Interdisciplinary challenges and promising theranostic effects of nanoscience in Alzheimer's disease
Sophie
Laurent
a,
Mohammad Reza
Ejtehadi
b,
Meisam
Rezaei
b,
Patrick G
Kehoe
*c and
Morteza
Mahmoudi
*de
aDepartment of General, Organic, and Biomedical Chemistry, NMR and Molecular Imaging Laboratory, University of Mons, Avenue Maistriau, 19, B-7000, Mons, Belgium
bDepartment of Physics, Sharif University of Technology, Tehran, 11155-8639, Iran
cDementia Research Group, John James Laboratories, Frenchay Hospital, Institute of Clinical Neurosciences, School of Clinical Sciences, University of Bristol, BS16 1LE, Bristol, England, UK. E-mail: Patrick.Kehoe@bristol.ac.uk
dNational Cell Bank, Pasteur Institute of Iran, Tehran, Iran
eNanotechnology Research Centre, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran. E-mail: Mahmoudi@biospion.com; Web: www.biospion.com
First published on 9th March 2012
Abstract
During the last decade, reports show that the incidence and prevalence of Alzheimer's disease (AD) and other dementias have significantly increased. AD poses an enormous escalating threat to health services and resources. Early diagnosis of AD is recognized as one of the major challenges and primary aims in scientific communities. With the arrival of nanoscience and nanotechnology to medicine, hopes for early diagnosis and treatment of AD have considerably increased. To this end, nanobioresearchers are focused on three major areas consisting of early detection and recognition, biological markers and diagnosis, and pharmacotherapy. Several efforts are in progress for the discovery of new nanodrugs with a sufficient effort on the fast therapy of AD. In this review, a comprehensive overview of the conventional theories together with new conflicts and challenges on the major causes of AD is provided. In addition, the opportunities and challenges of nanoscience and nanotechnology as theranostic agents for AD are discussed in detail.
 Sophie Laurent | Dr Sophie Laurent was born in 1967. Her studies were performed at the University of Mons-Hainaut (Belgium) where she received her PhD in Chemistry in 1993. She then joined Prof. R. N. Muller's team and was involved in the development (synthesis and physicochemical characterization) of paramagnetic Gd complexes and superparamagnetic iron oxide nanoparticles as contrast agents for MRI. She is currently working on the vectorization of contrast agents for molecular imaging. She is a lecturer and co-author of around 120 publications and more than 200 communications at international meetings. |
 Mohammad Reza Ejtehadi | Dr Mohammad Reza Ejtehadi obtained his PhD in Physics from Sharif University of Technology, Tehran, in 1998. He has worked in the Max-Planck Institute for Polymer Research in Mainz, University of British Columbia in Vancouver, and in 2004 he joined Sharif University of Technology, where he has held an associate professor position since 2008. He applies statistical physics and simulation to various problems in soft matter and biological systems. He has also been elected a board member of the Physics Society of Iran since 2009. |
 Meisam Rezaei | Meisam Rezaei was born in 1987. He started his Msc in the Sharif University of Technology under the supervision of Dr Mohammad Reza Ejtehadi in the Soft Condensed Matter group (SCM) (2009–2012). He joined the laboratory of Nano-Bio Interactions at the National Cell Bank, Pasteur Institute of Iran as a senior research assistant under the supervision of Dr Morteza Mahmoudi (2010–present). Since his Msc studies he has been working on all-atom simulation and studying biomolecules especially DNA and proteins. Also, he is researching into protein and nanoparticle interactions. |
 Patrick G Kehoe | Dr Kehoe has over 16 years experience on Alzheimer's disease, related dementias and mental health research. His initial interests in Alzheimer's disease developed from his undergraduate degree studies at University College Dublin, Ireland where he double majored in Pharmacology and Molecular Genetics and subsequently from undertaking his PhD on “Molecular Genetics Studies of Alzheimer's disease” at the University of Cardiff, Wales, United Kingdom. In the course of his PhD studies he developed one of his main areas of research interest, the molecular involvement of the Renin Angiotensin System Pathway in Alzheimer's disease and molecular mechanisms underlying the well established links between cardiovascular risk factors and disease, their treatments and Alzheimer's disease. |
 Morteza Mahmoudi | Dr Morteza Mahmoudi obtained his PhD in 2009 from Sharif University of Technology with specialisation on the cytotoxicity of superparamagnetic iron oxide nanoparticles (SPIONs). He has received many awards such as the Shahid Chamran Award (National Endowment for the Elite; 2011), Distinguished Researcher at Pasteur Institute of Iran (2010), the Dr Mojtahedi Innovation Award for Distinguished Innovation in Research and Education at Sharif University of Technology (2010) and Kharazmi Young Festival Award (2009). His current research involves magic SPIONs for simultaneous diagnosis and therapeutic applications (http://www.biospion.com). |
1. Introduction
Alzheimer's disease (AD) is the most common form of dementia. Dementia refers to a group of progressive diseases that affect memory and cognitive function, which over time result in a complete loss of independent living and the ability to care for oneself. According to the 2009 World Alzheimer Report,1 the global prevalence of dementia is predicted to double every 20 years, rising from 35 million in 2010 to 115.4 million in 2050. This anticipated unchecked growth is based on the current assumption that no cure for AD will be found. Thus, the quest to develop a cure or a long-term effective treatment for AD is an urgent healthcare priority.There has been significant progress in defining the main pathology of AD but, whilst the progression is better understood, the underlying cause of AD remains elusive. One of the most widely accepted characteristic hallmarks of AD is the accumulation/aggregation of insoluble amyloid beta (Aβ) peptides in the brain. These Aβs are produced in a soluble form from the amyloid precursor protein (APP) and form aggregations of various sizes and levels of solubility. Over time they fibrillize, which is the process by which misfolded (i.e. toxic folding) proteins form large linear aggregates or amyloid.2 These in turn form insoluble protein fibrils that go on to form senile plaques and contribute to neuronal cell death.
Until recently, two of the molecular-hypotheses believed to explain the pathology of AD are those that included the misfolding of some brain proteins/peptides (e.g. Aβ) and the effects of multivalent cations (e.g. Zn2+, Cu2+, Mn2+, Fe2+, Fe3+, and Al3+) in modulating the formation of plaques and tangles in the brain.3,4 It is noteworthy that, in addition to elderly people (i.e. where the majority of sporadic (non-genetic) forms of AD cases occur), younger people (e.g. 30 years and above) can also develop rare forms of AD due to autosomal dominant inherited mutations which give rise to what is commonly referred to as familial AD (see below). Interestingly, the genes causing familial AD are also involved in the dysregulation of calcium signaling pathways, altering intracellular levels of Ca2+.5 Furthermore, a large array of zinc metallo-proteases appear to have a pivotal role in either the production or degradation of Aβ.6
During the last decade, nanoscience and nanotechnologies have become widely considered as promising tools for the diagnosis and treatment of AD. Due to their ultra-small size, nanoparticles have distinct properties compared with the bulk form of the same materials.7 Improved understanding of these properties are rapidly informing and revolutionizing many areas of medicine and technology. One of the major effects of nanoparticles in the treatment of AD is their potential ability to inhibit the protein fibrillization process, which is an important step in the pathogenesis of AD.8 Recent results have confirmed not only that molecular events could cause the fibrillization but also that a number of intermediate oligomeric states of different association numbers and structures could interfere with the fibrillization process.8 The use of some agents that interfere with these processes can help to elucidate the molecular mechanism of fibril formation whilst some strategies could also have therapeutic potential for the treatment of amyloidosis. In this case, nanoparticles can have a significant effect on the fibrillization of proteins; for instance, the presence of nanoparticles could lead to a shortening of the lag phase for nucleation of the fibrillization process.8
In this review, a comprehensive overview of the current developments on AD will be given. In addition, we will also cover major reported effects of nanoparticles on the fibrillation process.
2. General background of Alzheimer's disease (AD)
Alzheimer's disease (AD) was first described in 1906 after examining the brain of one patient who had previously presented with clinical symptoms now commonly associated with dementia. These include memory loss (recent events in the first instance) and problems in communication, behavior, and orientation in space and time. AD is a progressive and incurable neurodegenerative disease, for which age is the major risk factor. It was previously thought that industrialized countries, where longevity of the population increases with improving living conditions, also suffered a rise of senile dementia, of which as much of 50% was AD. According to the USA national statistics from the Aging, Demographics and Memory Study (ADAMS),9 there were well over 2 million people over the age of 71 afflicted with the disease. This number is projected to reach 7.7 million people in 2030 and to increase to 16 million people in 2050 according to some estimates.10 In terms of mortality, AD was the fifth leading cause of death in the United States in 2006 for those older than 65 years old.11 However, with the rise of developing countries, these too are now seeing increases in the incidence and prevalence of the disease and so what was previously seen as a disease of the developed world is now more recognized as a global problem.12 Several of the current treatments only provide temporary alleviation of the disease symptoms but some also induce undesirable side effects. In the absence of a cure, AD will continue to serve as a current and future major public health problem.2.1. Clinical aspects of AD
The clinical diagnosis of AD is difficult and variable between primary and specialist centres. Cognitive tests of limited sensitivity are used although there are now promising developments in brain imaging and the use of cerebrospinal fluid biomarkers intended to help inform more accurate diagnoses. At present the most common imaging techniques used to support diagnosis are based on magnetic resonance imaging (MRI) or nuclear medicine (positron emission tomography (PET)). AD has many symptoms that can be sub-grouped into three main domains.13First, there is cognitive impairment that causes: loss of short-term memory and learning abilities; the gradual deterioration of language that leads to difficulty in finding words and names and, eventually, evolving into an aphasia; disruption of visual and spatial acuity that result in disorientation in the environment and difficulties in copying figures; inability to perform motor tasks despite motor functions being intact (apraxia); and inability to recognize people or objects despite an intact sensory function.
Second, are neuropsychiatric and behavioral disturbances leading to personality disorders, hallucinations, delusions or misidentifications. Apathy appears early and results in reduced interest and involvement. Agitation increases with the progression of the disease and is often a factor that contributes to the institutionalization of patients. Half of the patients present depression.
Third, are difficulties in activities of daily life, such as money management, conduct, using the telephone and, later, the capacity to dress, eat or bathe.
Life expectancy following a diagnosis of dementia is usually 7 to 10 years and often, particularly in the more advanced stages become very susceptible to infections such as pneumonia. The terminal stage leads irrevocably to a coma and then death.
As yet, a confirmed diagnosis of AD is only possible following autopsy by histological examination of the brain. Key features along with a prior clinical history of memory impairment and other features as described are the detection of an excess of amyloid plaques (AP) and neurofibrillary tangles (NFT) and atrophy of the cerebral cortex and hippocampus. In addition, some patients can present a cerebrovascular disease (cerebral amyloid angiopathy, CAA) that affects the cerebral blood vessels.14 Moreover, the decrease in synaptic density, loss of neurons, and degeneration of granulovascular hippocampal neurons are other factors. Finally, a deficit in neurotransmitters, mainly acetylcholine, is the main neurochemical criteria.
2.2. Spatiotemporal evolution of AD
It is estimated that the pathological processes underlying AD begin to develop a few decades before the clinical symptoms present which prompt the need for a diagnosis. Many AD patients likely present an early stage pre-dementia syndrome called mild cognitive impairment (MCI). MCI is thought to have a high likelihood of developing AD and to a lesser degree other dementias,15,16 which differ from a mild dysfunction of the limbic system (Fig. 1) resulting in slight loss of memory in healthy persons. Generally, AD develops when the damage extends to the temporal cortex and then to the frontal and parietal associative cortexes. The progression rate of MCI conversion to AD is estimated to be 10–15% of affected individuals. However it is important to note that about 20 to 30% of MCI cases do not develop into AD. Therefore, a diagnosis of MCI does not imply an early diagnosis of AD.17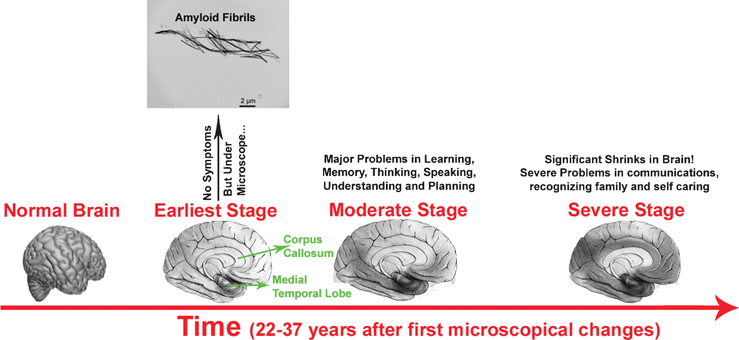 | ||
| Fig. 1 Manifestation of pathology and its progression in AD. In the early stages, there may be no obvious symptoms; however detectable amyloid fibrils, plaques and tangles may be present. The darker areas show the affected parts of the brain. Typically, there is a hierarchical and gradual expansion of the lesions over time. Braak stages I and II (defined by neurofibrillary tangle pathology),18 involving the entorhinal cortex and transentorhinal and the anterior part of the hippocampus, are not reflected by clinical symptoms. Subsequently, the disease progresses to the limbic system (Braak stages III and IV) where mild problems with memory and cognition appear. Finally, the disease spreads to the neocortex (Braak stages V and VI) where moderate problems appear and then leads to severe dementia and AD is very apparent. The hippocampus, particularly the entorhinal cortex, is the first brain region to be affected in AD, it expresses the molecular profile most susceptible to neurotoxic factors in AD.17 In addition, the pathology would spread in the brain as a retrogenesis sequence.19 Indeed, those structures that have matured later during the growth of the individual (myelinated at the end of adolescence), such as the prefrontal cortex, would be the most fragile and the first affected. Areas involved in orientation and language that became mature at puberty would follow. By contrast, the first myelinated structures, such as the primary sensitive areas, would remain intact in AD. | ||
2.3. Macroscopic and microscopic changes in the AD brain
AD is a unique disease of protein malformation because it simultaneously involves two proteins that cause two distinct histopathological changes in some regions of the cortex and the limbic system: AP and NFT.20,21 The pathological molecular processes surrounding them are complex and still only partially understood, but they are associated, inter alia, in an inflammatory response and oxidative stress. These produce the synaptic dysfunction, pronounced neuronal death and associated neurochemical loss that cause dementia. The brain atrophy resulting is so marked it is usually visible macroscopically at autopsy and by volumetric imaging techniques.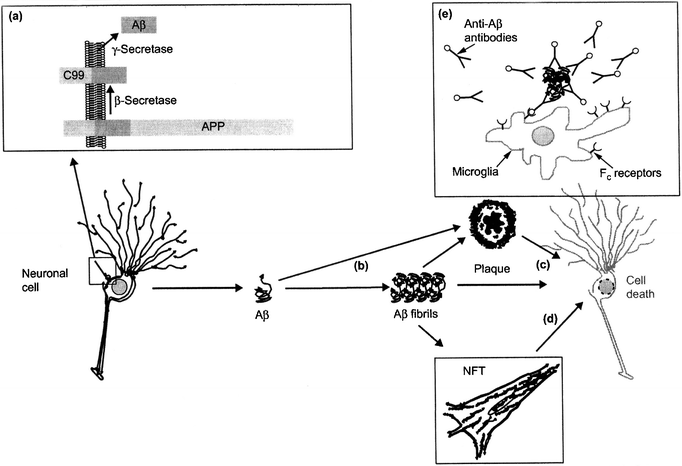 | ||
| Fig. 2 Illustration of the amyloid cascade hypothesis. (a) After processing of APP by β- and γ-secretase, Aβ is secreted and (b) aggregates into fibrils that deposit in senile plaques. Fibrils and plaques can induce neurotoxicity (c) directly or (d) indirectly, e.g. by the formation of NFTs. (e) A possible treatment concept is immunization with Aβ, which can induce an Fc receptor mediated clearance process by phagocytic microglia cells (reproduced with permission from ref 23) | ||
The amyloid hypothesis has been adapted to type 2 diabetes mellitus, which is also characterized by amyloidosis. Indeed, the pancreatic cells express a small co-amyloid protein, called islet amyloid polypeptide (IAPP), with insulin. This protein, which is overexpressed in pathological conditions, proved to be cytotoxic. Its toxic mechanisms, especially related to its oligomeric forms, are also very similar to those of Aβ.
2.3.2.1 Familial Alzheimer's disease, FAD. Unusually, approximately 5% of AD cases may develop in young people which is directly caused by an autosomal dominant mode of inheritance of gene mutations. Twenty mutations in the gene encoding the amyloid precursor protein (APP) have been identified and cause 10% of FAD. APP is a transmembrane glycoprotein of type 1 that is present throughout the body and exists essentially in three brain specific isoforms: APP695, APP751, and APP770. Its endoproteolytic cleavage can produce Aβ. Mutations that affect the gamma-secretase (γs) are located near the cleavage site of the enzyme complex residues 714 and 717 of the protein. This suggests that intramembrane γs is critical for the development of AD.
More than 140 different mutations in the presenilin 1 (PSEN1) gene have been identified. PSEN1 encodes for a part of the γs enzyme and plays a major role in the production of Aβ and the determination of its final size.
Ten mutations of the homologous PSEN2 that encodes for presenilin 2 have been also identified. PSEN2 mutations causes a more slow AD development than the mutations in PSEN1 and APP.
These mutations on three different chromosomes alter the same biochemical pathway and lead to an overproduction of Aβ or to an increase of the ratio Aβ42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Aβ40. Other cases of FAD do not appear to be caused by mutations of these three genes but it is postulated that mutations yet to be found may also interact with Aβ or be related to the phosphorylation of the tau protein but as yet these genes have yet to be discovered.
:Aβ40. Other cases of FAD do not appear to be caused by mutations of these three genes but it is postulated that mutations yet to be found may also interact with Aβ or be related to the phosphorylation of the tau protein but as yet these genes have yet to be discovered.
2.3.2.2 Sporadic Alzheimer's disease, SAD. SAD is much more complex because it is thought to be caused by a number of interacting genetic and nongenetic interactions. Indeed, the events that trigger the amyloid cascade are not yet understood. The main risk factor for SAD is aging, which induces a decrease in the efficiency of cellular mechanisms that regulate proteins. The clearance of Aβ would be reduced resulting in its accumulation in the central nervous system (CNS) and, thus, AD pathogenesis.
In terms of genetic risk factors, APOE, which encodes the ApoE protein is a well recognized risk factor and exists in three major allelic forms: ε2, ε3 and ε4.25 These give rise to the substitution of residues 112 and 158 of the ApoE. APOE ε4 (Arg/Arg) increases the risk of AD by a factor of 15 compared to APOE ε2 and 5 times over APOE ε3. This substitution is not sufficient, nor necessary for developing AD in contrast to the mutations discussed for FAD. Gender, high cholesterol, high blood pressure, and education are often cited as other potential risk factors. Trisomy 21 (Down syndrome) is a major risk factor for AD because chromosome 21, which carries the gene for APP, is present in triplicate. People with this syndrome inevitably develop AD in their 50s.22
More recently Genome Wide Association (GWA) studies have pronounced a number of new candidate genes (http://www.alzgene.org/)26 which now require in depth study to understand the nature of their potential involvement in AD.
2.4. Amyloid-beta
Aβ is produced in small amounts following the cleavage of APP following sequential cleavage by βs and γs. Its primary structure is amphiphilic, it contains no cysteine and thus cannot form a disulfide bridge. The 28 N-terminal amino acids are polar, with the exception of the hydrophobic heart (LVFFA17-21), and the C-terminal domain is apolar. This peptide has a tendency to polymerize and form amyloid fibrils (AF).The amyloidogenic cleavage of APP is also counterbalanced by its cleavage by the alpha-secretase (αs) which cleaves APP within the Aβ region thereby precluding full-length Aβ formation and releasing a soluble N-terminal fragment (called APPSa) and C-terminal fragments (called C83 or CTF) in the membrane. In the amyloidogenic pathway the cleavage of APP by βs forms the N-terminal end of the prospective Aβ peptide, releasing a soluble N-terminal fragment, called APPSb, and the remaining part of an intermediate membrane-bound fragment called b-CTF or C99. Both C83 or C99 are then cleaved in the membrane by γs to release either Aβ (i.e. amyloidogenic) or extracellular p3 and an intracellular “APP intracellular domain” (AICD) by the αs-nonamyloidogenic route). Furthermore γs can produce Aβ of different lengths ranging from 39 to 43 residues with isoforms of 40 and 42 residues being the most common. Thus, the specificity of γs seems to be very important in the amyloid cascade.
Post-translational modifications of APP occur at the cell surface in the secretory and the endosomal pathways. This results in the release of APP residues in the extracellular medium or in vesicles. The cleavage by αs occurs late in the secretory pathway (at the trans-Golgi network and secretory vesicles) and in the cell membrane (Fig. 3). βs and γs cleavages occur all along the secretory pathway as well as in the cell membrane and the endosomes. Aβ seems to be trapped in the intracellular environment if it is produced in the endoplasmic reticulum (ER); but it is released into the extracellular medium when produced later in the secretory pathway. Aβ40 is the isoform most produced at the late secretory pathway and the plasma membrane, whereas Aβ42 is at the early secretory pathway and endosomes. Aβ42 is more toxic than Aβ40 and aggregates more readily, so it is the initiator of the formation of AF and AP.
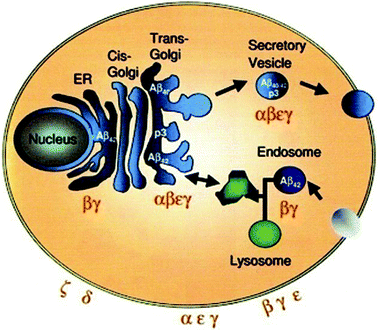 | ||
| Fig. 3 Schematic illustration of the intracellular itinerary of APP, cellular localization of APP processing. Aβ42 is the predominant product in the ER and endosome. Aβ40 is predominantly produced in the secretory pathway and at the plasma membrane. Reproduced with permission from reference.27 | ||
The location of γs does not always correspond with areas of activity because other factors appear to be involved. γs Cleaves the Notch transmembrane protein and allows the release of a cytoplasmic fragment that acts as a transcription factor. Similarly, the intracellular domain of APP is supposed to act as a transcriptional factor after cleavage by γs. Different compounds inhibit the activity of γs. Some compounds selectively reduce the production of Aβ42 in favor of the less fibrillogenic Aβ38.28 Non-steroidal anti-inflammatory drugs (NSAIDS) modulate its activity.29
γs Seems to cleave APP successively at different positions: e49, t46 and then g42, g40 and g38. APP can form dimers through dimerization motifs GxxxG.30 In this case, γs is blocked at residue 42 and therefore releases highly amyloidogenic Aβ42 forms. Presenilin 1 also seems to be involved in the pathogenesis by mechanisms independent of the production of Aβ. Indeed, it seems to influence synaptic plasticity through mechanisms modulating the response of the N-methyl-D-aspartate (NMDA) receptor.31 In addition, a mutant form of PSEN1 increases the phosphorylation of tau and decreases that of GSK-3b.
Presenilins seem to have other roles outside that related to the γs complex. For example, they appear to allow the regulation of beta-catenin or be involved in calcium homeostasis. γs Is a potential therapeutic target for AD; but because of its many physiological roles, possible severe side effects resulting from its inhibition are likely to be important.
The process of fiber formation seems to depend on a nucleation mechanism, where the nucleus grows by the addition of other protein molecules. The nature of the nuclei is not determined, but their growth is accelerated with the addition of preformed fibrils. The kinetics of fibril extension has been studied by surface plasmon resonance where fibrillar Aβ is immobilized on a chip.35,36
The relatively recent discovery of the involvement of Aβ oligomers in AD has addressed some of the perceived limitations of the amyloid hypothesis.37 These were that the spatial locations of AP and disturbed neural circuits do not always correlate. Similarly, the level of AP has never been robustly demonstrated to correlate with the severity of cognitive problems. Transgenic mice that overproduce Aβ develop problems before the onset of cognitive AD.37 Oligomers, which are formed more rapidly than AF, were found to be more neurotoxic. These species are soluble and can diffuse into the brain and spread the disease.
The fibrillogenesis is difficult to study because synthetic Aβ has a non-reproducible behavior.38,39 Indeed, for different batches of Aβ, the kinetics of fibrillogenesis and cytotoxicity are different. Aβ is produced either by solid phase synthesis or by recombinant techniques.40 The preparations may be contaminated with salts or other factors that affect the dissolution. Fibrillogenesis is affected by the preparation conditions such as pH, temperature, solvent, concentration, etc. In addition, Aβ is often dissolved in organic solvent as it is poorly soluble in physiological solvent. However, they favor the conformation of the alpha helix under physiological conditions.41 In addition, Aβ is metastable, it co-exists in solution in a variety of monomeric, oligomeric, and polymeric forms. Oligomerization occurs very quickly, in less than a second. The difficulty in reproducing results has thus lead to deriving the initial structure of the peptide in the solid state and then proceeding to the dissolution. Therefore, complex methods of reproducible preparations of Aβ are being developed.
Monomeric Aβ . Aβ cannot be easily studied because of its rapid aggregation in non-organic solvent. Nevertheless, liquid-state NMR has succeeded in giving information on monomers where X-ray crystallography had failed. According to this information, it seems that monomers are mostly disordered.Alpha-helix Aβ . Circular dichroism spectroscopy (CDS) has shown an intermediate state Hα between the unfolded state and the rich state in β-pleated sheet (β sheet) during fibrillogenesis. Aβ also adopts a soluble monomeric structure when dissolved in an organic solvent or in a lipid environment.42,43
ADDLs (Alzheimer's derived diffusible ligands) . ADDLs are non fibrillar oligomers that diffuse easily and bind to ApoJ (apolipoprotein J). They are only formed with Aβ42 and their structure is still undetermined. They are found in the AD brain and inhibit long-term potentiation (LTP) by damaging synapses. They appear more toxic than the fibrillar structure. Cleavage agents have been developed against the IAPP oligomers as therapy for type 2 diabetes. They have also proved effective against Aβ oligomers.44 Therefore, these oligomers probably possess similar micro-domains that are recognized by these agents.
Paranuclei Aβ . Paranuclei are penta- or hexameric units of Aβ42 that self-associate to form larger assemblies. Aβ40 does not form paranuclei, it is limited by rapid equilibrium structures with no higher structure than the tetramer.
Protofibrils and AF . AFs are structures of about ten nanometres in diameter and of indefinite length well over several hundred nm (Fig. 4). Electron microscopy has revealed a fibrillar sub-organization of the fiber in an association of 5 or 6 helical protofibrils of 2.5 to 3 nm in diameter. The protofibrils are thinner and shorter than the AF and appear earlier in the process of fibrillation and disappear at the onset of AF. This suggests that protofibrils are part of AF.45 At least eight other proteins form amyloid protofibrils, such as synuclein which are involved in Parkinson's disease.
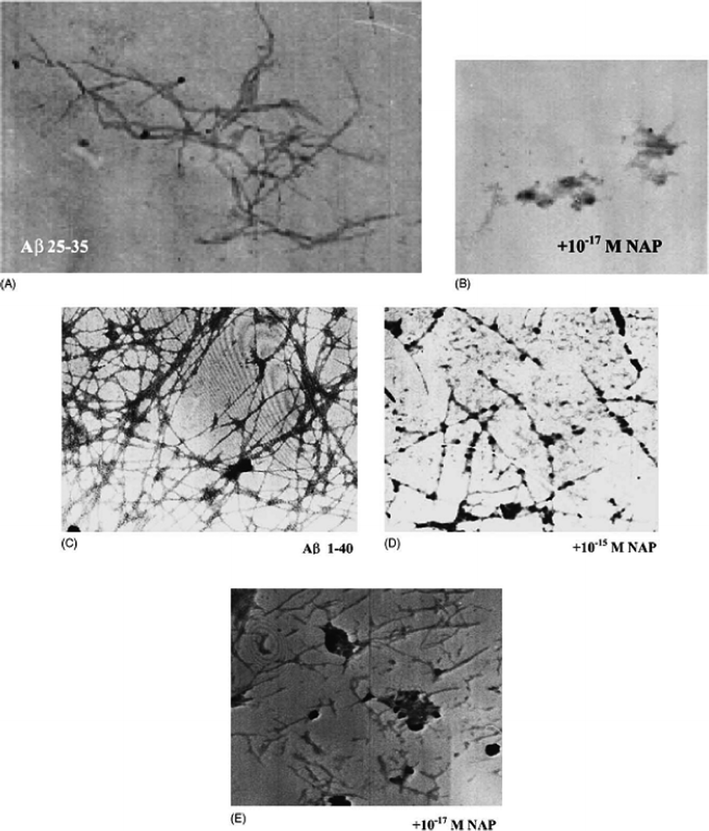 | ||
| Fig. 4 Electron microscopy: (A) Aβ (25–35) solution (100 μM); (B) Aβ (25–35) with the addition of NAP at 10−17 M; (C) Aβ (1–40) solution (250 μM); (D) Aβ (1–40) with the addition of NAP at 10−15 M and (E) 10−17 M concentrations. Reproduced with permission from reference.46 | ||
Despite the insolubility of AF, the 3D structure of Aβ42 fibers has been determined through various techniques, including NMR,47 X-ray diffraction, and CDS. Aβ organization adopts a hairpin containing two antiparallel beta-strands separated by a loop (Fig. 5). The first 10 amino acids at the N-terminal are disordered, residues 11-20 form the first beta-strand, residues 21–27 form a loop, residues 28 to 38 are the second beta-strand, and finally C-terminal residues 39 to 42 are disordered. The residues of the two beta-strands are mostly hydrophobic, in contrast to those of the loop and the N-terminus. Electrostatic interactions between Asp23 and Lys28 stabilize the loop. The design of this hairpin is perpendicular to the axis of the fiber. The peptides are arranged against each other and are stabilized by intermolecular hydrogen bonds that allow the formation of two β-sheets parallel to the axis of the fiber. A different organization of Aβ in the fiber seems possible. The plane will then have the pins in parallel to the axis of the fiber. There is then alternation between inter- and intramolecular H bonds.
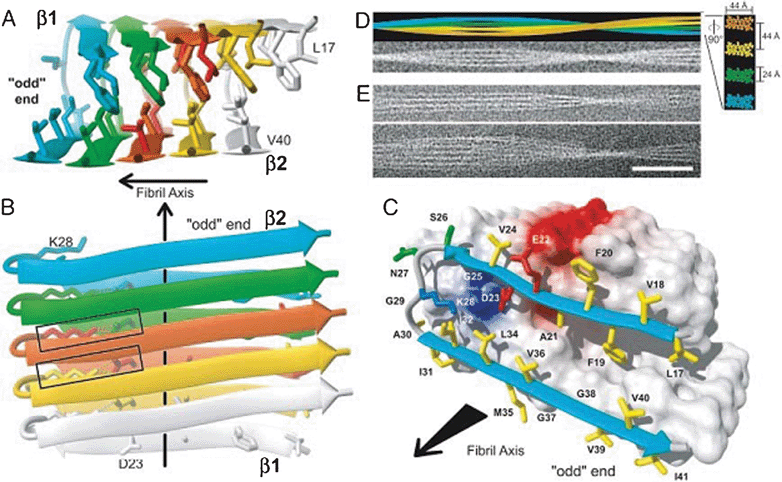 | ||
| Fig. 5 3D structure of one of the Aβ protofibrils. A, B and C show a different protofibrils along an axis (black arrow). The disordered N-terminal tail (1–16) is not represented. Each monomer within the protofibrils is represented by a different color. The beta strands are represented by an expanded arrow. Each peptide forms two beta-strands separated by a loop. There is a link between salt and D23 and K28 represented by a rectangle on B. The two beta strands form with their respective neighboring 2FPb perpendicular to the axis of the protofibrils. D shows an AF composed of four protofibrils. E shows cryoelectron micrographs of protofibrils. Reproduced with permission from reference.47 | ||
The sequence of five AA "KLVFF16-20" is crucial to initiate the aggregation of Aβ4245 and the two beta strands that are important for fibrillization.48 Simulations of molecular dynamics have shown the existence of hydrated cavities along the fiber and tight intermolecular bonds between the beta-pleated sheets.49 It seems that the side chain of Met35 is pointing outwards. There are a series of Met along the protofibrils. These Met35 allow the combination of two protofibrils by functioning as a zipper.
Amyloid deposits . Being insoluble, AFs are deposited in the brain parenchyma and form AP aggregates. Two types of plaques can be distinguished by their morphology. Diffuse APs are not as densely arranged and are not surrounded by dystrophic neurites and activated glial cells. On the contrary, neuritic APs (or senile plaques, SPs) contain a dense core surrounded by a fibrillar region, dystrophic neurites and activated glial cells. They also include a variety of macromolecules such as complement components, ApoE, glyco-amino-glycans (GAG), and serum amyloid P-component (SAP).15
Amyloid deposits can also form in the lining of the cerebral vessels, usually in the arterioles, in a condition known as cerebral amyloid angiopathy (CAA). These consist mainly of Aβ40- instead of AP-rich parenchymal lesions.50 This enrichment in Aβ40 probably contributes to its greater solubility, allowing it to be more widely distributed than Aβ42. CAA is very common in AD and can cause cerebral hemorrhage.
Other known forms of Aβ . a. Micelles: Aβ is an amphiphilic peptide with surfactant properties. The elongation of AF is proportional to the concentration of Aβ up to a point, beyond which the elongation reaches a plateau. Micelle formation is suggested to explain this phenomenon.
b. Channels: Aβ can form selective calcium channels in a lipid bilayer. These are among the neurotoxic mechanisms involved in the pathogenesis of AD. There are currently only theoretical models of these channels.
c. Beta-amyloid balls: Aβ40 can form at high concentrations of several hundred μM of fibers that spontaneously assemble into a sphere of 20 to 200 microns in diameter.
d. Amylospheroids (ASPD): these are spherical assemblies of Aβ40 and Aβ42 formed during slow rotations in vitro. ASPDs of Aβ42 are formed faster and are more toxic. These structures are not inhibited if peptide breakers are used instead of the AF formation.
2.4.5.1 Proteins. Many proteins influence the fibrillogenesis of Aβ. The alpha1-antichymotrypsin, which is produced by astrocytes, is found in AD and seems to influence the aggregation of Aβ. Acetylcholinesterase (AChE) is an enzyme that degrades acetylcholine and is found close to the AP. In senile plaques, it is particularly associated with the heart and may play a role in the formation of mature fibers. It seems to favor the conformational change of Aβ in β-sheets and thus its fibrillization.
Laminin is a glycoprotein of basement membranes which is associated in heterodimers. It accumulates in the AP and regulates the metabolism of APP in fibroblasts and neurons. Various studies show that it inhibits AF formation, but it does not interfere with the effect of AChE.52,53
Serum amyloid P-component (SAP) is a plasma glycoprotein which is found in all deposits of different amyloid proteins in vivo including the amyloid plaque and the neurofibrillary tangles. It appears to inhibit proteolysis of Aβ. Proteoglycans, which are glycoproteins that are anchored on the glyco-amino-glycans (GAG), are associated with SAP. GAG appear to be responsible for the binding of prostaglandins to Aβ and accelerating the formation and stabilization of AF.
The ApoE protein has its highest expression levels in the liver and then in the CNS where it is produced by astrocytes and microglia. ApoE ε3 and ε4 bind to Aβ to form very stable complexes with denaturants such as sodium dodecyl sulfate. One hypothesis suggests that ApoE acts as a chaperone that promotes the conformational transition from the soluble state to a structure rich in β-sheet. They seem to have more effect on the formation of amyloid deposits in CAA than AP. It would act as chaperones that modulate the clearance, transport and fibrillogenesis of Aβ.
2.4.5.2 Fats and lipids. AFs are closely linked to neuronal membranes, microglia, and endothelial cells. Some mechanisms of the neurotoxic Aβ involve membranes where it forms calcium channel blockers. Some sphingolipids appear to influence the conformation and promote AF formation.
Cholesterol modulates the metabolism of APP and increases the fibrillogenesis of Aβ. It is an essential component of cell membranes and a precursor of many hormones. It is also a risk factor for many diseases, such as cardiovascular disease. A minor change in its homeostasis affects many cellular events. The brain is rich in cholesterol compared to the rest of the body; approximately 25% of cholesterol is in the CNS. The majority of cholesterol (80%) is in the myelin membranes and the rest is distributed in glial cells and neurons. Cholesterol metabolism in the CNS is largely independent of systemic metabolism because the majority of CNS cholesterol is synthesized. Receptors in low density lipoprotein (LDL) and apolipoprotein are expressed in the brain.
Cholesterol is a major component of lipid rafts with sphingolipids and proteins related to glycosylphosphatidyl-inositol. Lipid rafts are membrane microdomains that are able to selectively include or exclude proteins, and are synthesized in the Golgi system and constantly endocytosed. αs Is located preferentially in areas of low cholesterol. Therefore, the cleavage of APP is influenced by its location in the membrane and by cholesterol. In addition, it appears that Aβ can bind to lipids that facilitate its aggregation.
2.4.5.3. Metal ions. An increase in the concentration of metal ions (Cu2+, Zn2+, and Fe3+) is observed in the AD brain. These ions are concentrated in the AP and appear able to induce aggregation and precipitation of soluble Aβ.54–56 Aβ may be linked to 3.5 equivalents of zinc and copper whilst multiple metal ions are coordinated in the formation of oligomers. The interaction of synthetic peptides with transition metals is so sensitive that self-aggregation of Aβ in solution is initiated at nanomolar levels of metals that contaminate common buffers.57 Therefore, the presence of metals, even at trace levels, accelerates the aggregation of Aβ. Cu2+ is more capable of forming covalent bonds between peptides. The release of Zn2+ and Cu2+ by the synaptic vesicles promotes the formation of AP near the synapses. Aluminum also appears to participate in the aggregation of Aβ and its neurotoxicity.58
The expression of APP is stimulated by neurotrophic factors and neuronal response to stress. APP also stimulates cell adhesion and protects neurons. APP is released in areas as a response to presynaptic electrical activity and regulates neuronal excitability, synaptic plasticity, learning, and memory. APPs is also involved in neuritogenesis and neuroprotection in cell culture and neural stem cells. AICD seems to act as a transcription factor because it is involved in gene regulation.
Aβ also has no known physiological role; it is perhaps a linker between APPs and AICD and there is some in vitro evidence that it might have vaso-modulatory properties.60 It is a high-affinity chelator of transition metal ions such as Cu2+, Fe3+, and Zn2+. Its role in oxidation is not clearly described.
:Aβ40 ratio are responsible for FAD. In the case of SAD, the mechanisms initiating the amyloid cascade remain unclear. One hypothesis suggests that the accumulation of Aβ does not initially come from overproduction, but rather from a decrease in the efficiency of cellular mechanisms for its clearance.61,62 Aβ is removed from the brain in several ways: by microglial phagocytosis, by enzymatic digestion, and by its transport through the BBB (blood brain barrier) allowing its degradation in the liver and kidneys (Fig. 6). Furthermore, it has been shown that chaperone proteins inhibit the formation of AFs.63
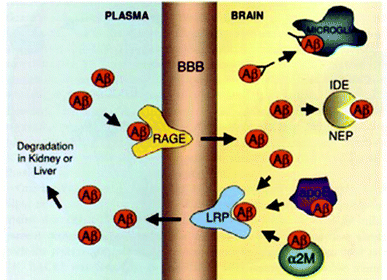 | ||
| Fig. 6 Mechanisms of Aβ clearance from the brain: (i) microglial phagocytosis, (ii) enzymatic digestion by insulin degrading enzyme (IDE) or neprilysin (NEP), (iii) receptor mediated clearance across the BBB via a receptor related protein (LRP) or a receptor for advanced glycation end-products (RAGE), and (iv) chaperone assisted (ApoE or α2-macroglobulin) clearance across the BBB. Reproduced with permission from reference.27 | ||
2.4.7.1. Proteolytic cleavage. A variety of enzymes can degrade Aβ in vivo. The elimination of these proteases increases the levels in Aβ.
Neprilysin (NEP) is a zinc-metalloendopeptidase with mass equal to 93 kDa which is involved in the catabolism of a wide range of peptides. This glycoprotein is related to the membrane with an extracellular active site on the side where Aβ is secreted. It is located in cellular compartments (Golgi, ER, etc.) where Aβ is produced. In addition, in neurons, it is mainly located near the synaptic terminals where Aβ is secreted. Its expression decreases with age primarily in the dentate gyrus and is more sensitive to oxidative damage. Some mutations of Aβ in FAD increase their resistance to NEP. It degrades Aβ in the oligomeric state but is unable to degrade AF. The expression of NEP is highly regulated, suggesting its potential for treatment of AD since its overexpression reduces Aβ.
The insulin degrading enzyme (IDE) is a zinc metalloprotease that degrades Aβ and AICD. It is found in the cytoplasm, endosomes and peroxisomes. Its expression decreases with age and is mainly located in the hippocampus. Its dysfunction also appears to contribute to type 2 diabetes, showing again a pathological similarity between the two diseases.
These two enzymes are located in two distinct compartments and catalyze different pools of Aβ. They are able to degrade monomers and possibly oligomers similar to NEP. AFs are therefore resistant to both proteases.
Furthermore, plasmin, a proteolytic enzyme that cleaves fibrin, has been reported to catalyze Aβ but its activity is reduced in people with AD.64 In transgenic mice, Aβ seems to stimulate the expression of the tissue plasminogen activator, matrix metalloproteinases also have the ability to degrade Aβ and their expression is stimulated by it as well.65
2.4.7.2. Phagocytosis. In the CNS, microglia and astrocytes have a trophic role and are involved in inflammatory responses. Aβ42 activates microglia and creates a localized inflammatory response with release of cytokines, chemokines, ROS, and complements. Microglia express several receptors, like RAGE, and are able to phagocytose. However, they are not able to degrade AFs due to insufficient enzyme units.66
2.4.7.3. Transport. The BBB does not normally allow the exchange of polar compounds such as Aβ between blood and brain. Thus, Aβ passes from one compartment to the other via two carriers, the LRP-1 and RAGE receptors.67 This transport is critical to regulate the concentration of Aβ and to prevent the formation of oligomers and AFs. Under physiological conditions, the concentration of Aβ is approximately 100 times higher in brain interstitial fluid than in plasma, but the total amount of Aβ is 10 times higher in plasma and FCS52 fluids in the body than in the brain. The few studies that have analyzed Aβ in the plasma in AD patients, showed no change as compared to those of healthy controls however there is a variability in methods.68–74 In contrast, the level of Aβ in the CNS was 10,000 times higher in these patients than in controls.
RAGE binds multiple ligands and Aβ. The accumulation of Aβ increases RAGE expression in blood vessels, neurons, and microglia. As the carrier conveys Aβ only in one direction, from the blood to the brain tissue, it contributes to its accumulation in the CNS and, thus, to cellular dysfunction.75 The transport of Aβ across the BBB involves the expression of proinflammatory cytokines in neurovascular cells and elaboration of endothelin-1, a vasoconstrictor peptide.
LRP-1 aids clearance of Aβ across the BBB. It is part of the family of LDL receptors and transports cholesterol and ApoE. When Aβ adopts a β-sheet conformation, its transcytosis by LRP-1 is reduced. Aβ appears to increase the degradation of the carrier by the proteasome, which promotes further accumulation in the brain.
2.4.8.1. Non-lethal mechanisms. Aβ influences critical processes that modulate neuronal plasticity involved in memory and learning. Interference of Aβ with signal transduction by two key pathways has been found: activation of the NMDA (N-methyl-D-aspartate) receptor and of the TrkB receptor by BDNF (brain-derived neurotrophic factor).78–83 The NMDA receptor is essential for some forms of memory and learning via long-term potentiation. BDNF and TrkB are critical for neuronal survival, protection of lesions, and neuronal plasticity of certain activities including LTP, neurogenesis, and synaptic growth. Av42 oligomers promote the endocytosis of NMDA receptors and therefore reduce their activity. They also reduce the ability of BDNF to regulate transcription factors (CREB and Eik-1) and compromise the ability of BDNF to protect neurons. Aβ does not seem to act directly on the receptor, but has been described to interact with some anchoring proteins. These mechanisms are similar to those involved in type 2 diabetes. Indeed, Aβ generates a state of resistance to neurotrophins by mechanisms involving anchoring proteins in a manner similar to the mechanisms leading to insulin resistance in type 2 diabetes. In addition, Aβ protein can inhibit kinase A and, consequently, the way of reporting the AMPc (cyclic adenosine monophosphate) that influences LTP.84 Aβ also disrupts synapses by changing, among others, the neuronal calcium involved in the release of synaptic vesicles. In addition, it damages the mitochondria, thereby reducing the axonal transport of vesicles and mitochondria themselves to the synapses.85 Furthermore, the microtubule network is destabilized by the hyperphosphorylation of tau.
2.4.8.2. Neuritic dystrophy. AFs induce neurite dystrophy that leads to synaptic loss and, later, to neurodegeneration. APs and their microenvironment induce a number of dendrite anomalies, such as loss of dendritic spines, atrophy of the main axis, twisting branches, and deposits around the “abrupt termination of branches” or “abrupt termination of branches in the deposits”.86 In addition, the accumulation of Aβ causes degeneration of axons independent of the extracellular intraneuronal Aβ deposits.87
AFs bind to the APP membrane whose level is over-regulated in the presence of Aβ. APP would bring the AF in contact with the focal adhesion sites, which are multiprotein complexes consisting primarily of integrins and involved in cell adhesion. The sites would then be activated and could initiate a cascade of multiple signal transductions. Some would lead to the hyperphosphorylation of tau, destabilization of microtubules, and neuritic dystrophy. Others involve the activation of focal adhesion kinase, which leads to cell death. Therefore, brain regions with the most plasticity are most vulnerable.
2.4.8.3. Oxidative stress. Aβ causes damage to neurons adjacent to the SP by the generation of ROS. Neurons are particularly vulnerable to ROS because of their large oxygen metabolism. Enzymes, which among other things convert ROS produced by mitochondria, are less active in AD. Aβ increases the levels of hydrogen peroxide that become cytotoxic because they saturate cellular antioxidant protection.88 Fe2+ and Cu2+ ions are oxidized by H2O2 to produce OH˙ which damages the cell.89 Zinc appears to suppress the formation of H2O2.90 This ion has an ambiguous role because, on the one hand, it promotes aggregation and, on the other hand, it limits oxidative stress. Studies on transgenic mice have been done.91 In the extracellular medium, the ROS produced by Aβ in combination with metal ions have been shown to produce deposits of Aβ responsible for lipid peroxidation and production of an aldehyde, 4-hydroxynonenal. The latter, is more stable than ROS and can diffuse away from the site of reaction, covalently modify proteins, and contribute to the pathogenesis. This inhibits dephosphorylation of tau and neuronal death.92
2.4.8.4. Calcium. Ca2+ plays a fundamental role in synaptic function and neuronal survival and its homeostasis is disturbed in AD. Aβ is capable of forming membrane pores that allow the influx of Ca2+ and consequently disrupt its homeostasis. The excitotoxicity of glutamate also leads to a massive influx of calcium into the neuron. This increase in the concentration of Ca2+ disrupts many cellular mechanisms that lead to neuronal death. Among these, there is activation of calpain, disruption of synapses, and the generation of oxidative stress in mitochondria. Aβ also disrupts the regulation of calcium in the ER, which activates a metabolic process specific to UPR (unfolded protein response) that can trigger apoptosis. It seems, however, that Aβ triggers apoptosis by mechanisms independent of the UPR.93 The role of caspases, which are proteases that regulate apoptosis in mammals, appears to be limited in AD.94
2.4.8.5. Localized inflammation. The neurons of the brain are “served” by the glial cells that are classified into two groups: microglia and macroglies (astrocytes and oligodendrocytes). Microglia have a phagocytic function similar to macrophages. Normally, they are few, but they multiply in response to injury or infection. In AD, they gather around amyloid plaques with a subset of reactive astrocytes. These cells are involved in the removal of Aβ deposits and residues of dystrophic neurites, but they are also actively involved in inflammatory and neurodegenerative processes of AD.
As for the tau protein, the Aβ form crosslinks with AGE. These bonds stabilize the APs and accelerate the training of FPb. Microglia have RAGE which potentiate the inflammatory response when activated.95 The activation of glial cells by Aβ leads to the production of pro-inflammatory cytokines, such as IL-1b (interleukin-1b) and TNF-α (tumor necrosis - factor alpha), but it inhibits the expression of inhibitory cytokines.95,96 This disruption results in runaway activation of glial cells and neurotoxicity.
The neurotoxic mechanisms associated with this route of inflammation are complex and numerous. Glial cells stimulated by Aβ overproduce free radicals (ROS and reactive nitrogen intermediates, RNI) that generate oxidative stress and damage cells. The pro-inflammatory cytokines are also involved in neurodegenerative mechanisms. The bonding of IL-1 to SP (i) induces the production of Aβ favoring the synthesis and the treatment of APP, (ii) stimulates the production of binding proteins to Aβ (e.g. ApoE) by astrocytes, (iii) triggers the production of other cytokines, and (iv) by its interaction with Aβ, it induces the production of RNI by astrocytes.97 Overexpression of IL-1 close to SP may promote phosphorylation of the tau protein, leading to the formation of NFT and death of neurons.98 NFT-α is indirectly toxic by inducing the production of RNI by glial cells.
Chemokines are also produced by these cells in response to Aβ. These are chemotactic for microglia, but not for astrocytes. Therefore, local expression near the AP will increase microglia. These cells will then in turn recruit and activate other phagocytes, thereby promoting inflammation and neurodegeneration.
The complement system is a complex system of inactive plasma factors. The activation of this system is secondary to the introduction of an antigen. It results in the appearance of activated factors and soluble factors activated by a particle-bound antigen, which account for three major functions of this system: opsonization of antigen particles, a localized pro-inflammatory activity related to an influx of the immune system, and a lytic activity of certain cells. The complement, which is directly activated by Aβ in AD, is deeply involved in the destruction of neurons by the membrane attack complex.
This hyperphosphorylation is secondary to different mechanisms many of which are triggered by Aβ. This increases the activity of the kinase GSK-3b (glycogen synthase kinase-3) that hyperphosphorylates tau.99 Aβ-derived diffusable ligands (ADLLS) stimulate the phosphorylation of tau by mechanisms involving Src family kinases.100 Oxidative stress created by inflammatory response and Aβ activates many kinases. The cascade of phosphorylation kinase MAPK (mitogen-activated protein kinase) is overactive in AD by the AF.101 The increase in neuronal calcium activates calpain, which will then cleave/activate the kinase that phosphorylates cdk5 (cyclin-dependent kinase)/p35 into cdk5/p25 and also the tau protein. Finally, MARK (microtubule affinity-regulating kinase) phosphorylates tau at binding sites for microtubules. Oligomeric forms of Aβ inhibit the activity of proteasomes and promote, therefore, its accumulation and that of tau.102
The hyperphosphorylation of tau reduces its ability to bind to microtubules and promote their assembly. The microtubule network is therefore destabilized and axonal transport is disturbed. The decrease in organelles and vesicles in the axonal and dendritic compartments weakens them and disrupts the synapse functions. In addition, the lack of peroxisomes increases the sensitivity of cells to oxidative stress. Finally, training of NFT obstructs the cytoplasm and thus causes mechanical damage to the cell. In addition, the hyperphosphorylation of tau increases its resistance to calpain and thus prevents its degradation. Indeed, tau self-assembles at the sites of binding to microtubules and therefore reduces the accessibility of these sites to calpain. The self-association is favored in an oxidative environment. Furthermore, depletion of tau on different models reduces the neurotoxic effects of Aβ.103,104
In addition to phosphorylation, tau protein can undergo glycation due to it being lysine rich. Glycation is the nonenzymatic addition of a reducing sugar to a protein (nonenzymatic glycosylation). This results in the formation of Schiff bases, an aldimine bond between a residue of aldehyde, an arabinose, and a nitrogen ε of a lysine of the protein. The end products of glycation are called AGE. These AGE then induce the formation of covalent ‘cross-link’ bonds between the tau proteins. These neurofibrillary tangles are then much more stable and have less affinity for microtubules. The cross-links increase resistance to proteolytic cleavage of tau (by calpain, for example) and contribute to the accumulation of ubiquitin conjugated to neurofibrillary tangles. Indeed, high levels of ubiquitin have been reported in several neurodegenerative diseases.105–108
2.5. Therapy for AD
AD is incurable at present, because the poor understanding of its molecular mechanisms makes it impossible to develop effective therapies. They currently have only short-term effects reducing the symptoms and are often accompanied by significant side effects. The amyloid cascade is considered responsible for the dysfunction of neurons and cell death and, therefore, is the basis for the pathogenesis of AD.109,110The compounds currently approved for the treatment of AD111 unfortunately do not treat the causes; they only treat the symptoms, allowing patients to regain or temporarily preserve some temporary cognitive and behavioral abilities. However, they also sometimes lead to serious side effects.
Acetylcholinesterase (AChE) inhibitors are used to temporarily prevent the breakdown of acetylcholine and, therefore, produce higher levels of acetylcholine. The most commonly prescribed drugs are donepezil, rivastigmine and galantamine. Tacrine, the first to be developed, is hardly used because of its hepatotoxicity. Memantine is an NMDA receptor antagonist that is used in more severe forms of AD. It reduces glutamate excitotoxicity, which is present in excessive amounts in AD, and therefore slows the progression of the disease.
There are a number of other therapeutic routes that have been proposed for AD. However the approaches that aim to regulate Aβ to try and decrease its toxic effects may have the greatest therapeutic value. Several routes are being considered: promoting its degradation, preventing its aggregation, or preventing its overproduction.112
Immunotherapy, which involves passive or active immunization against Aβ, is promising as a therapeutic approach for promoting the clearance of Aβ. Passive immunization involves the direct administration of anti-Aβ, while active immunization stimulates the immune system to produce its own anti-Aβ. Active immunization of transgenic mice resulted in a significant reduction in amyloidosis and restoration of cognitive deficits.113 Antibodies, which have been generated by immunization, targeted the soluble N-terminal portion of the hydrophilic Aβ. Dramatically, some patients developed meningoencephalitis in phase II clinical trials.
Aggregation of Aβ has been also inhibited by ligands targeting the KLVFF16 motif of Aβ.114–116 Molecules binding to Aβ and recruiting chaperones, increase their steric hindrance and inhibit their agglomeration.117 γs and βs inhibitors are also currently being studied because they could reduce the production of Aβ. Amongst these, a βs inhibitor anchored in the membranes has shown good efficacy.118 However, the possible inhibition of physiological processes arouses fear of severe side effects and hinders research in this direction. Indeed, a recent trial on semagacestat had to be terminated prematurely because of unwanted effects.119 A similar but alternative approach would be the stimulation of αs which has also been considered. Activators of protein kinase C, such as bryostatin, seem to favor this path and significantly reduce the production of Aβ on cellular models and transgenic animals.120
Stimulation of the physiological pathways of Aβ elimination (proteases, transport, phagocytosis) is also being considered. These strategies focus on the monomers or oligomers as the AF are very resistant. In addition, oligomers were found to be the most neurotoxic species. However, reducing the concentration of these species would tip the balance with the AF and promote their disassembly. Curcumin, a compound that crosses the BBB and has anti-oxidative, anti-inflammatory, and anti-fibrillogenic properties, appears to disrupt the formation of SP. Colloidal particles composed of organic molecules inhibit the formation of AF by mechanisms of sequestration.121
Statins may have a preventive effect on AD. They are already used in the treatment of cardiovascular diseases where their role is to reduce the level of plasma LDL and cholesterol and increase the high density lipoprotein (HDL). Recent statin trials in AD have been disappointing.122
Chelation therapy proposes the administration of a chelate for zinc and copper in particular. The results illustrated that stabilization of cognitive abilities and of the level of plasma Aβ42. Vitamin B12 is co-administered with these chelating agents to reduce their severe side effects.
Anti-inflammatory drugs (NSAIDS) also seem to reduce the propensity to develop AD123, but there is no clear explanation as to how they work. Studies of certain anti-inflammatory drugs have shown mixed findings and some have been discontinued due to toxicity.123
BDNF has been suggested to counteract the pathological effects of Aβ.124 Indeed, its administration in different animal models allows the restoration of their cognitive and various cellular mechanisms. Finally, the tau protein is also being studied for therapeutic purposes.120
Antioxidants (e.g. vitamin E) may have a beneficial effect on AD in fighting the ROS. Patients are also often treated with antipsychotics and antidepressants since various forms of mental health and behavioural problems occur in AD.
In recent times the results of a number of phase III trials of candidate drugs have been disappointing including semagacestat as mentioned and also DiMEBON which was very promising at phase II.125 Of the 77 compounds reviewed by Mangialasche and colleagues in 2010 which were in pre-clinical development or in various phases of clinical trial up to hase III level, all but 10 (except for already licensed drugs) targeted various aspects of Aβ or tau related pathology or aspects of neurochemical deficits. Of the remaining 10, these target a variety of other recognised and important aspects of AD pathogenesis including oxidative stress; mitochondrial dysfunction; serotonergic imbalance; neuronal preservation; fatty acid maintenance; the regulation of cGMP signalling; and cholesterol lowering.188
2.6. Diagnosis
The most effect treatment for AD would be one of prevention and as such it would need to be administered before the onset of dementia, when patients are asymptomatic. However, the course of dementia development (AD as well as some others) is such that once it is identified, the brain has already suffered significant damage which is irreversible. Therefore, early diagnosis of AD and is crucial is likely to be the main determinant of the efficacy of existing and new therapies.Cognitive tests currently allow the diagnosis of AD with varying degrees of “probable” accuracy however it is dependent on these cognitive deficits which occur at a quite an advanced stage of the disease to be possible. At this point, the sensitivity is reasonalby high, the specificity very low but damage well advanced. The MMSE test (mini mental state examination)126 is commonly used to help screen for dementia, but it does have limitations and is not very specific to AD.
A far preferable scenario is to be able to detect MCI as early as possible. The number of people with MCI developing AD is 3–6 times higher than that of healthy people.14 In addition, it is clear that the molecular and cellular mechanisms underlying this trajectory of cognitive decline is triggered in young adults long before the MCI is evident (Fig. 7). However the often subtle symptoms of MCI make it very difficult to distinguish from normal aging or other forms neurodegeneration.
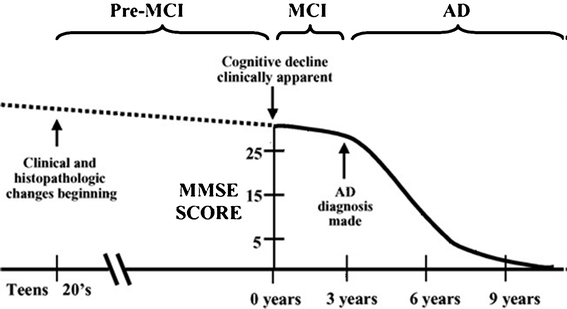 | ||
| Fig. 7 Time evolution of senile dementia to AD. The decline in cognitive abilities (represented by the MMSE) occurs very late. Reproduced with permission from reference.16 | ||
In plasma, as yet, there have been less robust biomarkers identified. Indeed, the amount of Aβ in plasma seems to increase slightly with the pathogenesis of AD, but the results are quite variable and it is not clear if this variability is due to the Aβ or the methods used to measure them.130,131 Plasma levels of CSF and Aβ are not correlated because of many complex mechanisms involved in the regulation of the BBB. A new 79-AA amyloid protein was discovered recently: ALZAS67 (Alzheimer's associated protein).130 It contains the sequence of Aβ42, the signal sequence of intramembranal APP, and 12 C-terminal original residues. ALZAS67 seems to compete with APP for proteolytic pathways and be very neurotoxic. It was detected in AD patients at early and late stages and thus has potential as a biomarker in blood.
In the CSF, the biomarker most sensitive and most specific to distinguish AD from other dementias is the combined study of the level of Aβ, total tau and p-tau (hyperphosphorylated tau). Indeed, the level of soluble Aβ42 decreases, while levels in total tau and p-tau increase. Taken separately or indeed together, these three biomarkers are not strong enough to make a diagnosis in isolation—information on the clinical history of these patients is still important. The reduction of soluble Aβ42 is explained by the increase in aggregates and the "reservoir" role of AP. It should be noted that the level of CSF Aβ40 is not affected by the disease; and that, despite its decline, the level of Aβ42 in CSF is well above that in plasma.132 These biomarkers are present in the early clinical stages of AD and change soon after.133 They were detected in aged healthy people at rates similar to those of AD patients, but not in a group of younger healthy people,134 showing that these markers allow early diagnosis of AD before the onset of severe dementia. The VLP-1 (visinin-like protein 1) was proposed as a biomarker because its level increases significantly in the CSF in AD patients. The VLP-1 protein is a calcium sensor that is expressed in abundance by neurons, but is not detectable in peripheral tissues.134 It is overexpressed in neuronal death and is therefore a biomarker sensitive enough to neurodegeneration, but it is not specific to AD. The VLP-1 is the only biomarker that correlates (inversely) to MMSE scores.135 In addition, its expression is twice that seen in people homozygous for APOE ε4 compared with those homozygous for APOE ε3.
In AD, ROS are produced in excess and the body struggles to limit their damaging effects. ROS cause oxidative damage to proteins, nucleic acids, and lipids. Among these molecules, unsaturated fatty acids are abundant in the brain and are very sensitive to damage. The concentration of certain products of this oxidation, such as isoprostanes, in plasma and CSF is higher in AD patients than in healthy people.136
An important consideration in the analysis of CSF is that it requires the practice of an invasive lumbar puncture in people which is considered with varying degrees of receptiveness even amongst clinicians. No useful biomarker has yet been validated in urine or systemically.
3. Effects of nanoparticles on inhibition/acceleration of amyloid fibrillation
Since nanoparticles (NPs) with proteins can cause perturbation of protein structure and function, it is conceivable that the interaction of nanoparticles and amyloid proteins or peptides could inhibit or facilitate amyloid formation. Therefore engineered nanoparticles could be potential drugs for controlling or curing amyloid-related diseases. Two opposing scenarios can be envisaged:137(i) if nanoparticles can inhibit the process of fibrillogenesis or disaggregate amyloid fibrils,138 they have potential to be used as ‘drugs’ to control neurodegenerative amyloid diseases like AD. In addition, detailed information about the interactions between nanoparticles and amyloid proteins could help to elucidate the mechanism of fibril formation.
(ii) if nanoparticles facilitate the process of protein fibrillogenesis, there is a serious risk to human health, and great care would have to be taken to ensure their safe use.
Aβ peptides are regularly used as model proteins to investigate the effect of nanoparticles on fibrillogenesis and the potential application of nanoparticles in treating neurodegenerative diseases. Recently, antibodies against Aβ and surface-modified proteins with Aβ residues were applied to prevent Aβ aggregation and reduce toxicity.139,140
It is interesting to investigate the self-organization processes between NPs and peptides; the assembly behavior of NPs reveals similarities with those of biological species.141–145 Such studies also reveal new aspects of NP toxicology and provide an alternative methodology for preventing the agglomeration of Aβ peptides.146–148 The literature data on the effects of both organic and inorganic NPs on peptide assembly are controversial. The presence of NPs has typically promoted aggregation of Aβ, which was explained in terms of a condensation-ordering mechanism.149–151 Since the fibrillization occurs by nucleation dependent kinetics, the increased local concentration of peptides in the vicinity of NPs as a result of electrostatic attraction greatly accelerates the fibril formation.
For example, copolymeric NPs of N-isopropylacrylamide and N-tertbutylacrylamide with diameters of 70 and 200 nm, cerium oxide NPs with a diameter of 16 nm, polymer-coated quantum dots (QD) with a diameter of 16 nm, and carbon nanotubes were shown to catalyze the fibrillation of the amyloid protein of b2 microglobulin.149
In the presence of PEGylated phospholipid micelles, Aβ42 exists in a predominantly α-helical conformation thereby reducing its aggregation potential.152 Fullerene can significantly inhibit the amyloid formation of Aβ40 by specifically binding to its central hydrophobic KLVFF motif.153 In the presence of fluorinated nanoparticles, Aβ40 has a helical structure without peptide aggregation, but hydrogen nanoparticles accelerate the conversion of Aβ40 from random coil to β-sheet resulting in fibrillogenesis.154 Triulzi et al.138 have designed nanoparticles which could be used as drugs to inhibit fibril formation or disrupt fibrillar aggregates; in vitro experiments indicated that some nanoparticles actually accelerate fibril assembly kinetics.
Cabaleiro-Lago et al.155 have shown that copolymeric NiPAM–BAM nanoparticles promote β-amyloid fibril formation in vitro. These nanoparticles possess a strong adsorption capacity and have a high local concentration of Aβ on the nanoparticle surface. The extension of the lag phase in the fibrillization of Aβ is strongly dependent on both the amount and surface character of the nanoparticles. Surface plasmon resonance studies have shown that Aβ binds to the nanoparticles and have provided rate and equilibrium constants for the interaction.156 Analysis of the kinetic data for fibrillisation suggested that binding of monomeric Aβ and prefibrillar oligomers to the nanoparticles prevents fibrillation. However, a high local concentration of protein on the nanoparticle surface can also be used to explain why some nanoparticles inhibit fibril formation. The bond between nanoparticles and amyloid proteins could block active sites for fibril formation.
Similarly, the strong absorption capacity of TiO2 NPs (20 nm in diameter) for the amyloid peptide has promoted fibril formation.151 It has been reported also that biological molecules,157,158 large colloidal particles,159 and liquid–air and liquid–solid interfaces160,161 can promote fibril formation. Some polymeric NPs (40 nm in diameter) have been found to slow down the rate of Aβ fibrillisation by depleting the amount of free monomeric peptides, although the fibril formation could still not be prevented.155 Surface-modified nanogels,162 micelles,152 or fullerenes153 were able to alter the conformation of the peptides and reduce the toxic activity. N-Acetyl-L-cysteine capped quantum dots can promote assembly of amyloid peptides. The formation of dense NP coatings on fibrils was observed and a computer model of a kinetic model based on nucleation and growth inhibition has been developed.163
Cabaleiro-Lago et al.164 studied the fibrillation kinetics of the amyloid β peptide in the presence of cationic polystyrene nanoparticles. Depending on the ratio between the peptide and particle concentration, the kinetic effects vary from acceleration of the fibrillation process, by reducing the lag phase at low particle surface area in solution, to inhibition of the fibrillisation process at high particle surface area. The kinetic behavior involved in this has been proposed to be a balance between two different pathways: fibrillisation of free monomer in solution and nucleation and fibrillation promoted at the particle's surface. The overall rate of fibrillation will depend on the interplay between these two pathways, and the predominance of one mechanism over the other will be determined by the relative equilibrium and rate constants.
Thakur et al.165 have demonstrated that the Aβ mixed with and conjugated to dihydrolipoic acid- capped CdSe–ZnS QD of size approximately 2.5 nm can be used to reduce the fibrillation process. Transmission electron microscopy (TEM) and atomic force microscopy (AFM) were used for analysis of fibrillation. They report a significant change in morphology of fibrils when Aβ(1–42) is mixed or conjugated to the QD. The length and the width of the fibrils were found to vary under modified conditions. Thioflavin T fluorescence supported the decrease in fibril formation in presence of dihydrolipoic acid capped QD.
Yoo et al.145 have reported the strong inhibition of Aβ fibrillisation by thioglycolic acid stabilized CdTe NPs. These NPs were selected because they resemble some proteins in terms of the size, charge, and association behavior. The inhibited fibrillisation by CdTe NPs was confirmed by TEM and AFM (Fig. 8). The authors consider that it is possible that a minor fraction of NPs would have the proper local geometry and conformation of stabilizers to specifically self-assemble with (mis)folded peptides that can accelerate or inhibit fibrillisation. This mechanism of NP–peptide interaction was found to be different from those considered previously for any kind of NPs,149–151,162–164 but very similar to that found for protective proteins inhibiting peptide fibrillation in the body.166–168
![TEM (a–j) and AFM (k–o) images of Aβ1-40 incubated with and without CdTe NPs. [CdTe] : [Aβ1-40] molar ratios: (a, f, k) 0, (b, g, l) 0.001, (c, h, m) 0.005, (d, i, n) 0.01, and (e, j, o) 0.05. In the TEM images, NPs appear as bright spots that can be easily distinguished from the fibrils even for small concentrations of the NPs. Reproduced with permission from reference.145](/image/article/2012/RA/c2ra01374f/c2ra01374f-f8.gif) | ||
| Fig. 8 TEM (a–j) and AFM (k–o) images of Aβ1-40 incubated with and without CdTe NPs. [CdTe]:[Aβ1-40] molar ratios: (a, f, k) 0, (b, g, l) 0.001, (c, h, m) 0.005, (d, i, n) 0.01, and (e, j, o) 0.05. In the TEM images, NPs appear as bright spots that can be easily distinguished from the fibrils even for small concentrations of the NPs. Reproduced with permission from reference.145 | ||
In order to examine the influence of CdTe NPs on the fibril formation of Aβ peptides, incubation solutions of Aβ1-40 with and without NPs have been prepared. The kinetics of fibrillisation can be monitored by a dye-binding assay with thioflavin T, the fluorescent spectrum of which can be altered with the growth of fibrils.149,151,155 As for the neat peptide, the fibrillisation follows a conventional nucleated-growth mechanism that can be described by a lag phase, a rapid exponential growth, and a final equilibrium state.
Skaat et al.169 have designed nanoparticles that modulate the formation of toxic amyloid plaques. Fluorescent maghemite nanoparticles have been coated with Aβ40 or LPFFD peptide. The kinetics of the Aβ40 fibrillisation process in the absence and presence of varying concentrations of the Aβ40 or LPFFD conjugated nanoparticles were elucidated. The non-peptide conjugated fluorescent nanoparticles were found not to affect the Aβ40 fibrillisation process significantly. However, the Aβ40-grafted nanoparticles accelerated the fibrillisation process while the LPFFD-conjugated nanoparticles inhibited it. These bioactive fluorescent magnetic iron oxide nanoparticles can be used as an efficient tool to study, and control, the Aβ40 fibril formation process by MRI and fluorescence imaging.
4. Multivalent cationic signaling pathways
Multivalent cations can accelerate amyloid fibrillisation both in vitro and in vivo. However, the main claim of this section is that the acceleration effects of multivalent cations on Aβ fibrillisation are strongly related to the cell-type.4.1. All-atom molecular dynamics simulation
We have used the molecular dynamic simulation method to achieve an in-depth understanding of the effect of multivalent cations on amyloid aggregation. Two simulations were performed as follows.We simulated two amyloids in the presence of single-valence (i.e. Na+) and divalent cations (i.e. Ca2+) in canonical ensemble (NVT) conditions. The initial atomic coordinates of amyloids were constructed from the crystal structure (code: 1IYT). We used the OPLS-AA force field170 and SPC water model171 to describe the solvent. The GROMACS version 4.5.3 software was employed. In both simulations, two amyloids were immersed in a rectangular water box 70 × 70 × 90 Å (Angstroms), with about 14200 water molecules. In one simulation the system was neutralized by 6 sodium cations and in another by 3 calcium cations. The systems were minimized using the steepest descent method. Electrostatic interactions were treated by the particle mesh Ewald method.172 The LINC algorithm173 was used to constrain bonds involving hydrogen atoms so that a 2 fs (femto-second) time step was used. The system was equilibrated at a constant temperature (T) = 300 K (Kelvin) with the help of the Berendsen coupling procedure171 during 0.6 ns (nano-second). The total length of each trajectory was 50 ns; snapshots were taken every 1 ps (pico-second).
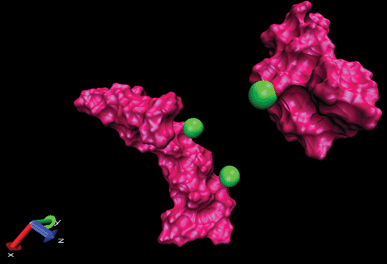 | ||
| Fig. 9 Display of two amyloids in the presence of Ca2+ where two cations are placed on one amyloid and another cation placed on another amyloid (snapshots were taken by VMD software). Unpublished data by the authors. | ||
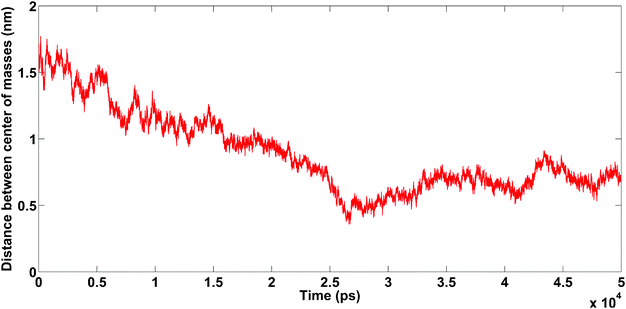
The DCMA in the presence of Na+ changes permanently during simulation (50 ns). This shows that the binding between amyloids is not stable. On the other hand, in the presence of Ca2+, the amyloids are close to each other at about t = 25 ns, and DCMA remains in a stable state.
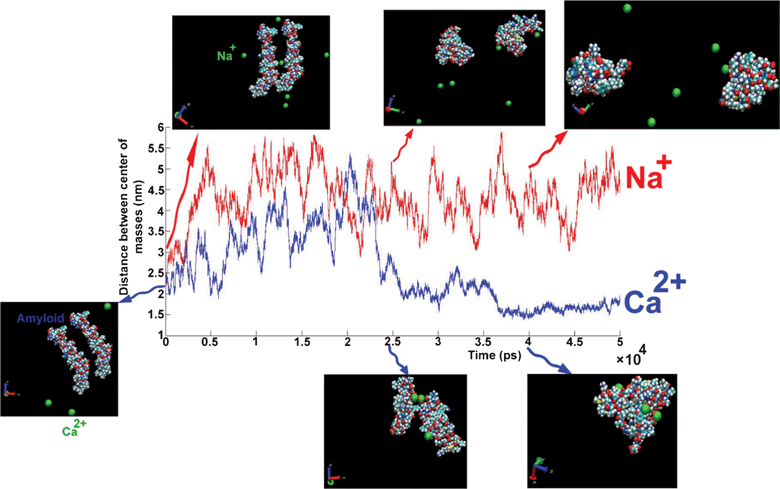 | ||
| Fig. 10 The distance between the centers of mass of amyloids as a function of simulation length for different cations: Na+ (red line), Ca2+ (blue line). Snapshots at three different times for each trajectory plotted on curve. Unpublished data by the authors. | ||
In the Fe3+ and Ca2+ simulations, parts of one amyloid bind to another amyloid at the beginning (because of the short-range interaction (hydrophobic interaction)), then cations bind to the amyloids and a stable binding between amyloids is created (see Fig. 11 and 12).
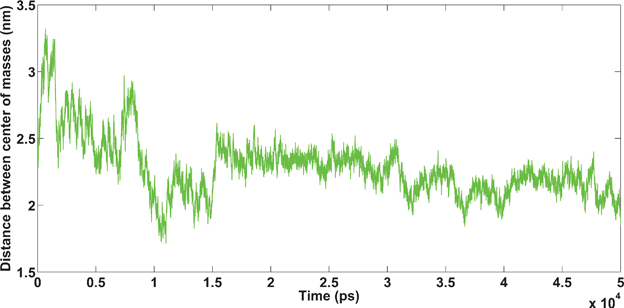 | ||
| Fig. 11 Amyloids in the presence of Ca2+ cations. Unpublished data by the authors. | ||
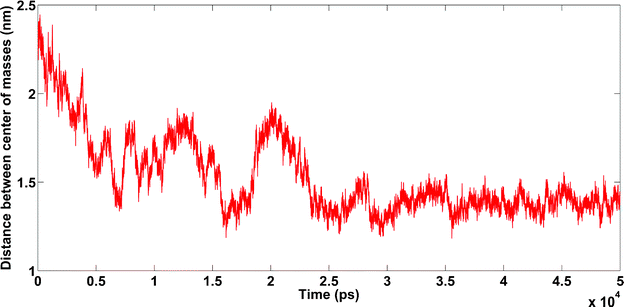 | ||
| Fig. 12 Amyloids in the presence of Fe3+ cations. Unpublished data by the authors. | ||
4.1.4.1. Amyloids in the presence of K+. In this simulation, amyloids did not bind to each other (see Fig. 13).
 | ||
| Fig. 13 Amyloids in the presence of K+ cations. Unpublished data by the authors. | ||
4.1.4.2. Amyloids in the presence of Na+. The system was neutralized by six Na+ cations. Hydrophobic interactions between the amyloids caused them to bind to each other, showing the importance of amyloid concentration in the solution (see Fig. 14).
 | ||
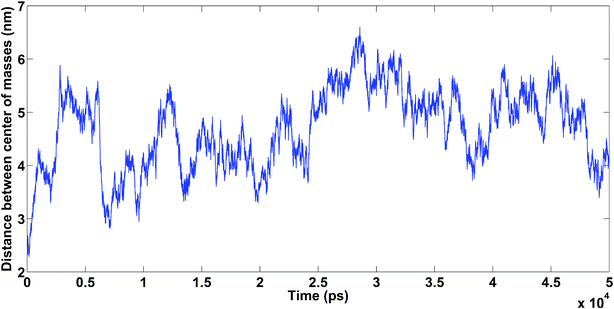
| Fig. 14 Amyloids in the presence of Na+. Unpublished data by the authors. | ||
4.2. Importance of ignored matters in multivalent cations
As already mentioned, dysregulation of calcium signaling is an important role in the development and pathogenesis of AD; however, the underlying mechanisms remain unclear. While there are several hypotheses for AD, many do not satisfactorily incorporate the elements of protein misfolding and of multivalent cations. This might explain inconsistencies in the literature on Aβ formation that can be due to various technical limitations (e.g. the variation of cell medium used, in vitro and in vivo models). Thus, it is possible, if not probable, in the absence of a greater understanding of these other factors, that there may be wasteful repetition of various experimental models which will not make any progress because they continue to involve inherently flawed or inappropriate methods or will be interpreted incorrectly. Both in vitro and in vivo studies to investigate more closely these variables and factors involved in the unbalancing of multivalent cation concentrations may therefore be a necessary objective for the future. Such ‘back-to-first principles’ experiments are not glamorous and are often hard to get funding for, yet they may help resolve some of the fundamental confounding variables involved in model systems to maximize the outcome of future investigations and in the long term reduce further unnecessary waste of valuable research resources (i.e. time and money).In order to have a comprehensive knowledge of multivalent cationic sources in the body, the distribution of elements (i.e. iron, copper, zinc, magnesium, and calcium) in the normal brain can serve as a useful starting point. In the study of diseases such as AD, where there is region-specific neuropathology, more in-depth knowledge of regional distribution of the elements could also be highly relevant. In AD the amygdala, hippocampus, caudate nucleus, cortex, corpus mamillare, gyruscinguli, and hypothalamus are all affected by Aβ-related pathology. Fig. 15a presents the distributions of a number of vital metals in these same brain regions in a postmortem normal brain.174–178 For comparison, Fig. 15b shows the concentrations of copper, iron, and zinc measured using micro particle-induced X-ray emission in the rims and cores of senile plaques and also in the neuropil of the amygdala of nine AD patients. Data on the distribution of the same ions in the neuropil of the amygdala of five neurologically normal control subjects is also presented illustrating the enrichment of these elements in plaque formation.174–178 There are no significant differences between the observed levels of individual elements in either the rim or the core of the senile plaques. What is worth noting, however, is the prominence of both zinc and iron in the senile plaque-related measures, which is not so evident in the AD neuropil measurements. In addition, copper is significantly elevated in the rim of senile plaques compared with AD neuropil (P < 0.05). Comparison of AD and control neuropil revealed a significant elevation of Zn in AD subjects (P < 0.05).
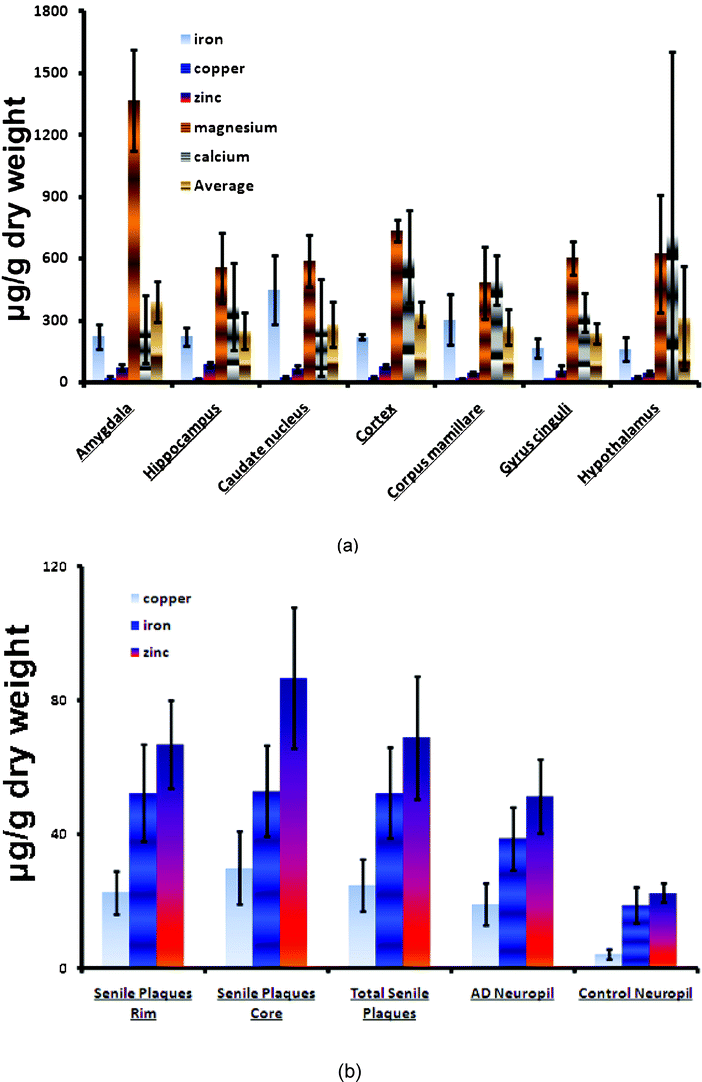 | ||
| Fig. 15 (a) Case series data and results of the analysis of various metals in postmortem human normal brains; (b) case data and results of copper, iron, and zinc concentrations in various plaque-related measures in AD subjects and in neuropil in both AD and normal subjects.174,178–180 | ||
While these data reveal some interesting trends, there is still a paucity of knowledge on the distribution of elements in the brain and, likewise, of cellular receptors that can modulate multivalent cationic signaling pathways. Improving the information available in both of these areas may provide major insights that could help develop modes of intervention to inhibit the development or enable the early detection of AD.
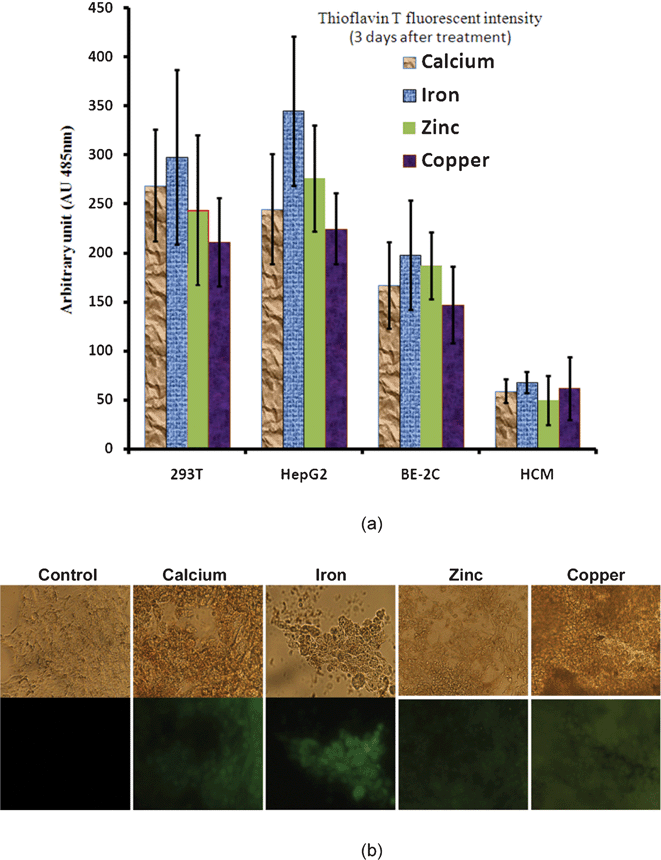 | ||
| Fig. 16 Amyloid formation in the presence of cationic ions. (a) Thioflavin T fluorescence measured in various cell lines exposed to the same amount of cationic ions at a 5 μM concentration (b) Corresponding light and fluorescence microscope images for control and treated kidney cells. Unpublished data by the authors. | ||
5. Future perspectives—application of nanotechnology principles to AD
On the basis of recent observations and developments in technical approaches, there may be considerable value in attempting to better understand the relationship between intracellular and extracellular matrix (ECM) levels of multivalent cations with respect to the malfunction of various biological systems. In relation to AD, the fact that Aβ can be formed in both intracellular and ECM environments and that those disturbances to the balance of multivalent cations have already been observed in AD supports this hypothesis.3,6,88,90,174,175,178,180,182–186We would encourage researchers, in the context of various experimental approaches to dissect the underlying mechanisms of AD, and to also consider the simultaneous measurement of cations. In doing so, they should explore the nature of possible associations between cation levels and Aβ misfolding, levels in the ECM, and dysregulation of corresponding signaling pathways in the intracellular environment. Further information yielded by such efforts could provide very valuable insights into possible mechanisms for the prevention or treatment or indeed the diagnosis of AD. In achieving these ambitious aims, new and exciting opportunities are presented for nano-bioscientists. These can contribute their valuable expertise to the development of smart brain penetrating nano-sensors, which might be suitable for the early detection of the disturbances to multivalent systems that are thought to play a pivotal role in the development and progression of AD.
A further facet of the cation measurement is the emergence over the last decade or so of a whole array of Aβ degrading enzymes which are zinc metalloproteases. Neprilysin 1 & 2, endothelin converting enzymes, angiotensin converting enzyme and aminopeptidase A, all of which coincidentally are also vasomodulatory enzymes and can contribute to increased blood pressure and reduced cerebral blood flow are all elevated in AD.62 Interestingly, many of these are also found to be elevated in AD60 while the peptides they all generate are also likely to be elevated. However as yet accurate measures of these peptides remains a challenge in plasma or serum because of their often rapid transformation to other bioactive or inactive metabolites. Nanoscience could help with these challenges of measurement which could be of considerable benefit not only in helping to provide new information for AD but also provide new data that will also be of relevance in the hypertension, stroke and kidney disease fields.
Another area in which nanotechnology can aid dementia research is potentially bringing increased levels of sensitivity and specificity to the endeavours to find new biomarkers for AD which will allow earlier diagnosis. While various CSF measures of Aβ and tau are the most advanced, there remains discomfort with regard to their universal use both from the perspective of the clinical procedure (i.e. lumbar puncture) needed to obtain the necessary sample to the sensitivity of the measure. At present the Alzheimer's disease imaging initiative (ADNI) has been undertaking extensive imaging based studies to better understand the natural course of brain changes and atrophy in aging, MCI and AD. From these endeavours and others there have been significant developments in the identification of more robust objective measures (i.e. brain atrophy) of disease progression than the currently used clinical assessment tools. In doing so, these studies while bringing greater stringency to the outcome measurements are also helping to reduce the necessary sample sizes for clinical trials and in doing so reduce costs.
Nanotechnology may help to improve on the current endeavours which attempt to combine both the imaging and CSF biomarker measurement strategies to help diagnose patients sooner but also increase the accuracy of subjects recruited to clinical trials. Improved sensitivity in CSF biosampling would be of benefit. However given the invasive nature of CSF sampling, the application of nanotechnological techniques to improve the sensitivity of plasma measures of Aβ may be of benefit. This is particularly relevant as the lack of satisfaction of Aβ as a plasma biomarker may be due to its low levels in plasma and the variable sensitivity of methods used to quantify it. Similar work could relate to proposals that AChE187 may also be a potential plasma biomarker for AD.
Indeed, while nanotechnology can certainly help with remaining diagnostic limitations at present, the added flexibility that nanotechnology has in being able to offer therapeutic promise whilst at the necessary site of action brings an extra dimension to things. In other words the use of sensitive methods to either diagnose patients clinically or for trials is really only most helpful if there is also a long-term effective treatment that can then begin to be administered. However, if one can develop a method that not only allows for earlier diagnosis and at the same time begins to treat, which is a potential that nanoscience offers, then the bleak picture predicted for the increase of AD in the future could be significantly altered.
References
- http://www.alz.co.uk/research/world-report. , Date of access 20.02.2012.
- C. M. Dobson, Protein Folding and Misfolding, Nature, 2003, 426, 884–890 CrossRef CAS.
- C. J. Frederickson, J.-Y. Koh and A. I. Bush, The neurobiology of zinc in health and disease, Nat. Rev. Neurosci., 2005, 6, 449–462 CrossRef CAS.
- L. Mucke, Neuroscience: Alzheimer's disease, Nature, 2009, 461, 895–897 CrossRef CAS.
- F. M. LaFerla, Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease, Nat. Rev. Neurosci., 2002, 3, 862–872 CrossRef CAS.
- M. Gough, C. Parr-Sturgess and E. Parkin, Zinc Metalloproteinases and Amyloid Beta-Peptide Metabolism: The Positive Side of Proteolysis in Alzheimer's Disease, Biochem. Res. Int., 2011, 721463, 13 Search PubMed.
- M. Mahmoudi, P. Stroeve, A. S. Milani and A. Arbab, Superparamagnetic iron oxide nanoparticles for biomedical applications,Nova Science Publishers, Inc., New York, 2010 Search PubMed.
- R. Von Bernhardi and J. Eugenín, Alzheimer’s disease: Redox dysregulation as a common denominator for diverse pathogenic mechanisms, Antioxidants and Redox Signaling, 2012, 16, 974–1031 CrossRef CAS.
- B. L. Plassman, K. M. Langa, G. G. Fisher, S. G. Heeringa, D. R. Weir, M. B. Ofstedal, J. R. Burke, M. D. Hurd, G. G. Potter, W. L. Rodgers, D. C. Steffens, R. J. Willis and R. B. Wallace, Prevalence of Dementia in the United States: The Aging, Demographics, and Memory Study, Neuroepidemiology, 2007, 29, 125–132 CAS.
- L. E. Hebert, P. A. Scherr, J. L. Bienias, D. A. Bennett and D. A. Evans, Alzheimer disease in the US population: prevalence estimates using the 2000 census, Arch. Neurol., 2003, 60, 1119–1122 CrossRef.
- M. Heron, D. L. Hoyert, S. L. Murphy, J. Xu, K. D. Kochanek and B. Tejada-Vera, Deaths: final data for 2006., National Vital Statistics Report, 2009, 57, 1–134 Search PubMed.
- C. P. Ferri, M. Prince, C. Brayne, H. Brodaty, L. Fratiglioni, M. Ganguli, K. Hall, K. Hasegawa, H. Hendrie, Y. Huang, A. Jorm, C. Mathers, P. R. Menezes, E. Rimmer and M. Scazufca, Global prevalence of dementia: a Delphi consensus study, Lancet, 2005, 366, 2112–7 CrossRef.
- P. A. Broderick, D. N. Rahni and E. H. Kolodny, Bioimaging in neurodegeneration, Humana Press, New York, 2005 Search PubMed.
- D. Soffer, Cerebral amyloid angiopathy—a disease or age-related condition, Isr Med Assoc J., 2006, 8, 803–806 CAS.
- R. C. Petersen, Mild cognitive impairment: transition between aging and Alzheimer's disease, Neurologia (Barcelona, Spain), 2000, 15, 93–101 CAS.
- S. Russell H, Is aging part of Alzheimer's disease, or is Alzheimer's disease part of aging?, Neurobiol. Aging, 2007, 28, 1465–1480 CrossRef.
- J. L. Cummings, R. Doody and C. Clark, Disease-modifying therapies for Alzheimer disease, Neurology, 2007, 69, 1622–1634 CrossRef.
- H. Braak and E. Braak, Neuropathological staging of Alzheimer-related changes, Acta Neuropathol., 1991, 82, 239–59 CrossRef CAS.
- P. M. Thompson, K. M. Hayashi, R. A. Dutton, M.-C. Chiang, A. D. Leow, E. R. Sowell, G. De Zubicaray, J. T. Becker, O. L. Lopez, H. J. Aizenstein and A. W. Toga, Tracking Alzheimer's Disease, Ann. N. Y. Acad. Sci., 2007, 1097, 183–214 CrossRef.
- K. Sambamurti, N. Greig and D. Lahiri, Advances in the cellular and molecular biology of the beta-amyloid protein in Alzheimer's disease, NeuroMol. Med., 2002, 1, 1–31 CrossRef CAS.
- H. A. Lashuel, D. Hartley, B. M. Petre, T. Walz and P. T. Lansbury, Neurodegenerative disease: Amyloid pores from pathogenic mutations, Nature, 2002, 418, 291–291 CrossRef CAS.
- L. Haataja, T. Gurlo, C. J. Huang and P. C. Butler, Islet Amyloid in Type 2 Diabetes, and the Toxic Oligomer Hypothesis, Endocr. Rev., 2008, 29, 303–316 CrossRef CAS.
- B. Sommer, Alzheimer's disease and the amyloid cascade hypothesis: ten years on, Curr. Opin. Pharmacol., 2002, 2, 87–92 CrossRef CAS.
- S. S. Sisodia, Beta-amyloid precursor protein cleavage by a membrane-bound protease, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 6075–6079 CrossRef CAS.
- A. L. Brunkan and A. M. Goate, Presenilin function and γ-secretase activity, J. Neurochem., 2005, 93, 769–792 CrossRef CAS.
- L. Bertram, M. B. McQueen, K. Mullin, D. Blacker and R. E. Tanzi, Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database, Nat. Genet., 2007, 39, 17–23 CrossRef CAS.
- J. D. Sipe, Amyloid proteins—the beta sheet conformation and disease, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed.
- K. Uemura, C. M. Lill, X. Li, J. A. Peters, A. Ivanov, Z. Fan, B. DeStrooper, B. J. Bacskai, B. T. Hyman and O. Berezovska, Allosteric Modulation of PS1/γ-Secretase Conformation Correlates with Amyloid β42/40 Ratio, PLoS One, 2009, 4, e7893 Search PubMed.
- A. P. Carvalho, L. C. Gursky, R. T. Rosa, A. U. M. Rymovicz, P. M. S. Campelo, A. M. T. Grégio, C. Y. Koga-Ito, L. P. Samaranayake and E. A. R. Rosa, Non-steroidal anti-inflammatory drugs may modulate the protease activity of Candida albicans, Microb. Pathog., 2010, 49, 315–322 CrossRef CAS.
- L.-M. Munter, P. Voigt, A. Harmeier, D. Kaden, K. E. Gottschalk, C. Weise, R. Pipkorn, M. Schaefer, D. Langosch and G. Multhaup, GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of A[beta]42, EMBO J., 2007, 26, 1702–1712 CrossRef CAS.
- I. Dewachter, L. Ris, S. Croes, P. Borghgraef, H. Devijver, T. Voets, B. Nilius, E. Godaux and F. Van Leuven, Modulation of synaptic plasticity and Tau phosphorylation by wild-type and mutant presenilin1., Neurobiol. Aging, 2008, 29, 639–652 CrossRef CAS.
- D. Walsh, I. Klyubin, J. V. Fadeeva, W. K. Cullen, R. Anwyl, M. S. Wolfe, M. J. Rowan and D. J. Selkoe, Naturally secreted oligomers of amyloid b protein potently inhibit hippocampal long-term potentiation in vivo, Nature, 2002, 416, 535–539 CrossRef CAS.
- D. M. Walsh, D. M. Hartley, Y. Kusumoto, Y. Fezoui, M. Margaret, M. M. Condron, A. Lomakin, G. B. Benedek, D. J. Selkoe and D. B. Teplow, Amyloid betaprotein fibrillogenesis. Structure and biological activity of protofibrillar intermediates, J. Biol. Chem., 1999, 274, 25945–25952 CrossRef CAS.
- D. M. Walsh and D. J. Selkoe, Abeta Oligomers a decade of discovery, J. Neurochem., 2007, 101, 1172–1184 CrossRef CAS.
- M. Ramakrishnan, K. K. Kandimalla, T. M. Wengenack, K. G. Howell and J. F. Poduslo, Biochemistry, 2009, 48, 10405–10415 CrossRef CAS.
- K. Brännström, A. Öhman and A. Olofsson, PLoS One, 2010, 6, e25157 Search PubMed.
- M. C. Montalto, G. Farrar and C. T. Hehir, Fibrillar and Oligomeric β-Amyloid as Distinct Local Biomarkers for Alzheimer's Disease, Ann. N. Y. Acad. Sci., 2007, 1097, 239–258 CrossRef CAS.
- Y. Fezoui, D. M. Hartley, J. D. Harper, R. Khurana, D. M. Walsh, M. M. Condron, D. J. Selkoe, P. T. Lansbury, A. L. Fink and D. B. Teplow, An improved method of preparing the amyloid β-protein for fibrillogenesis and neurotoxicity experiments, Amyloid, 2000, 7, 166–178 CrossRef CAS.
- T. B. David, Preparation of Amyloid β-Protein for Structural and Functional Studies, in Methods in Enzymology, ed. K. Indu and W. Ronald, Academic Press, 2006, vol. 413, pp 20–33 Search PubMed.
- K. Wiesehan, S. A. Funke, M. Fries and D. Willbold, Purification of recombinantly expressed and cytotoxic human amyloid-beta peptide 1–42, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 856, 229–233 CrossRef CAS.
- H. Wei and Z. Xie, Anesthesia, calcium homeostasis and Alzheimer's disease, Curr. Alzheimer Res., 2009, 6, 30–35 CrossRef CAS.
- A. M. D'Ursi, M. R. Armenante, R. Guerrini, S. Salvadori, G. Sorrentino and D. Picone, Solution Structure of Amyloid β-Peptide (25–35) in Different Media, J. Med. Chem., 2004, 47, 4231–4238 CrossRef CAS.
- T. Kohno, K. Kobayashi, T. Maeda, K. Sato and A. Takashima, Three-Dimensional Structures of the Amyloid β Peptide (25–35) in Membrane-Mimicking Environment, Biochemistry, 1996, 35, 16094–16104 CrossRef CAS.
- J. Suh and W. S. Chei, Metal complexes as artificial proteases: toward catalytic drugs, Curr. Opin. Chem. Biol., 2008, 12, 207–213 CrossRef CAS.
- J. D. Harper and P. T. Lansbury, Models of Amyloid Seeding in Alzheimer’s Disease and Scrapie: Mechanistic Truths and Physiological Consequences of the Time-Dependent Solubility of Amyloid Proteins, Annu. Rev. Biochem., 1997, 66, 385–407 CrossRef CAS.
- O. Ashur-Fabian, Y. Segal-Ruder, E. Skutelsky, D. E. Brenneman, R. A. Steingart, E. Giladi and I. Gozes, Peptides, 2003, 24, 1413–1423 CrossRef CAS.
- T. Lührs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Döbeli, D. Schubert and R. Riek, 3D structure of Alzheimer's amyloid-β(1–42) fibrils, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef.
- L. O. Tjernberg, A. Tjernberg, N. Bark, Y. Shi, B. P. Ruzsicska, Z. Bu, J. Thyberg and D. J. Callaway, Assembling amyloid fibrils from designed structures containing a significant amyloid beta-peptide fragment, Biochem J., 2002, 366, 343–351 CAS.
- J. Zheng, H. Jang, B. Ma, C.-J. Tsai and R. Nussinov, Modeling the Alzheimer A²17-42 Fibril Architecture: Tight Intermolecular Sheet-Sheet Association and Intramolecular Hydrated Cavities, Biophys. J., 2007, 93, 3046–3057 CrossRef CAS.
- J. Van Dorpe, L. Smeijers, I. Dewachter, D. Nuyens, K. Spittaels, C. Van den Haute, M. Mercken, D. Moechars, I. Laenen, C. Kuiperi, K. Bruynseels, I. Tesseur, R. Loos, H. Vanderstichele, F. Checler, R. Sciot and F. Van Leuven, Prominent Cerebral Amyloid Angiopathy in Transgenic Mice Overexpressing the London Mutant of Human APP in Neurons, Am. J. Pathol., 2000, 157, 1283–1298 CrossRef CAS.
- C. S. Atwood, R. D. Moir, X. Huang, R. C. Scarpa, N. M. E. Bacarra, D. M. Romano, M. A. Hartshorn, R. E. Tanzi and A. I. Bush, Dramatic Aggregation of Alzheimer Aβ by Cu(II) Is Induced by Conditions Representing Physiological Acidosis, J. Biol. Chem., 1998, 273, 12817–12826 CrossRef CAS.
- N. C. Inestrosa, J. P. Sagal and M. Colombres, Acetylcholinesterase interaction with Alzheimer amyloid β Alzheimer's disease, ed. J. R. Harris and F. Fahrenholz, Springer, US, 2005, vol. 38, pp 299–317 Search PubMed.
- C. Morgan, M. P. Bugueño, J. Garrido and N. C. Inestrosa, Laminin affects polymerization, depolymerization and neurotoxicity of Aβ peptide, Peptides, 2002, 23, 1229–1240 CrossRef CAS.
- L. Guilloreau, L. Damian, Y. Coppel, H. Mazarguil, M. Winterhalter and P. Faller, Structural and thermodynamical properties of Cu(II) amyloid-β 16/28 complexes associated with Alzheimer's disease, J. Biol. Inorg. Chem., 2006, 11, 1024–1038 CrossRef CAS.
- L. Hou and M. G. Zagorski, NMR Reveals Anomalous Copper(II) Binding to the Amyloid Aβ Peptide of Alzheimer's Disease, J. Am. Chem. Soc., 2006, 128, 9260–9261 CrossRef CAS.
- M. L. Steven, Iron deposits in multiple sclerosis and Alzheimer's disease brains, Brain Res., 1997, 760, 298–303 CrossRef.
- C. Glabe and A. I. Bush, Aβ Structure and aggregation Alzheimer's disease. ed. S. S. Sisodia and R. E.Tanzi, Springer, US, 2007, pp 113–131 Search PubMed.
- A. C. Miu and O. Benga, Aluminum and Alzheimer's disease: A new look, Journal of Alzheimer's Disease, 2006, 10, 179–201 CAS.
- S. L. Sabo, A. F. Ikin, J. D. Buxbaum and P. Greengard, The Amyloid Precursor Protein and Its Regulatory Protein, FE65, in Growth Cones and Synapses In Vitro and In Vivo, The Journal of Neuroscience, 2003, 23, 5407–5415 CAS.
- J. C. Palmer, S. Baig, P. G. Kehoe and S. Love, Endothelin-Converting Enzyme-2 Is Increased in Alzheimer's Disease and Up-Regulated by Aβ, Am. J. Pathol., 2009, 175, 262–270 CrossRef CAS.
- I. Dewachter, J. Van Dorpe, L. Smeijers, M. Gilis, C. Kuipéri, I. Laenen, N. Caluwaerts, D. Moechars, F. Checler, H. Vanderstichele and F. Van Leuven, Aging Increased Amyloid Peptide and Caused Amyloid Plaques in Brain of Old APP/V717I Transgenic Mice by a Different Mechanism than Mutant Presenilin1, The Journal of Neuroscience, 2000, 20, 6452–6458 CAS.
- J. S. Miners, Z. Van Helmond, K. Chalmers, G. Wilcock, S. Love and P. G. Kehoe, Decreased Expression and Activity of Neprilysin in Alzheimer Disease Are Associated With Cerebral Amyloid Angiopathy, J. Neuropathol. Exp. Neurol., 2006, 65, 1012–1021 CrossRef CAS.
- Y. C. Kudva, H. J. Hiddinga, P. C. Butler, C. S. Mueske and N. L. Eberhardt, Small heat shock proteins inhibit in vitro Aβ1–42 amyloidogenesis, FEBS Lett., 1997, 416, 117–121 CrossRef CAS.
- W. E. Van Nostrand and M Porter, Plasmin Cleavage of the Amyloid β-Protein: Alteration of Secondary Structure and Stimulation of Tissue Plasminogen Activator Activity, Biochemistry, 1999, 38, 11570–11576 CrossRef CAS.
- C. Reitz, F. J. A. van Rooij, H. D. Soares, M. P. M. de Maat, A. Hofman, J. C. M. Witteman and M. M. B. Breteler, Matrix metalloproteinase 3 haplotypes and plasma amyloid beta levels: The Rotterdam Study, Neurobiol. Aging, 2010, 31, 715–718 CrossRef CAS.
- A. Majumdar, H. Chung, G. Dolios, R. Wang, N. Asamoah, P. Lobel and F. R. Maxfield, Degradation of fibrillar forms of Alzheimer's amyloid β-peptide by macrophages, Neurobiol. Aging, 2008, 29, 707–715 CrossRef CAS.
- R. Deane and B. V. Zlokovic, Role of the Blood-Brain Barrier in the Pathogenesis of Alzheimers Disease, Curr. Alzheimer Res., 2007, 4, 191–197 CrossRef CAS.
- F. Licastro, S. Pedrini, L. Caputo, G. Annoni, L. J. Davis, C. Ferri, V. Casadei and L. M. E. Grimaldi, Increased plasma levels of interleukin-1, interleukin-6 and α-1-antichymotrypsin in patients with Alzheimer's disease: peripheral inflammation or signals from the brain?, J. Neuroimmunol., 2000, 103, 97–102 CrossRef CAS.
- S. Craft, E. Peskind, M. W. Schwartz, G. D. Schellenberg, M. Raskind and D. Porte, Cerebrospinal fluid and plasma insulin levels in Alzheimer's disease, Neurology, 1998, 50, 164–168 CAS.
- R. Mayeux, L. S. Honig, M.-X. Tang, J. Manly, Y. Stern, N. Schupf and P. D. Mehta, Plasma Aβ40 and Aβ42 and Alzheimer's disease, Neurology, 2003, 61, 1185–1190 CAS.
- S. Lorenzl, D. S. Albers, N. Relkin, T. Ngyuen, S. L. Hilgenberg, J. Chirichigno, M. E. Cudkowicz and M. F. Beal, Increased plasma levels of matrix metalloproteinase-9 in patients with Alzheimer's disease, Neurochem. Int., 2003, 43, 191–196 CrossRef CAS.
- R. Clarke, A. D. Smith, K. A. Jobst, H. Refsum, L. Sutton and P. M. Ueland, Folate, Vitamin B12, and Serum Total Homocysteine Levels in Confirmed Alzheimer Disease, Arch. Neurol., 1998, 55, 1449–1455 CrossRef CAS.
- P. D. Mehta, T. Pirttila, B. A. Patrick, M. Barshatzky and S. P. Mehta, Amyloid β protein 1–40 and 1–42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease, Neurosci. Lett., 2001, 304, 102–106 CrossRef CAS.
- T. Nishikimi, Y. Saito, K. Kitamura, T. Ishimitsu, T. Eto, K. Kangawa, H. Matsuo, T. Omae and H. Matsuoka, Increased plasma levels of adrenomedullin in patients with heart failure, J. Am. Coll. Cardiol., 1995, 26, 1424–1431 CrossRef CAS.
- R. Deane, S. Du Yan, R. K. Submamaryan, B. LaRue, S. Jovanovic, E. Hogg, D. Welch, L. Manness, C. Lin, J. Yu, H. Zhu, J. Ghiso, B. Frangione, A. Stern, A. M. Schmidt, D. L. Armstrong, B. Arnold, B. Liliensiek, P. Nawroth, F. Hofman, M. Kindy, D. Stern and B. Zlokovic, RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain, Nat. Med., 2003, 9, 907–913 CrossRef CAS.
- V. Calabrese, R. Lodi, C. Tonon, V. D'Agata, M. Sapienza, G. Scapagnini, A. Mangiameli, G. Pennisi, A. M. G. Stella and D. A. Butterfield, Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich's ataxia, J. Neurol. Sci., 2005, 233, 145–162 CrossRef CAS.
- I. G. Onyango and S. M. Khan, Oxidative Stress, Mitochondrial Dysfunction, and Stress Signaling in Alzheimers Disease, Curr. Alzheimer Res., 2006, 3, 339–349 CrossRef CAS.
- P.-C. Suen, K. Wu, E. S. Levine, H. T. J. Mount, J.-L. Xu, S.-Y. Lin and I. B. Black, Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-D-aspartate receptor subunit 1, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8191–8195 CrossRef CAS.
- E. S. Levine and J. E. Kolb, Brain-derived neurotrophic factor increases activity of NR2B-containing N-methyl-D-aspartate receptors in excised patches from hippocampal neurons, J. Neurosci. Res., 2000, 62, 357–362 CrossRef CAS.
- A. M. Marini, S. J. Rabin, R. H. Lipsky and I. Mocchetti, Activity-dependent Release of Brain-derived Neurotrophic Factor Underlies the Neuroprotective Effect of N-Methyl-D-aspartate, J. Biol. Chem., 1998, 273, 29394–29399 CrossRef CAS.
- S. Vaynman, Z. Ying and F. Gomez-Pinilla, Interplay between brain-derived neurotrophic factor and signal transduction modulators in the regulation of the effects of exercise on synaptic-plasticity, Neuroscience, 2003, 122, 647–657 CrossRef CAS.
- T. Tanaka, H. Saito and N. Matsuki, Inhibition of GABAA Synaptic Responses by Brain-Derived Neurotrophic Factor (BDNF) in Rat Hippocampus, The Journal of Neuroscience, 1997, 17, 2959–2966 CAS.
- R. H. Lipsky, K. Xu, D. Zhu, C. Kelly, A. Terhakopian, A. Novelli and A. M. Marini, Nuclear factor κB is a critical determinant in N-methyl-D-aspartate receptor-mediated neuroprotection, J. Neurochem., 2001, 78, 254–264 CrossRef CAS.
- B. E. Bisel, K. M. Henkins and K. D. Parfitt, Alzheimer Amyloid β-Peptide A-β25-35 Blocks Adenylate Cyclase-Mediated Forms of Hippocampal Long-Term Potentiation, Ann. N. Y. Acad. Sci., 2007, 1097, 58–63 CrossRef CAS.
- K. J. De Vos, A. J. Grierson, S. Ackerley and C. C. J. Miller, Role of Axonal Transport in Neurodegenerative Diseases, Annu. Rev. Neurosci., 2008, 31, 151–173 CrossRef CAS.
- J. Grutzendler, K. Helmin, J. Tsai and W.-B. Gan, Various Dendritic Abnormalities Are Associated with Fibrillar Amyloid Deposits in Alzheimer's Disease, Ann. N. Y. Acad. Sci., 2007, 1097, 30–39 CrossRef.
- O. Wirths, J. Weis, R. Kayed, T. C. Saido and T. A. Bayer, Age-dependent axonal degeneration in an Alzheimer mouse model, Neurobiol. Aging, 2007, 28, 1689–1699 CrossRef CAS.
- M. P. Cuajungco and K. Y. Fagét, Zinc takes the center stage: its paradoxical role in Alzheimer's disease, Brain Res. Rev., 2003, 41, 44–56 CrossRef CAS.
- M. A. Smith, P. L. R. Harris, L. M. Sayre and G. Perry, Iron accumulation in Alzheimer disease is a source of redox-generated free radicals, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 9866–9868 CrossRef CAS.
- X. Huang, M. P. Cuajungco, C. S. Atwood, R. D. Moir, R. E. Tanzi and A. I. Bush, Alzheimer's Disease, β-Amyloid Protein and Zinc, The Journal of Nutrition, 2000, 130, 1488S–1492S CAS.
- M. A. Smith, K. Hirai, K. Hsiao, M. A. Pappolla, P. L. R. Harris, S. L. Siedlak, M. Tabaton and G. Perry, Amyloid-β Deposition in Alzheimer Transgenic Mice Is Associated with Oxidative Stress, J. Neurochem., 1998, 70, 2212–2215 CrossRef CAS.
- K. R. Brunden, J. Q. Trojanowski and V. M. Y. Lee, Advances in tau-focused drug discovery for Alzheimer's disease and related tauopathies, Nat. Rev. Drug Discovery, 2009, 8, 783–793 CrossRef CAS.
- M.-S. Yu, K.-C. Suen, N.-S. Kwok, K.-F. So, J. Hugon and R. Chuen-Chung Chang, Beta-amyloid peptides induces neuronal apoptosis via a mechanism independent of unfolded protein responses, Apoptosis, 2006, 11, 687–700 CrossRef CAS.
- K. A. Roth, Caspases, Apoptosis, and Alzheimer Disease: Causation, Correlation, and Confusion, Journal of Neuropathology & Experimental Neurology, 2001, 60, 829–838 CAS.
- S. D. Yan, X. Chen, J. Fu, M. Chen, H. Zhu, A. Roher, T. Slattery, L. Zhao, M. Nagashima, J. Morser, A. Migheli, P. Nawroth, D. Stern and A. M. Schmidt, RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease., Nature, 1996, 382, 685–691 CrossRef CAS.
- J. Rogers and L.-F. Lue, Microglial chemotaxis, activation, and phagocytosis of amyloid β-peptide as linked phenomena in Alzheimer's disease, Neurochem. Int., 39, 333–340 CrossRef CAS.
- L. Meda, P. Baron and G. Scarlato, Glial activation in Alzheimer's disease: the role of Aβ and its associated proteins, Neurobiol. Aging, 2001, 22, 885–893 CrossRef CAS.
- J. G. Sheng, R. A. Jones, X. Q. Zhou, J. M. McGinness, L. J. Van Eldik, R. E. Mrak and W. S. T. Griffin, Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer's disease: potential significance for tau protein phosphorylation, Neurochem. Int., 2001, 39, 341–348 CrossRef CAS.
- D. Terwel, D. Muyllaert, I. Dewachter, P. Borghgraef, S. Croes, H. Devijver and F. Van Leuven, Amyloid Activates GSK-3β to Aggravate Neuronal Tauopathy in Bigenic Mice., Am. J. Pathol., 2008, 172, 786–798 CrossRef CAS.
- F. G. De Felice, D. Wu, M. P. Lambert, S. J. Fernandez, P. T. Velasco, P. N. Lacor, E. H. Bigio, J. Jerecic, P. J. Acton, P. J. Shughrue, E. Chen-Dodson, G. G. Kinney and W. L. Klein, Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers, Neurobiol. Aging, 2008, 29, 1334–1347 CrossRef CAS.
- M. Rapoport and A. Ferreira, PD98059 prevents neurite degeneration induced by fibrillar beta-amyloid in mature hippocampal neurons, J. Neurochem., 2000, 74, 125–133 CrossRef CAS.
- B. P. Tseng, K. N. Green, J. L. Chan, M. Blurton-Jones and F. M. LaFerla, Aβ inhibits the proteasome and enhances amyloid and tau accumulation., Neurobiol. Aging, 2008, 29, 1607–1618 CrossRef CAS.
- M. Rapoport, H. N. Dawson, L. I. Binder, M. P. Vitek and A. Ferreira, Tau is essential to β-amyloid-induced neurotoxicity, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6364–6369 CrossRef CAS.
- E. D. Roberson, K. Scearce-Levie, J. J. Palop, F. Yan, I. H. Cheng, T. Wu, H. Gerstein, G.-Q. Yu and L. Mucke, Reducing Endogenous Tau Ameliorates Amyloid β-Induced Deficits in an Alzheimer's Disease Mouse Model, Science, 2007, 316, 750–754 CrossRef CAS.
- N. F. Bence, R. M. Sampat and R. R. Kopito, Impairment of the Ubiquitin-Proteasome System by Protein Aggregation, Science, 2001, 292, 1552–1555 CrossRef CAS.
- J. Lowe, H. McDermott, M. Landon, R. J. Mayer and K. D. Wilkinson, Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases, J. Pathol., 1990, 161, 153–160 CrossRef CAS.
- A. Ciechanover and P. Brundin, The Ubiquitin Proteasome System in Neurodegenerative Diseases: Sometimes the Chicken, Sometimes the Egg, Neuron, 2003, 40, 427–446 CrossRef CAS.
- J. Lowe, R. J. Mayer and M. Landon, Ubiquitin in Neurodegenerative Diseases, Brain Pathol., 1993, 3, 55–65 CrossRef CAS.
- D. E. Ehrnhoefer, B. K. Y. Wong and M. R. Hayden, Convergent pathogenic pathways in Alzheimer's and Huntington's diseases: shared targets for drug development, Nat. Rev. Drug Discovery, 2011, 10, 853–867 CrossRef CAS.
- E. Karran, M. Mercken and B. D. Strooper, The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics, Nat. Rev. Drug Discovery, 2011, 10, 698–712 CrossRef CAS.
- E. D. Roberson and L. Mucke, 100 Years and Counting: Prospects for Defeating Alzheimer's Disease, Science, 2006, 314, 781–784 CrossRef.
- D. L. Brody and D. M. Holtzman, Active and Passive Immunotherapy for Neurodegenerative Disorders, Annu. Rev. Neurosci., 2008, 31, 175–193 CrossRef CAS.
- E. M. Sigurdsson, E. Knudsen, A. Asuni, C. Fitzer-Attas, D. Sage, D. Quartermain, F. Goni, B. Frangione and T. Wisniewski, An Attenuated Immune Response Is Sufficient to Enhance Cognition in an Alzheimer's Disease Mouse Model Immunized with Amyloid-β Derivatives, J. Neurosci., 2004, 24, 6277–6282 CrossRef CAS.
- L. O. Tjernberg, J. Näslund, F. Lindqvist, J. Johansson, A. R. Karlström, J. Thyberg, L. Terenius and C. Nordstedt, Arrest of β-Amyloid Fibril Formation by a Pentapeptide Ligand, J. Biol. Chem., 1996, 271, 8545–8548 CrossRef CAS.
- L. O. Tjernberg, C. Lilliehöök, D. J. E. Callaway, J. Näslund, S. Hahne, J. Thyberg, L. Terenius and C. Nordstedt, Controlling Amyloid β-Peptide Fibril Formation with Protease-stable Ligands, J. Biol. Chem., 1997, 272, 12601–12605 CrossRef CAS.
- K.-I. Watanabe, K. Nakamura, S. Akikusa, T. Okada, M. Kodaka, T. Konakahara and H. Okuno, Inhibitors of Fibril Formation and Cytotoxicity of β-Amyloid Peptide Composed of KLVFF Recognition Element and Flexible Hydrophilic Disrupting Element, Biochem. Biophys. Res. Commun., 2002, 290, 121–124 CrossRef CAS.
- J. E. Gestwicki, G. R. Crabtree and I. A. Graef, Harnessing Chaperones to Generate Small-Molecule Inhibitors of Amyloid β Aggregation, Science, 2004, 306, 865–869 CrossRef CAS.
- L. Rajendran, A. Schneider, G. Schlechtingen, S. Weidlich, J. Ries, T. Braxmeier, P. Schwille, J. B. Schulz, C. Schroeder, M. Simons, G. Jennings, H.-J. Knölker and K. Simons, Efficient Inhibition of the Alzheimer's Disease β-Secretase by Membrane Targeting, Science, 2008, 320, 520–523 CrossRef CAS.
- N. F. Schor, What the halted phase III γ-secretase inhibitor trial may (or may not) be telling us, Ann. Neurol., 2011, 69, 237–239 CrossRef CAS.
- R. Etcheberrigaray, M. Tan, I. Dewachter, C. Kuipéri, I. Van der Auwera, S. Wera, L. Qiao, B. Bank, T. J. Nelson, A. P. Kozikowski, F. Van Leuven and D. L. Alkon, Therapeutic effects of PKC activators in Alzheimer's disease transgenic mice, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11141–11146 CrossRef CAS.
- M. Garcia-Alloza, L. A. Borrelli, A. Rozkalne, B. T. Hyman and B. J. Bacskai, Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model, J. Neurochem., 2007, 102, 1095–1104 CrossRef CAS.
- M. Sano, K. L. Bell, D. Galasko, J. E. Galvin, R. G. Thomas, C. H. van Dyck and P. S. Aisen, A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease, Neurology, 2011, 77, 556–563 CrossRef CAS.
- B. Marchetti and M. P. Abbracchio, To be or not to be (inflamed)—is that the question in anti-inflammatory drug therapy of neurodegenerative disorders?, Trends Pharmacol. Sci., 2005, 26, 517–525 CrossRef CAS.
- A. H. Nagahara, D. A. Merrill, G. Coppola, S. Tsukada, B. E. Schroeder, G. M. Shaked, L. Wang, A. Blesch, A. Kim, J. M. Conner, E. Rockenstein, M. V. Chao, E. H. Koo, D. Geschwind, E. Masliah, A. A. Chiba and M. H. Tuszynski, Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease, Nat. Med., 2009, 15, 331–337 CrossRef CAS.
- R. S. Doody, S. I. Gavrilova, M. Sano, R. G. Thomas, P. S. Aisen, S. O. Bachurin, L. Seely and D. Hung, Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer's disease: a randomised, double-blind, placebo-controlled study, Lancet, 2008, 372, 207–215 CrossRef CAS.
- M. N. Lopez, R. A. Charter, B. Mostafavi, L. P. Nibut and W. E. Smith, Psychometric Properties of the Folstein Mini-Mental State Examination, Assessment, 2005, 12, 137–144 CrossRef.
- B. Vellas, S. Andrieu, C. Sampaio and G. Wilcock, Disease-modifying trials in Alzheimer's disease: a European task force consensus, Lancet Neurol., 2007, 6, 56–62 CrossRef.
- H. Hampel, R. Frank, K. Broich, S. J. Teipel, R. G. Katz, J. Hardy, K. Herholz, A. L. W. Bokde, F. Jessen, Y. C. Hoessler, W. R. Sanhai, H. Zetterberg, J. Woodcock and K. Blennow, Biomarkers for Alzheimer's disease: academic, industry and regulatory perspectives, Nat. Rev. Drug Discovery, 2010, 9, 560–574 CrossRef CAS.
- M. Citron, Alzheimer's disease: strategies for disease modification, Nat. Rev. Drug Discovery, 2010, 9, 387–398 CrossRef CAS.
- K. A. Jellinger, B. Janetzky, J. Attems and E. Kienzl, Alzheimer Review Series: Biomarkers for early diagnosis of Alzheimer disease: ‘Alzheimer Associated gene’—a new blood biomarker?, J. Cell. Mol. Med., 2008, 12, 1094–1117 CrossRef CAS.
- I. Blasko, K. Jellinger, G. Kemmler, W. Krampla, S. Jungwirth, I. Wichart, K. H. Tragl and P. Fischer, Conversion from cognitive health to mild cognitive impairment and Alzheimer's disease: Prediction by plasma amyloid beta 42, medial temporal lobe atrophy and homocysteine, Neurobiol. Aging, 2008, 29, 1–11 CrossRef CAS.
- A. M. Fagan, C. M. Roe, C. Xiong, M. A. Mintun, J. C. Morris and D. M. Holtzman, Cerebrospinal Fluid tau/beta-Amyloid42 Ratio as a Prediction of Cognitive Decline in Nondemented Older Adults, Arch. Neurol., 2007, 64, 343–349 CrossRef.
- N. S. M. Schoonenboom, W. M. van der Flier, M. A. Blankenstein, F. H. Bouwman, G. J. Van Kamp, F. Barkhof and P. Scheltens, CSF and MRI markers independently contribute to the diagnosis of Alzheimer's disease, Neurobiol. Aging, 2008, 29, 669–675 CrossRef CAS.
- F. H. Bouwman, N. S. M. Schoonenboom, N. A. Verwey, E. J. van Elk, A. Kok, M. A. Blankenstein, P. Scheltens and W. M. van der Flier, CSF biomarker levels in early and late onset Alzheimer's disease, Neurobiol. Aging, 2009, 30, 1895–1901 CrossRef CAS.
- J.-M. Lee, K. Blennow, N. Andreasen, O. Laterza, V. Modur, J. Olander, F. Gao, M. Ohlendorf and J. H. Ladenson, The Brain Injury Biomarker VLP-1 Is Increased in the Cerebrospinal Fluid of Alzheimer Disease Patients, Clin. Chem., 2008, 54, 1617–1623 CAS.
- P. Montuschi, P. J. Barnes and L. J. Roberts, Isoprostanes: markers and mediators of oxidative stress, FASEB J., 2004, 18, 1791–1800 CrossRef CAS.
- L. Fei and S. Perrett, Effect of Nanoparticles on Protein Folding and fibrillogenesis, Int. J. Mol. Sci., 2009, 10, 646–655 CrossRef CAS.
- R. C. Triulzi, Q. Dai, J. Zou, R. M. Leblanc, Q. Gu, J. Orbulescu and Q. Huo, Photothermal ablation of amyloid aggregates by gold nanoparticles, Colloids Surf., B, 2008, 63, 200–208 CrossRef CAS.
- T. Takahashi and H. Mihara, Acc. Chem. Res., 2008, 41, 1309 CrossRef CAS.
- I. Klyubin, D. M. Walsh, C. A. Lemere, W. K. Cullen, G. M. Shankar, Y. Betts, E. T. Spooner, L. Jiang, R. Anwyl, D. J. Selkoe and M. J. Rowan, Nat. Med., 2005, 11, 556 CrossRef CAS.
- Y. Bae, N. H. Kim, M. Kim, K. Y. Lee and S. W. Han, J. Am. Chem. Soc., 2008, 130, 5432 CrossRef CAS.
- G. A. DeVries, Y. Hu, B. Long, B. T. Neltner, O. Uzun, B. H. Wunsch and F. Stellacci, Science, 2007, 315, 358 CrossRef CAS.
- M. Klokkenburg, A. J. Houtepen, R. Koole, J. W. J. de Folter, B. M. Ern, E. van Faassen and D. Vanmaekelbergh, Nano Lett., 2007, 7, 2931 CrossRef CAS.
- K. S. Cho, D. V. Talapin, W. Gaschler and C. B. Murray, J. Am. Chem. Soc., 2005, 127, 7140 CrossRef CAS.
- S. Yoo, M. Yang, J. R. Brender, V. Subramanian, K. Sun, N. E. Joo, S. H. Jeong, A. Ramamoorthy and N. A. Kotov, Inhibition of Amyloid Peptide Fibrillation by Inorganic Nanoparticles: Functional Similarities with Proteins, Angew. Chem., Int. Ed., 2011, 50, 5110–5115 CrossRef CAS.
- Y. Xia, Nat. Mater., 2008, 7, 758 CrossRef CAS.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40 CrossRef CAS.
- M. Sarikaya, C. Tamerler, A. K. Y. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577 CrossRef CAS.
- S. Linse, C. Cabaleiro-Lago, W. F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8691 CrossRef CAS.
- S. Auer, A. Trovato and M. A. Vendruscolo, PLoS Comput. Biol., 2009, 5, e1000458 Search PubMed.
- W. H. Wu, X. Sun, Y. P. Yu, J. Hu, L. Zhao, Q. Liu, Y. F. Zhao and Y. M. Li, Biochem. Biophys. Res. Commun., 2008, 373, 315 CrossRef CAS.
- A. S Pai, I. Rubinstein and H. Onyuksel, Peptides, 2006, 7, 2858–2866 CrossRef.
- J. E. Kim and M. Lee, Biochem. Biophys. Res. Commun., 2003, 303, 576–579 CrossRef CAS.
- S. Rocha, A. F. Thunemann, C. Pereira Mdo, M. Coelho, H. Mohwald and G. Brezesinski, Biophys. Chem., 2008, 137, 35–42 CrossRef CAS.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, S. Lindman, A. M. Minogue, E. Thulin, D. M. Walsh, K. A. Dawson and S. Linse, J. Am. Chem. Soc., 2008, 130, 15437–15443 CrossRef CAS.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Biosensing with plasmonic nanosensors, Nat. Mater., 2008, 7, 442–453 CrossRef CAS.
- A. Relini, C. Canale, S. De Stefano, R. Rolandi, S. Giorgetti, M. Stoppini, A. Rossi, F. Fogolari, A. Corazza, G. Esposito, A Gliozzi and V. Bellotti, J. Biol. Chem., 2006, 281, 16521 CrossRef CAS.
- S. L. Myers, S. Jones, T. R. Jahn, I. J. Morten, G. A. Tennent, E. W. Hewitt and S. E. Radford, Biochemistry, 2006, 45, 2311 CrossRef CAS.
- V. Sluzky, J. A. Tamada, A. M. Klibanov and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 9377 CrossRef CAS.
- J. R. Lu, S. Perumal, E. T. Powers, J. W. Kelly, J. R. P. Webster and J. Penfold, J. Am. Chem. Soc., 2003, 125, 3751 CrossRef CAS.
- E. T. Powers and J. W. Kelly, J. Am. Chem. Soc., 2001, 123, 775 CrossRef CAS.
- K. Ikeda, T. Okada, S. I. Sawada, K. Akiyoshi and K. Matsuzaki, FEBS Lett., 2006, 580, 6587 CrossRef CAS.
- L. Xiao, D. Zhao, W. H. Chan, M. M. F. Choi and H. W. Li, Biomaterials, 2010, 31, 91 CrossRef CAS.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, K. A. Dawson and S. Linse, ACS Chem. Neurosci., 2010, 1, 279–287 CrossRef CAS.
- G. Thakur, M. Micic, Y. Yang, W. Li, D. Movia, S. Giordani, H. Zhang and R. M. Leblanc, International Journal of Alzheimer's Disease, 2011 Search PubMed , Article ID 502386.
- J. Milojevic, A. Raditsis and G. Melacini, Biophys. J., 2009, 97, 2585 CrossRef CAS.
- J. Milojevic, V. Esposito, R. Das and G. Melacini, J. Am. Chem. Soc., 2007, 129, 4282 CrossRef CAS.
- B. Bohrmann, L. Tjernberg, P. Kuner, S. Poli, B. Levet-Trafit, J. N. Slund, G. Richards, W. Huber, H. Dbeli and C. Nordstedt, J. Biol. Chem., 1999, 274, 15990 CrossRef CAS.
- H. Skaat, G. Shafir and S. Margel, J. Nanopart. Res., 2011, 13, 3521–3534 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. Dinola and J. R. Haak, Molecular dynamics with coupling to an external bat, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, LINCS: A Linear Constraint Solver for molecular simulations, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, Copper, iron and zinc in Alzheimer's disease senile plaques, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS.
- G. Faa, M. Lisci, M. P. Caria, R. Ambu, R. Sciot, V. M. Nurchi, R. Silvagni, A. Diaz and G. Crisponi, Brain copper, iron, magnesium, zinc, calcium, sulfur and phosphorus storage in Wilson's disease, J. Trace Elem. Med. Biol., 2001, 15, 155–160 CAS.
- K. S. Jagannatha Rao, R. V. Rao, P. Shanmugavelu and R. B. Menon, Trace elements in Alzheimer's disease brain: A new hypothesis, Alzheimer's Reports, 1999, 2, 241–246 Search PubMed.
- K. S. J. Rao, P. Shanmugavelu, S. K. Shankar, R. P. R. Devi, R. V. Rao and S. P. R. B. Menon, Trace elements in the cerebrospinal fluid in Alzheimer's disease, Alzheimer's Reports, 1999, 2, 333–338 Search PubMed.
- J. Kornhuber, K. W. Lange, P. Kruzik, W. D. Rausch, E. Gabriel, K. Jellinger and P. Riederer, Iron, copper, zinc, magnesium, and calcium in postmortem brain tissue from schizophrenic patients, Biol. Psychiatry, 1994, 36, 31–34 CrossRef CAS.
- T. M. Michel, S. Frangou, D. Thiemeyer, S. Camara, J. Jecel, K. Nara, A. Brunklaus, R. Zoechling and P. Riederer, Evidence for oxidative stress in the frontal cortex in patients with recurrent depressive disorder—a postmortem study, Psychiatry Res., 2007, 151, 145–150 CrossRef CAS.
- T. Lech and J. K. Sadlik, Zinc in Postmortem Body Tissues and Fluids, Biol. Trace Elem. Res., 2011, 142, 11–17 CrossRef CAS.
- M. Mahmoudi, S. Laurent, M. A. Shokrgozar and M. Hosseinkhani, Toxicity Evaluations of Superparamagnetic Iron Oxide Nanoparticles: Cell Vision Versus Physicochemical Properties of Nanoparticles, ACS Nano, 2011, 5, 7263–7276 CrossRef CAS.
- N. T. Hettiarachchi, A. Parker, M. L. Dallas, K. Pennington, C. C. Hung, H. A. Pearson, J. P. Boyle, P. Robinson and C. Peers, B-Synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells, J. Neurochem., 2009, 111, 1192–1201 CrossRef CAS.
- S. Kaja, R. S. Duncan, S. Longoria, J. D. Hilgenberg, A. J. Payne, N. M. Desai, R. A. Parikh, S. L. Burroughs, E. V. Gregg, D. L. Goad and P. Koulen, Novel mechanism of increased Ca2+ release following oxidative stress in neuronal cells involves type 2 inositol-1,4,5-trisphosphate receptors, Neuroscience, 2011, 175, 281–291 CrossRef CAS.
- R. Piacentini, C. Ripoli, L. Leone, F. Misiti, M. E. Clementi, M. D'Ascenzo, B. Giardina, G. B. Azzena and C. Grassi, Role of methionine 35 in the intracellular Ca2+ homeostasis dysregulation and Ca2+-dependent apoptosis induced by amyloid B-peptide in human neuroblastoma IMR32 cells, Journal of Neurochemistry, 2008, 107, 1070–1082 CAS.
- J. R. Steinert, A. W. Wyatt, R. Jacob and G. E. Mann, Redox modulation of Ca2+ signaling in human endothelial and smooth muscle cells in pre-eclampsia., Antioxid. Redox Signaling, 2009, 11, 1149–1163 CrossRef CAS.
- G. Maulthaup, H. Mechler and C. L. Masters, Characterization of the high affinity heparin binding site of the Alzheimer's disease βA4 amyloid precursor protein (APP) and its enhancement by zinc(II), J. Mol. Recognit., 1995, 8, 247–257 CrossRef.
- M.-S. García-Ayllón, O. Cauli, M.-X. Silveyra, R. Rodrigo, A. Candela, A. Compañ, R. Jover, M. Pérez-Mateo, S. Martínez, V. Felipo and J. Sáez-Valero, Brain cholinergic impairment in liver failure, Brain, 2008, 131, 2946–2956 CrossRef.
- F. Mangialasche, A. Solomon, B. Winblad, P. Mecocci and M. Kivipelto, Aging Research Center, Karolinska Institutet, Stockholm, Sweden, PubMed.gov, http://www.ncbi.nlm.nih.gov/pubmed/20610346, Erratum in Lancet Neurol., 2011, 10(6), 501 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |