DOI:
10.1039/C2RA20061A
(Paper)
RSC Adv., 2012,
2, 3379-3386
Synthesis and fluorescence properties of N-methyl-1,2-dihyroquinoline-3-carboxylate derivatives: light-emitting compounds in organic solvent, in neat form, and in water†
Received
11th January 2012
, Accepted 11th January 2012
First published on 16th January 2012
Abstract
The reaction of N-methyl-3-(arylamino)acrylates with in situ generated HI in CH3CN gave N-methyl-1,2-dihydroquinoline-3-carboxylates, which exhibited fluorescence at 490–560 nm in THF solution with good quantum yields (ΦF = 0.5–0.9). Electron-withdrawing substituents at the 6-position promoted high quantum yields. N-Methyl-1,2-dihydroquinoline-3-carboxylates were also emitted in neat form. Photoluminescence was observed from the dicarboxylic acid hydrolysis products in water without any organic co-solvent.
Introduction
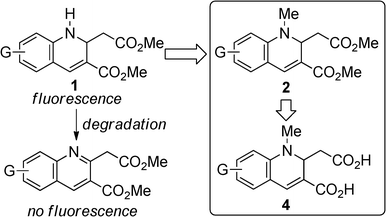
Luminescent materials have been utilized in various fields. For instance, emitting materials have been used as dopants in the emitting layer of organic light-emitting diodes (OLED), with promising application in display instruments.1 OLED devices with a non-doped emitting layer have also attracted considerable attention for device fabrication because the doping method requires complicated evaporation techniques with precise process control.2 In this case, luminescence from the emitting material in neat form is required. Recently, a variety of materials which fluoresce in the solid state have been reported.3 In the field of chemical and environmental sensors, differences in fluorescence characteristics of materials in solution and under aggregated conditions have been exploited.4–9 Some compounds were utilized as vapor sensors by using solid state.4b,e,i Furthermore, fluorescence from organic compounds has been utilized in bio-imaging and labeling techniques.4e,o,5f,9a,10 Various water-soluble emitting compounds have been developed,11,12 and have been utilized as sensors and bio-probes.4f,j,p,5d,f,h,k,10c,i,j,m Hence, it is important to explore organic compounds that emit not only in organic solutions but also in neat form and in water, because these fluorophores can be utilized in a variety of fields. In the design of light-emitting materials, some fluorophores are composed of a donor-acceptor structure connected to a rigid π-system to prevent non-radiative decay.13 1,2-Dihydroquinolines bearing an electron-withdrawing group at the 3-position possess such structural features. There are some reports concerning the fluorescence of 1,2-dihydroquinoline derivatives with electron-withdrawing substituent at the 3-position, but detailed investigations of their fluorescence properties are very few.14 We found that methyl 1,2-dihydroquinoline-3-carboxylates (1) emitted under UV irradiation both in solution and in neat form during our synthetic study of 1.15 The hydrolysis of 1 gives water-soluble emissive compounds that have two carboxylic acid moieties. However, 1 is relatively unstable under severe conditions, giving rise to quinoline derivatives that lose their fluorescence ability (Scheme 1). Therefore, we designed N-methylated compounds (2) to prevent oxidative aromatization. Here, we report the synthesis of 1,2-dihydroquinoline-3-carboxylate derivatives (2) from methyl N-methyl-3-(arylamino)acrylates (3), and the fluorescence properties of 2 and their hydrolyzed derivatives (4) in solution and in neat form. We found that 2 has a moderate-to-high fluorescence quantum yield (ΦF) and that 4 photoluminesces even in water, without an organic co-solvent to dissolve the compound.
 |
| | Scheme 1 Design for a stable fluorophore based on 1,2-dihydroquinoline structure. | |
Results and discussion
1. Synthesis of 2
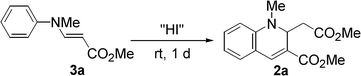
We first examined the synthesis of 2 by the reaction of methyl N-methyl-3-(arylamino)acrylates (3)16 with HI, analogous to the preparation of 1. However, the reaction was strongly inhibited when 3a was treated with 0.6 equivalent of 55% aq. HI in t-BuOH under the conditions reported for the synthesis of 1 (Table 1).15 By changing from t-BuOH to an aprotic solvent, the reaction proceeded to give 2a. We then tried to prepare 2a using in situ generated HI. When HI generation was completed using 0.6 equiv. of EtSH and 0.3 mol equiv. of I2 for 2 h before the addition of substrate 3a,17 the yield of 2a slightly decreased in CH3CN (58%). However, an increase in the yield was achieved by changing the addition order and the reaction time. When a mixture of 3a and I2 was treated with EtSH in CH3CN or CHCl3, 2a was obtained in 81% or 78% yield, respectively. Therefore, the formation of 2a was more efficient with in situ generated HI which was immediately available for reaction. As expected, the N-methylated product 2a was more stable than the non-methylated parent 1, which oxidized to the corresponding quinoline in 15% yield after 1 day of refluxing CH3CN under aerobic conditions. No reaction of 2 occurred under the same conditions. Compounds bearing various substituents were obtained in moderate to good yields (Scheme 2). Longer reaction times were required for compounds with electron-withdrawing substituents (3d, 3e, and 3f).
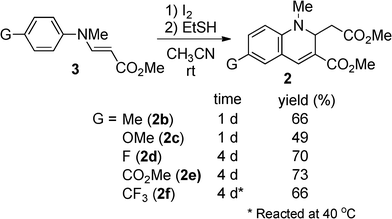 |
| | Scheme 2 Formation of 2b–f. | |
Table 1 Formation of 2a under various conditions
| HI source |
Addition order |
Solventa |
Yield of 2a (%) |
|
Concentration: 0.1 M based on 3a.
0.6 equiv. of HI was used.
0.6 equiv. of EtSH and 0.3 mol-equiv. of I2 were used.
The mixture of EtSH and I2 was stirred for 2 h before adding 3a.
|
| 55% aq. HIb |
— |
t-BuOH |
trace |
| |
|
CHCl3 |
11 |
| |
|
CH3CN |
64 |
| EtSH + I2c |
I2→EtSH→3ad |
CH3CN |
58 |
| |
3a→I2→EtSH |
CH3CN |
81 |
| |
|
CHCl3 |
78 |
The plausible reaction mechanism is depicted in Scheme 3. HI combines with 3a to provide the cationic intermediate A. Intermediate A undergoes addition to another 3a to give the intermediate B. The intramolecular nucleophilic addition occurred at the phenyl ring to give the intermediate C. The reason for requiring longer reaction times is to decrease the nucleophilicity of the phenyl ring in the case of compounds bearing electron-withdrawing substituents. The 1,2-dihydroquinoline derivative 2a is formed by elimination of proton and N-methylaniline, and HI is recovered.
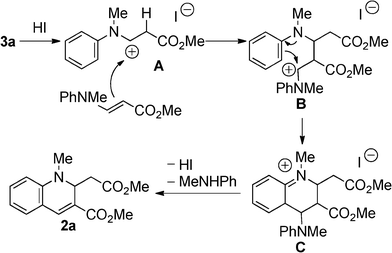 |
| | Scheme 3 Plausible reaction mechanism for the formation of 2a. | |
2. Optical properties of 2
Absorption and fluorescence spectra were measured in THF solution, and the results are summarized in Table 2. Maximum absorptions (λmax) were observed around 400–440 nm (Fig. 1). Increasing the electron-donating ability of substituents (G) attached at the 6-position of the 1,2-dihydroquinoline resulted in bathochromic shifts (2avs.2b and 2c). The compound bearing fluorine (2d) exhibited a red shift in λmax, even though the other compounds with electron-withdrawing substituents (2e and 2f) showed hypsochromic shifts. This is due to the character of fluorine atom, which possesses both inductive (-I) and mesomeric (-M) effects. Halogens have -I effect observed in the deactivation in the aromatic electrophilic substitution.18 However, fluorobenzenes show a reactivity approximating that of benzene itself. This can be explained from the unique energy level of the fluorine lone-pair orbital, which is lower than the adjacent orbital of benzene.19 Therefore, the electron-withdrawing character of a fluorine substituent will be reduced in the case of 2f. Furthermore, the tendency of the absorption shift by the substituents is closely related to the energy gap between HOMO and LUMO estimated by time-dependent density functional theory (TD-DFT) calculation with B3LYP/6-31+G* (Table S1†). By introducing electron-donating substituents such as methyl and methoxy group (2b and 2c), energy levels of HOMO were efficiently increased compared to that of 2a, which remained unchanged for energy level of LUMO. In the case of 2d, 2e, and 2f, both HOMO and LUMO level were lowered. And, in the case of 2e and 2f, the energy of LUMO was largely decreased. Therefore, the hypsochromic shift would be observed in the absorption spectra of 2e and 2f.
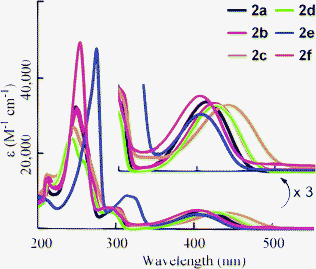 |
| | Fig. 1 Absorption spectra of 2a–f in THF (3.0 × 10−5 M). Inset: Vertical scale is expanded threefold. | |
Table 2 Optical properties of 2 in THF and in neat form
| Compound |
G |
In THF |
In neat form |
| |
|
λ
max (nm) [ε (M−1 cm−1)]a |
λ
em (nm)b [ΦF]c |
λ
em (nm)d |
|
Measured in THF (3.0 × 10−5 M).
Measured in THF (3.0 × 10−7 M). Excited at λmax.
Determined by fluorescein as a standard (ΦF = 0.90) excited at 436 nm.
Excited at λmax in THF.
|
|
2a
|
H |
411.5 [4,700] |
513 [0.80] |
526 |
|
2b
|
Me |
422.0 [4,600] |
529 [0.48] |
523 |
|
2c
|
OMe |
439.5 [4,500] |
552 [0.45] |
540 |
|
2d
|
F |
424.0 [4,500] |
531 [0.92] |
565 |
|
2e
|
CO2Me |
405.5 [3,800] |
494 [0.88] |
507 |
|
2f
|
CF3 |
403.0 [5,100] |
493 [0.70] |
490 |
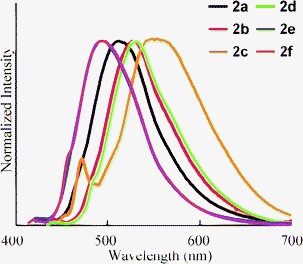
The fluorescence peaks (λem) of 2 were observed in the range 490–560 nm (Fig. 2). The changes in λem resembled the shifts in λabs according to the nature of the substituent: the electron-donating substituents showed red shifts in λem, whereas the electron-withdrawing substituents showed blue shifts. A side peak was observed in 2c. Although its origin is not clear, this peak is essential in 2c because the same peak was observed by changing the wavelength of excitation. The quantum yield (ΦF) of 2a was estimated as 0.80. The value of ΦF decreased when an electron-donating substituent was introduced (2b and 2c). Compound 2f also exhibited a slight decrease in ΦF. However, increases in ΦF for 2d and 2e were observed. Therefore, an efficient fluorophore can be obtained by the introduction of an electron-withdrawing substituent at the 6-position.
To gain further insight into the excited state, we examined solvent effects on the optical properties of 2a. The absorption and fluorescence spectra of 2a were measured in polar aprotic CH3CN and protic EtOH (Fig. 3). The results are summarized in Table 3. The absorption spectra were not affected by varying the solvents. However, the fluorescence peaks moved to longer wavelengths with increasing solvent polarity. This suggests that the excited state of these compounds has a polar structure. This also supports the fact that the HOMO orbital lies in the nitrogen atom, whereas the LUMO orbital is elongated to the carboxylate moiety (Table S1†). The quantum yield (ΦF) decreased with increasing solvent polarity upon going from THF to CH3CN. However, ΦF was 0.28 in EtOH. Therefore, 2 has the ability to fluoresce both in protic and polar solvents.
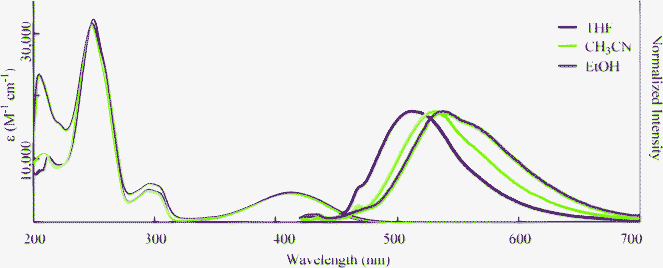 |
| | Fig. 3 Absorption (narrow line; 3.0 × 10−5 M) and fluorescence (bold line; 3.0 × 10−7 M, Excited at λmax) spectra of 2a in various organic solvents. | |
Table 3 Optical properties of 2a in various solvents
| Solvent |
λ
max (nm) [ε (M−1 cm−1)]a |
λ
em (nm)b [ΦF]c |
|
Concentration: 3.0 × 10−5 M.
Concentration: 3.0 × 10−7 M. Excited at λmax.
Determined by fluorescein as a standard (ΦF = 0.90) excited at 436 nm.
|
| THF |
412.0 [4,700] |
513 [0.80] |
| CH3CN |
411.5 [4,400] |
532 [0.52] |
| EtOH |
414.0 [4,600] |
539 [0.28] |
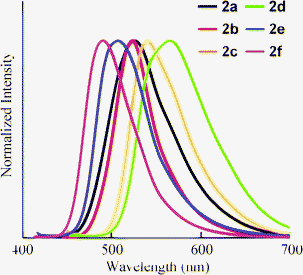
Furthermore, we studied the fluorescence characteristics of 2 in neat form. The compounds were obtained as solids, except for 2a, which was an oil at ambient temperature. Fluorescence in neat form was observed in 2a–f (Fig. 4), and the optical data are listed in Table 2. There was no correlation between the tendency of the shift of λem and the electronic character of the substituents, unlike the solution results. To clarify the difference in the luminescence intensity between aggregated conditions and in solution, we measured the absolute fluorescence quantum yield of 2b in crystalline form (ΦF = 0.34), which is close to the value in solution (ΦF = 0.48). Therefore, these compounds maintain their emission efficiencies to some extent in neat form.
3. Optical properties of 4
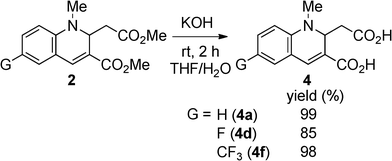
As mentioned above, 2a exhibited fluorescence in EtOH solution. We conjectured that compounds having the 1,2-dihyroquinoline-3-carboxylate structure could fluoresce even in water. Therefore, we saponified the esters (2) to the carboxylic acids (4). To achieve intense luminescence, we chose 2a, 2d, and 2f as the compounds of interest. The hydrolyses proceeded easily in good yield by the reaction of 2 with aqueous KOH in THF (Scheme 4).
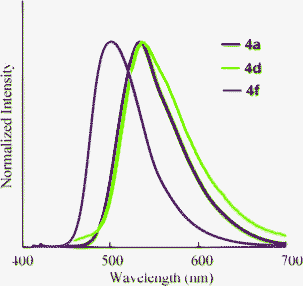
The optical properties of 4 in THF, water, and neat form are summarized in Table 4. It is clear that the dicarboxylic acids 4 retain the ability to fluoresce. The absorption and fluorescence peaks of 4 were comparable to 2 in THF (Fig. 5). However, the fluorescence quantum yields varied with the substituents. The non-substituted compounds (2avs.4a) and fluorinated compounds (2dvs.4d) showed decreases in ΦF from 0.80 and 0.92 to 0.62 and 0.46, respectively. In contrast, the ΦF value of 4f increased after hydrolysis. The reason for these differences is not clear, but might be explained by the differences in nitrogen basicity. From the investigation of the solvent effects for 2a, we found that these chromophores fluoresce in the polar excited state, which results from the interaction between the electron-donating nitrogen lone pair and the electron-accepting carboxyl moiety at the 3-position. Compound 4 has two acidic functional groups, and self-protonation of nitrogen is possible to decrease the population of the polar excited state, although we did not observe the protonated (zwitterionic) structure. When the strong electron-withdrawing trifluoromethyl group is introduced (Hammet value σp+ = –0.07 (F) and 0.61 (CF3)),20 protonation will be significantly inhibited, resulting in a high fluorescence quantum yield.
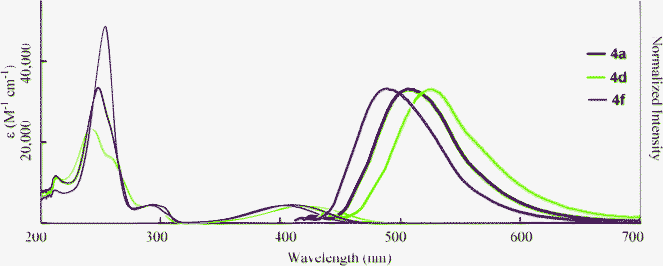 |
| | Fig. 5 Absorption (narrow line; 3.0 × 10−5 M in THF) and fluorescence (bold line; 3.0 × 10−7 M in THF, excited at λmax) spectra of 4. | |
Table 4 Optical properties of 4
| Compound |
G |
In THF |
In H2O |
In neat form |
| |
|
λ
max (nm) [ε (M−1 cm−1)]a |
λ
em (nm)b [ΦF]c |
λ
max (nm) [ε (M−1 cm−1)]a |
λ
em (nm)b [ΦF]d |
λ
em (nm)e |
|
Measured in 3.0 × 10−5 M.
Measured in 3.0 × 10−7 M. Excited at λmax.
Determined by fluorescein as a standard (ΦF = 0.90) excited at 436 nm.
Determined by quinine sulfate as a standard (ΦF = 0.55) excited at 366 nm.
Excited at λmax in THF.
|
|
4a
|
H |
410.0 [4,500] |
506 [0.62] |
391.0 [2,300] |
529 [0.12] |
534 |
|
4d
|
F |
419.5 [4,100] |
527 [0.46] |
396.5 [2,600] |
538 [0.06] |
537 |
|
4f
|
CF3 |
402.0 [4,400] |
488 [0.83] |
391.0 [3,700] |
508 [0.18] |
501 |
The fluorescence of 4 was also observed in neat form (Fig. 6). Unfortunately, we could not obtain a good crystal to investigate the solid-state structure using single crystal X-ray crystallography. However, since strong quenching was not observed, the donor-acceptor relationship for fluorescence exists.
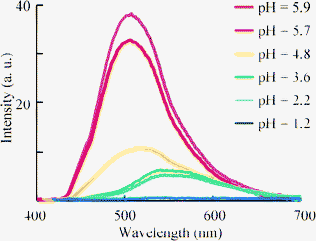
Finally, we examined the optical properties of 4 in water. Compound 4 could be dissolved in water without any organic co-solvent at a concentration of 3.0 × 10−5 M, which was sufficient to measure the absorption and fluorescence spectra (Fig. 7). λmax showed blue shifts compared to those in THF solution (Table 4), and the absorption coefficients in water decreased. This suggests an increase in protonation, which decreases the absorption based on the donor-acceptor interaction between the amino group and the carboxylic moiety of 4. However, the fluorescence from 4 was still observed in water with bathochromic shifts compared to those in THF. Therefore, protonation at the amino group of 4 in H2O did not fully quench the fluorescence from the excited state. The maximum fluorescence quantum yield (ΦF = 0.18) was obtained for 4f, which bore the strongly electron-withdrawing trifluoromethyl group that inhibited protonation of the amine, although pH of the aqueous solution of 4f was 5.9 caused by carboxylate moiety of 4f. And the polar character of H2O influenced the change in the fluorescence peaks. The protonation at nitrogen atom is influenced in pH value of solution. The pH dependence of fluorescence of dihydroquinolne derivatives was reported by Lygo and co-workers.21 They showed the decrease of fluorescence intensity by the protonation at nitrogen atom. Fluorescence character of 4f was also depended on pH. The intensity was decreased by reducing the pH value of solution with the bathochromic shift of fluorescence peak of 4f, which will be caused by the changing the polarity of the solution (Fig. 8).
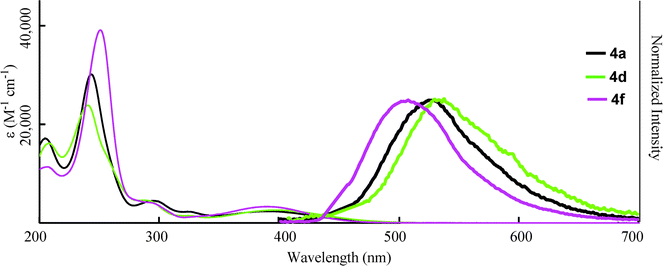 |
| | Fig. 7 Absorption (narrow line; 3.0 × 10−5 M in H2O) and fluorescence (bold line; 3.0 × 10−7 M in H2O, excited at λmax) spectra of 4. | |
Conclusions
In conclusion, we synthesized N-methyl-1,2-dihydroquinoline-3-carboxylates (2) in the presence of in situ generated HI in aprotic solvents. Organic solutions of 2 showed specific fluorescence with high quantum yields in the polar excited state. The introduction of an electron-withdrawing substituent at the 6-position efficiently increases the fluorescence quantum yield. Compound 2 also has the ability to fluorescence in neat form. Furthermore, compounds 4, bearing carboxylic acid moieties, could emit under UV irradiation in an organic solvent, in neat form, and even in water. Therefore, we found that the 1,2-dihydoroquinoline-3-carboxylate skeleton possesses fluorescence ability under various conditions. This result is expected to open a new direction in the design of fluorophores.
Experimental
General
Melting points were determined with Yanaco MP-J3 and values were uncorrected. 1H and 13C NMR measurements were performed on a Varian GEMINI 2000 (300 MHz) spectrometer. Chemical shifts (δ) of 1H NMR were expressed in parts per million downfield from tetramethylsilane (δ = 0) or DMSO-d5 (δ = 2.49) as an internal standard. Multiplicities are indicated as s (singlet), bs (broadened singlet), d (doublet), t (triplet), m (multiplet), and coupling constants (J) are reported in Hz unit. Chemical shifts (δ) of 13C NMR (75 MHz) are expressed in parts per million downfield or upfield from CDCl3 (δ = 77.0) or DMSO-d6 (δ = 39.6) (as an internal standard). IR spectra were recorded on a JASCO FT/IR-460 plus spectrometer in KBr disk or on NaCl plate. Absorption spectra were measured with quartz cell (1 cm × 1 cm) on a JASCO V570 spectrophotometer. Fluorescence spectra in solution were measured with quartz cell (1 cm × 1 cm) on a JASCO FP-6600 spectrofluorometer. Fluorescence spectra in neat form were measured with glass plates sandwiched the compound (oil or crystals). Absolute fluorescence quantum yield was measured with the integrating sphere unit. Elemental analyses (EA) were carried out on a Perkin-Elmer 2400CHN or an Exeter Analytical, Inc. CE-440 in Analytical Chemical Center of Chiba University. Mass spectra were carried out on a JEOL JMS-AX500, a JMS-HX110, or THERMO Scientific Exactive in the Analytical Chemical Center of Chiba University. Analytical thin-layer chromatography (TLC) was performed on glass plates pre-coated with silica gel (layer thickness 0.25 mm). Column chromatography was performed on 70–230 mesh silica gel. The commercially available materials were purchased from Aldrich Chemical Co., Tokyo Kasei Chemical Industry Co., Wako Pure Chemical Co., Kanto Chemical Co., and Nacalai Tesque Inc. Anhydrous CH3CN was distilled from sodium hydride and was stored with MS 3 Å. The reactions were performed under nitrogen atmosphere otherwise noted.
Typical procedure for the formation of methyl 1-methyl-1,2-dihydroquinoline-3-carboxylate derivatives (2)
Methyl 2-(Methoxycarbonylmethyl)-1-methyl-1,2-dihydroquinoline-3-carboxylate (2a).
To a solution of methyl 3-(methyl(phenyl)amino)acrylate (3a) (0.208 g, 1.09 mmol) in anhydrous CH3CN (10 mL) was added I2 (84.1 mg, 0.331 mmol) at room temperature under nitrogen atmosphere. To the mixture was added EtSH (39.0 mg, 0.628 mmol). The resultant mixture was stirred for 1 d at this temperature. EtOAc (10 mL) was added to the reaction mixture, and it was washed with saturated aqueous Na2SO3 (10 mL × 4). The aquous layer was extracted with EtOAc (5 mL × 2). The combined organic layer was dried with Na2SO4. After filtration and evaporation, the residue was subject to column chromatography on SiO2 (chloroform![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate = 15:1) to give methyl 2-(methoxycarbonylmethyl)-1-methyl-1,2-dihydroquinoline-3-carboxylate (2a) (0.121 g, 0.440 mmol) in 81% yield as yellow oil. 1H NMR (300 MHz, CDCl3): δ 2.40 (dd, 1H, J = 5.3 and 13.6 Hz), 2.52 (dd, 1H, J = 6.3 and 13.6 Hz), 3.02 (s, 3H), 3.53 (s, 3H), 3.81 (s, 3H), 4.94 (dd, 1H, J = 5.4 and 6.2 Hz), 6.56 (d, 1H, J = 8.2 Hz), 6.70 (dd, 1H, J = 6.7 and 7,4 Hz), 7.12 (dd, 1H, J = 1.4 and 7.5 Hz), 7.24 (dt, 1H, J = 1.5 and 6.9 Hz), 7.49 (s, 1H); 13C NMR (75 MHz, CDCl3): δ 37.3 (2C), 51.7, 51.8, 56.8, 111.9, 117.4, 120.2, 122.2, 129.7, 132.1, 135.3, 145.1, 165.7, 171.6; IR (neat): 2950, 1738, 1733, 1704, 1699, 1634, 1600, 1494, 1353, 1324, 1305, 1248, 1203, 1161, 1079, 1032, 1013, 986, 768, 750 cm−1. EA Calcd for C15H17NO4: C, 65.44; H, 6.22; N, 5.09%. Found: C, 65.39; H, 6.19; N, 5.03%.
:ethyl acetate = 15:1) to give methyl 2-(methoxycarbonylmethyl)-1-methyl-1,2-dihydroquinoline-3-carboxylate (2a) (0.121 g, 0.440 mmol) in 81% yield as yellow oil. 1H NMR (300 MHz, CDCl3): δ 2.40 (dd, 1H, J = 5.3 and 13.6 Hz), 2.52 (dd, 1H, J = 6.3 and 13.6 Hz), 3.02 (s, 3H), 3.53 (s, 3H), 3.81 (s, 3H), 4.94 (dd, 1H, J = 5.4 and 6.2 Hz), 6.56 (d, 1H, J = 8.2 Hz), 6.70 (dd, 1H, J = 6.7 and 7,4 Hz), 7.12 (dd, 1H, J = 1.4 and 7.5 Hz), 7.24 (dt, 1H, J = 1.5 and 6.9 Hz), 7.49 (s, 1H); 13C NMR (75 MHz, CDCl3): δ 37.3 (2C), 51.7, 51.8, 56.8, 111.9, 117.4, 120.2, 122.2, 129.7, 132.1, 135.3, 145.1, 165.7, 171.6; IR (neat): 2950, 1738, 1733, 1704, 1699, 1634, 1600, 1494, 1353, 1324, 1305, 1248, 1203, 1161, 1079, 1032, 1013, 986, 768, 750 cm−1. EA Calcd for C15H17NO4: C, 65.44; H, 6.22; N, 5.09%. Found: C, 65.39; H, 6.19; N, 5.03%.
Typical procedure for hydrolysis of 2
2-(Carboxymethyl)-1-methyl-1,2-dihydroquinoline-3-carboxylic acid (4a).
To a solution of methyl 2-(methoxycarbonylmethyl)-1-methyl-1,2-dihydroquinoline-3-carboxylate (2a) (55.0 mg, 0.200 mmol) in THF (1 mL) was added 1 M aqueous solution of KOH (1 mL). The mixture was stirred for 2 h at room temperature under air atmosphere. To the reaction mixture was added 5 mL water and washed with CHCl3 (5 mL × 4). To the aqueous layer was added 1 M aqueous HCl (2 mL) to acidify the solution to pH 1∼2. After extraction with CHCl3 (5 mL × 9), the extracted organic layer was dried with Na2SO4 and was concentrated under reduced pressure to give 2-(carboxymethyl)-1-methyl-1,2-dihydroquinoline-3-carboxylic acid (4a) (49.1 mg, 1.99 mmol) in 99% yield as a yellow solid. m.p. 120.8–121.1 °C (hexane–chloroform); 1H NMR (300 MHz, DMSO-d6): δ 2.16 (dd, 1H, J = 4.3 and 14.0 Hz), 2.32 (dd, 1H, J = 7.4 and 14.0 Hz), 2.94 (s, 3H), 4.80 (dd, 1H, J = 4.3 and 7.3 Hz), 6.59 (d, 1H, J = 8.6 Hz), 6.64 (t, 1H, J = 7.3 Hz), 7.19 (d, 1H, J = 7.4 Hz), 7.19 (t, 1H, J = 6.7 Hz), 7.38 (s, 1H), 12.33 (bs, 2H); 13C NMR (75 MHz, DMSO-d6): δ 37.1, 37.5, 56.2, 111.9, 116.9, 120.1, 123.7, 129.5, 131.9, 134.0, 144.9, 166.2, 172.3; IR (KBr): 2950(br), 2400(br), 1695, 1684, 1653, 1559, 1540, 1507, 1490, 1457, 1436, 1266, 1204, 1161, 750 cm−1. HRMS (ESI): Calcd for C13H13NNaO4 ([M+Na]+): 270.0737. Found: 270.0725.
Acknowledgements
This work was supported by Iodine Research Project in Chiba University and a Grant-in-Aid for Young Scientists (B) (20750026) from the Japan Society for the Promotion of Science.
References
-
(a)
Organic Electroluminescent Materials and Device, ed. S. Miyata and H. S. Nalwa, Gordon & Breach, Amsterdam, 1997 Search PubMed;
(b) Z. Liu, M. F. Qayyum, C. Wu, M. T. Whited, P. I. Djurovich, K. O. Hodgson, B. Hedman, E. I. Solomon and M. E. Thompson, J. Am. Chem. Soc., 2011, 133, 3700–3703 CrossRef CAS;
(c) P. Sonar, M. S. Soh, Y. H. Cheng, J. T. Henssier and A. Sellinger, Org. Lett., 2010, 12, 3292–3295 CrossRef CAS;
(d) Y. Wu, X. Hao, J. Wu, J. Jin and X. Ba, Macromolecules, 2010, 43, 731–738 CrossRef CAS;
(e) F. Liu, C. Tang, Q.-Q. Chen, F.-F. Shi, H.-B. Wu, L.-H. Xie, B. Peng, W. Wei, Y. Cao and W. Huang, J. Phys. Chem. C, 2009, 113, 4641–4647 CrossRef CAS;
(f) S.-H. Liao, J.-R. Shiu, S.-W. Liu, S.-J. Yeh, Y.-H. Chen, C.-T. Chen, T. J. Chow and C.-I. Wu, J. Am. Chem. Soc., 2009, 131, 763–777 CrossRef CAS;
(g) S. Tang, M. Liu, C. Gu, Y. Zhao, P. Lu, D. Lu, L. Liu, F. Shen, B. Yang and Y. Ma, J. Org. Chem., 2008, 73, 4212–4218 CrossRef CAS;
(h) L. Brauge, G. Vériot, G. Franc, R. Deloncle, A.-M. Caminade and J.-P. Majoral, Tetrahedron, 2006, 62, 11891–11899 CrossRef CAS;
(i) D. Gebeyehu, K. Walzer, G. He, M. Pfeiffer, K. Leo, J. Brandt, A. Gerhard, P. Stößel and H. Westweber, Synth. Met., 2005, 148, 205–211 CrossRef CAS;
(j) B. W. D'Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585–1595 CrossRef CAS;
(k) C. Xue and F.-T. Luo, Tetrahedron, 2003, 59, 5193–5198 CrossRef CAS;
(l) L.-H. Chan, R.-H. Lee, C.-F. Hsieh, H.-C. Yeh and C.-T. Chen, J. Am. Chem. Soc., 2002, 124, 6469–6479 CrossRef CAS.
-
(a) Z. Zhao, C. Deng, S. Chen, J. W. Y. Lam, W. Qin, L. Ping, Z. Wang, H. S. Kwok, Y. Ma, H. Qiu and B. Z. Tang, Chem. Commun., 2011, 47, 8847–8849 RSC;
(b) S. Matsumoto, B. Erdenebolor, M. Akazome, Y. Takata and M. Tamano, Org. Biomol. Chem., 2011, 9, 5941–5944 RSC;
(c) A. Thangthong, D. Meunmart, N. Prachumrak, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, Chem. Commun., 2011, 47, 7122–7124 RSC;
(d) J.-F. Gu, G.-H. Xie, L. Zhang, S.-F. Chen, Z.-Q. Lin, Z.-S. Zhang, J.-F. Zhao, L.-H. Xie, C. Tang, Y. Zhao, S.-Y. Liu and W. Huang, J. Phys. Chem. Lett., 2010, 1, 2849–2853 CrossRef CAS;
(e) S. Tao, Y. Zhou, C.-S. Lee, X. Zhang and S.-T. Lee, Chem. Mater., 2010, 22, 2138–2141 CrossRef CAS;
(f) C.-H. Wu, C.-H. Chien, F.-M. Hsu, P.-I. Shih and C.-F. Shu, J. Mater. Chem., 2009, 19, 1464–1470 RSC;
(g) S.-L. Lin, L.-H. Chan, R.-H. Lee, M.-Y. Yen, W.-J. Kuo, C.-T. Chen and R.-J. Jeng, Adv. Mater., 2008, 20, 3947–3952 CrossRef CAS;
(h) Y. Wei and C.-T. Chen, J. Am. Chem. Soc., 2007, 129, 7478–7479 CrossRef CAS;
(i) C. J. Tonzola, A. P. Kulkarni, A. P. Gifford, W. Kaminsky and S. A. Jenekhe, Adv. Funct. Mater., 2007, 17, 863–874 CrossRef CAS.
-
(a) S. H. Lim, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2011, 133, 10229–10238 CrossRef CAS;
(b) C. Muschelknautz, W. Frank and T. J. J. Müller, Org. Lett., 2011, 13, 2556–2559 CrossRef CAS;
(c) Z. Zhao, S. Chen, J. W. Y. Lam, P. Lu, Y. Zhong, K. S. Wong, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 2221–2223 RSC;
(d) M. Imoto, H. Ikeda, T. Fujii, H. Taniguchi, A. Tamaki, M. Takeda and K. Mizuno, Org. Lett., 2010, 12, 1940–1943 CrossRef CAS;
(e) F. Gao, Q. Liao, Z.-Z. Xu, Y.-H. Yue, Q. Wang, H.-L. Zhang and H.-B. Fu, Angew. Chem., Int. Ed., 2010, 49, 732–735 CAS;
(f) Z. Zhao, Z. Wang, P. Lu, C.Y. K. Chan, D. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Ma and B. Z. Tang, Angew. Chem., Int. Ed., 2009, 48, 7608–7611 CrossRef CAS;
(g) M. Shimizu, Y. Takeda, M. Higashi and T. Hiyama, Angew. Chem., Int. Ed., 2009, 48, 3653–3656 CrossRef CAS;
(h) S.-Y. Park, Y. Kubota, K. Funabiki and M. Matsui, Chem. Lett., 2009, 38, 162–163 CrossRef CAS.
-
(a) R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419–4476 CrossRef;
(b) B.-P. Jiang, D.-S. Guo and Y. Liu, J. Org. Chem., 2011, 76, 6101–6107 CrossRef CAS;
(c) K. Li, Y. Chen, W. Lu, N. Zhu and C.-M. Che, Chem. Eur. J., 2011, 17, 4109 CrossRef CAS;
(d) P. Sokkalingam and C.-H. Lee, J. Org. Chem., 2011, 76, 3820–3828 CrossRef CAS;
(e) B. Zhu, F. Yuan, R. Li, Y. Li, Q. Wei, Z. Ma, B. Du and X. Zhang, Chem. Commun., 2011, 47, 7098–7100 RSC;
(f) Y. Liu, C. Deng, L. Tang, A. Qin, R. Hu, J. Z. Sun and B. Z. Tang, J. Am. Chem. Soc., 2011, 133, 660–663 CrossRef CAS;
(g) Z. M. Hudson, X.-Y. Liu and S. Wang, Org. Lett., 2011, 13, 300–303 CrossRef CAS;
(h) Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951–13953 CrossRef CAS;
(i) C. He, Q. He, C. Deng, L. Shi, D. Zhu, Y. Fu, H. Cao and J. Cheng, Chem. Commun., 2010, 46, 7536–7538 RSC;
(j) Y. L. Jiang, P. Patel and S. M. Klein, Bioorg. Med. Chem., 2010, 18, 7034–7042 CrossRef CAS;
(k) H. N. Lee, N. J. Singh, S. K. Kim, J. Y. Kwon, Y. Y. Kim, K. S. Kim and J. Yoon, Tetrahedron Lett., 2007, 48, 169–172 CrossRef CAS;
(l) Y. M. Chung, B. Raman, D.-S. Kim and K. H. Ahn, Chem. Commun., 2006, 186–188 RSC;
(m) Z. Wang, D. Zhang and D. Zhu, Tetrahedron Lett., 2005, 46, 4609–4612 CrossRef CAS;
(n) K. Tanaka, T. Miura, N. Umezawa, Y. Urano, K. Kikuchi, T. Higuchi and T. Nagano, J. Am. Chem. Soc., 2001, 123, 2530–2536 CrossRef CAS;
(o) H. Kojima, N. Nakatsubo, K. Kikuchi, S. Kawahara, Y. Kirino, H. Nagoshi, Y. Hirata and T. Nagano, Anal. Chem., 1998, 70, 2446–2453 CrossRef CAS;
(p) V. Nardello and J.-M. Aubry, Tetrahedron Lett., 1997, 42, 7361–7364 CrossRef.
-
(a) Z. Li, L. Zhang, L. Wang, Y. Guo, L. Cai, M. Yu and L. Wei, Chem. Commun., 2011, 47, 5798–5800 RSC;
(b) S. Panda, P. B. Pati and S. S. Zade, Chem. Commun., 2011, 47, 4174–4176 RSC;
(c) B. Wang, Y. Gao, H.-W. Li, Z.-P. Hu and Y. Wu, Org. Biomol. Chem., 2011, 9, 4032–4034 RSC;
(d) J. Yuasa, A. Mitsui and T. Kawai, Chem. Commun., 2011, 47, 5807–5809 RSC;
(e) D. Ray and P.K. Bharadwaj, Inorg. Chem., 2008, 47, 2252–2254 CrossRef CAS;
(f) W. Liu, L. Xu, R. Sheng, P. Wang, H. Li and S. Wu, Org. Lett., 2007, 9, 3829–3832 CrossRef CAS;
(g) J.-S. Wu, W.-M. Liu, X.-Q. Zhuang, F. Wang, P.-F. Wang, S.-L. Tao, X.-H. Zhang, S.-K. Wu and S.-T. Lee, Org. Lett., 2007, 9, 33–36 CrossRef CAS;
(h) S. H. Kim, J. K. Choi, S. K. Kim, W. Sim and J. S. Kim, Tetrahedron Lett., 2006, 47, 3737–3741 CrossRef CAS;
(i) Y. Mei, P. A. Bentley and W. Wang, Tetrahedron Lett., 2006, 47, 2447–2449 CrossRef CAS;
(j) J. H. Bok, H. J. Kim, J. W. Lee, S. K. Kim, J. K. Choi, J. Vicens and J. S. Kim, Tetrahedron Lett., 2006, 47, 1237–1240 CrossRef CAS;
(k) J. V. Mello and N. S. Finney, J. Am. Chem. Soc., 2005, 127, 10124–10125 CrossRef.
-
(a) J. Feng, K. Tian, D. Hu, S. Wang, S. Li, Y. Zeng, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2011, 50, 8072–8076 CrossRef CAS;
(b) Y. Shiraishi, R. Miyamoto, X. Zhang and T. Hirai, Org. Lett., 2007, 9, 3921–3924 CrossRef CAS.
-
(a) C. Zhou, Y. Li, Y. Zheo, J. Zhang, W. Yang and Y. Li, Org. Lett., 2011, 13, 292–295 CrossRef CAS;
(b) C.-K. Koo, B. Lam, S. Leung, M. H.-W. Lam and W.-Y. Wong, J. Am. Chem. Soc., 2006, 128, 16434–16435 CrossRef CAS.
- S. Uchiyama, K. Takehira, T. Yoshihara, S. Tobita and T. Ohwada, Org. Lett., 2006, 8, 5869–5872 CrossRef CAS.
-
(a) M. K. Kuimova, G. Yahioglu, J. A. Levitt and K. Suhling, J. Am. Chem. Soc., 2008, 130, 6672–6673 CrossRef CAS;
(b) M. A. Haidekker, T. P. Brady, D. Lichlyter and E. A. Theodorakis, J. Am. Chem. Soc., 2006, 128, 398–399 CrossRef.
-
(a) L. D. Lavis and R. T. Ralnes, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS;
(b) A. Waggoner, Curr. Opin. Chem. Biol., 2006, 10, 62–66 CrossRef CAS;
(c) M. Kamiya, D. Asanuma, E. Kuranaga, A. Takeishi, M. Sakabe, M. Miura, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2011, 133, 12960–12963 CrossRef CAS;
(d) M. Kawaguchi, T. Okabe, S. Okudaira, K. Hanaoka, Y. Fujikawa, T. Terai, T. Komatsu, H. Kojima, J. Aoki and T. Nagano, J. Am. Chem. Soc., 2011, 133, 12021–12030 CrossRef CAS;
(e) F. Céspedes-Guirao, A. B. Ropero, E. Font-Sanchis, Á. Nadal, F. Fernández-Lázaro and Á. Sastre-Santos, Chem. Commun., 2011, 47, 8307–8309 RSC;
(f) K. Peneva, G. Mihov, A. Herrmann, N. Zarrabi, M. Börsch, T. M. Duncan and K. Müllen, J. Am. Chem. Soc., 2008, 130, 5398–5399 CrossRef CAS;
(g) R. Lalor, H. Baillie-Johnson, C. Redshaw, S. E. Matthews and A. Mueller, J. Am. Chem. Soc., 2008, 130, 2892–2893 CrossRef CAS;
(h) Y.-J. Lee, S. Datta and J.-P. Pellois, J. Am. Chem. Soc., 2008, 130, 2398–2399 CrossRef CAS;
(i) Y. Meyer, J.-A. Richard, M. Massonneau, P.-Y. Renard and A. Romieu, Org. Lett., 2008, 10, 1517–1520 CrossRef CAS;
(j) M. M. Pires and J. Chmielewski, Org. Lett., 2008, 10, 837–840 CrossRef CAS;
(k) W. M. Leevy, S. T. Gammon, H. Jiang, J. R. Johnson, D. J. Maxwell, E. N. Jackson, M. Marquez, D. Piwnica-Worms and B. D. Smith, J. Am. Chem. Soc., 2006, 128, 16476–16477 CrossRef CAS;
(l) K. Setsukinai, Y. Urano, K. Kakinuma, H. J. Majima and T. Nagano, J. Biol. Chem., 2003, 278, 3170–3175 CrossRef CAS;
(m) H. Takakusa, K. Kikuchi, Y. Urano, S. Sakamoto, K. Yamaguchi and T. Nagano, J. Am. Chem. Soc., 2002, 124, 1653–1657 CrossRef CAS.
-
(a) A. Romieu, D. Brossand, M. Hamon, H. Outaabout, C. Portal and R. -Y. Renard, Bioconjugate Chem., 2008, 19, 279–289 CrossRef CAS;
(b) G. Clavé, A. Bernardin, M. Massonneau, P.-Y. Renard and A. Romieu, Tetrahedron Lett., 2006, 47, 6229–6233 CrossRef;
(c) T. Mineno, T. Ueno, Y. Urano, H. Kojima and T. Nagano, Org. Lett., 2006, 8, 5963–5966 CrossRef CAS;
(d) P. Siejak and D. Frackowiak, J. Phys. Chem. B, 2005, 109, 14382–14386 CrossRef CAS;
(e) J. Zheng, J. T. Petty and R. M. Dickson, J. Am. Chem. Soc., 2003, 125, 7780–7781 CrossRef CAS.
- Water-soluble materials using organic cages:
(a) K. Ono, J. K. Klosterman, M. Yoshizawa, K. Sekiguchi, T. Tahara and M. Fujita, J. Am. Chem. Soc., 2009, 131, 12526–12527 CrossRef CAS;
(b) A. L. Koner and W. M. Nau, Supramol. Chem., 2007, 19, 55–66 CrossRef CAS;
(c) S. J. Dalgarno, S. A. Tucker, D. B. Bassil and J. L. Atwood, Science, 2005, 309, 2037–2039 CrossRef CAS.
-
(a) S. Matsumoto, D. Samata, M. Akazome and K. Ogura, Tetrahedron Lett., 2009, 50, 111–114 CrossRef CAS;
(b) G. Jones II, W. R. Jackson, C. Choi and W. R. Bergmark, J. Phys. Chem., 1985, 89, 294–300 CrossRef.
-
(a) I. A. Apple and O. Meth-Cohn, ARKIVOC, 2002,(vi), 4–14 CAS;
(b) N. S. Kozlov, L. F. Gladchenko and G. P. Korotyshova, Chem. Heterocycl. Compd., 1980, 16, 758–761 CrossRef;
(c) N. S. Kozlov, L. F. Gladchenko, G. V. Vorob'eva, V. A. Serzhanina and R. D. Sauts, Chem. Heterocycl. Compd., 1977, 13, 660–663 CrossRef.
- S. Matsumoto, T. Mori and M. Akazome, Synthesis, 2010, 3615–3622 CrossRef CAS.
- A. Padwa and L. Zhi, J. Am. Chem. Soc., 1990, 112, 2037–2038 CrossRef CAS.
-
(a)
A. J. Downs and C. J. Adams, in Comprehensive Inorganic Chemistry, ed. A. F. Trotman-Dickenson, Pergamon, Oxford, 1973, 2, 1107 Search PubMed;
(b) S. Matsumoto, T. Kishimoto and K. Ogura, Chem. Lett., 2002, 134–135 CrossRef CAS;
(c) S. M. Chervin, P. Abada and M. Koreeda, Org. Lett., 2000, 2, 369–373 CrossRef CAS.
-
M. B. Smith and J. March, in Advanced Organic Chemistry, 5th edition, John Wiley & Sons, Canada, 2001, 683 Search PubMed.
- S. Tomoda, K. Takamatsu and M. Iwaoka, Chem. Lett., 1998, 581–582 CrossRef CAS.
-
(a) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS;
(b) H. C. Brown and Y. Okamoto, J. Am. Chem. Soc., 1958, 80, 4979–4987 CrossRef CAS.
- O. N. Lygo, T. D. Nekipelova and E. N. Khodot, High Energy Chem., 2010, 44, 399–403 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procdures, characterization and NMR spectra for all new compounds, and a summary of TD-DFT calculations. |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.