E. Morris, Biopharm Drug Dispos, 2010, 31, 340 CAS.
- Y. Imai, S. Tsukahara, S. Asada and Y. Sugimoto, Cancer Res., 2004, 64, 4346 CrossRef CAS.
- H. Tamaki, H. Satoh, S. Hori, H. Ohtani and Y. Sawada, Drug Metab. Pharmacokinet., 2010, 25, 170 CrossRef CAS.
- A. Pick, H. Muller, R. Mayer, B. Haenisch, I. K. Pajeva, M. Weigt, H. Bonisch, C. E. Muller and M. Wiese, Bioorg. Med. Chem., 2011, 19, 2090 CrossRef CAS.
- H. C. Cooray, T. Janvilisri, H. W. van Veen, S. B. Hladky and M. A. Barrand, Biochem. Biophys. Res. Commun., 2004, 317, 269 CrossRef CAS.
- B. Ebert, A. Seidel and A. Lampen, Toxicol. Sci., 2007, 96, 227 CrossRef CAS.
- C. C. Wong, N. P. Botting, C. Orfila, N. Al-Maharik and G. Williamson, Biochem. Pharmacol., 2011, 81, 942 CrossRef CAS.
- X. Wang, A. W. Wolkoff and M. E. Morris, Drug Metab. Dispos., 2005, 33, 1666 CrossRef CAS.
- G. K. Dresser, D. G. Bailey, B. F. Leake, U. I. Schwarz, P. A. Dawson, D. J. Freeman and R. B. Kim, Clin. Pharmacol. Ther., 2002, 71, 11 CrossRef CAS.
- D. G. Bailey, G. K. Dresser, B. F. Leake and R. B. Kim, Clin. Pharmacol. Ther., 2007, 81, 495 CrossRef CAS.
- H. Glaeser, D. G. Bailey, G. K. Dresser, J. C. Gregor, U. I. Schwarz, J. S. McGrath, E. Jolicoeur, W. Lee, B. F. Leake, R. G. Tirona and R. B. Kim, Clin. Pharmacol. Ther., 2007, 81, 362 CrossRef CAS.
- B. Hagenbuch and P. J. Meier, Pfluegers Arch., 2004, 447, 653 CrossRef CAS.
- H. Satoh, F. Yamashita, M. Tsujimoto, H. Murakami, N. Koyabu, H. Ohtani and Y. Sawada, Drug Metab. Dispos., 2005, 33, 518 CrossRef CAS.
- R. Kojima, T. Sekine, M. Kawachi, S. H. Cha, Y. Suzuki and H. Endou, J Am Soc Nephrol, 2002, 13, 848 CAS.
- A. C. Whitley, D. H. Sweet and T. Walle, Drug Metab. Dispos., 2005, 33, 1097 CrossRef CAS.
- S. S. Hong, K. Seo, S. C. Lim and H. K. Han, Pharmacol. Res., 2007, 56, 468 CrossRef CAS.
- D. R. Ferry, A. Smith, J. Malkhandi, D. W. Fyfe, P. G. deTakats, D. Anderson, J. Baker and D. J. Kerr, Clin Cancer Res, 1996, 2, 659 CAS.
- G. Zhang, L. Qin and Y. Shi, J. Bone Miner. Res., 2007, 22, 1072 CrossRef CAS.
- Y. J. Moon and M. E. Morris, Mol. Pharmaceutics, 2007, 4, 865 CrossRef CAS.
- W. X. Peng, H. D. Li and H. H. Zhou, Eur. J. Clin. Pharmacol., 2003, 59, 237 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |