Solubility improvements in aromatic polyimides by macromolecular engineering
Anindita
Ghosh
ab,
Suman Kumar
Sen
a,
Susanta
Banerjee
*a and
Brigitte
Voit
b
aMaterials Science Centre, Indian Institute of Technology, Kharagpur-721302, India. E-mail: susanta@matsc.iitkgp.ernet.in(S. Banerjee); Fax: +91 (3222) 255303; Tel: +91 (3222) 283972
bLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Str. 6, 01069, Dresden, Germany
First published on 20th March 2012
Abstract
Polyimides are proved to be an important class of polymers due to their outstanding set of physical and chemical properties. However, this class of polymers suffers from poor processability, which limits their scope of application. This review article deals with approaches that have been undertaken by different researchers to improve the processability of this class of polymers. Mostly, these are synthetic approaches using new diamine and dianhydride monomers that reduce the interchain interaction of the final poyimide by flexibilizing the polymer chain or increasing the fraction free volume. Accordingly, the structural factors that are responsible for better processability are discussed and representative diamine and diahydride structures are tabulated under different categories. Major efforts towards development of soluble polyimides but maintaining excellent mechanical and thermal properties have been done by our group and are also covered in this article.
 Anindita Ghosh | Anindita Ghosh received her PhD in Polymer Science and Technology from the Materials Science Centre, Indian Institute of Technology, Kharagpur, India in 2009. She was the recipient of the Alexander von Humboldt Research Fellowship (AvH) and spent two years (2009-2011) in the group of Professor Brigitte Voit at the Leibniz Institute of Polymer Research (IPF), Dresden. After postdoctoral work in Germany she joined Dow Chemical International Pvt. Ltd., Pune, India as a Lead Chemist (Research Specialist). Her major research interest is in high performance polymers as well as hyperbranched polymers, polymeric membranes and coatings. |
 Suman Kumar Sen | Suman Kumar Sen received his PhD in Polymer Science and Technology from the Materials Science Centre, Indian Institute of Technology, Kharagpur, India in 2011. He did his masters in Organic Chemistry (M.Sc.) at Banaras Hindu University (India). His research interests include designing and synthesising new monomers, the development of high performance polymeric membrane materials with special emphasis on membrane separation processes. He is currently a postdoctoral fellow in the Department of Forest Biomaterials, North Carolina State University, USA. His current research activities focus on the development of biodegradable polymer films (bioplastic) and producing biofuels using naturally available biopolymers. |
 Susanta Banerjee | Susanta Banerjee received his PhD in Polymer Chemistry in 1992 from the Indian Institute of Technology, Kharagpur. He joined the Defence Research and Development Organisation (DRDO) as a Scientist untill April 2004. He was AvH Fellow at the Technical University, Munich between 1997–1999. In 2004 he joined the R&D Centre of the General Electric Company, Bangalore as Lead Scientist. He moved to the Indian Institute of Technology in 2006 and holds the position of Full Professor at the Materials Science Centre. His major research interest is synthesis and characterization of new high performance polymers for low-K and membrane based applications as well as dendritic polymers, conjugated polymers and nanocomposites. |
 Brigitte Voit | Brigitte Voit received her PhD in Macromolecular Chemistry in 1990 from the University of Bayreuth, Germany. After postdoctoral work in 1991/92 at Eastman Kodak in Rochester, USA, and habilitation at Technische Universität München on hyperbranched polymers, in 1997 Brigitte Voit was jointly appointed Professor for "Organic Chemistry of Polymers“ at the University of Technology, Dresden and head of the Institute of Macromolecular Chemistry at the Leibniz Institute of Polymer Research (IPF), Dresden which she has lead as Scientific Director since 2002. Her major research interest is synthesis of new functional polymer architectures such as dendritic polymers, functional block and graft copolymers, and high performance polymers and nanocomposites. |
1. Introduction
Polyimides are high performance polymer materials characterized by their outstanding thermal stability owing to the presence of rigid aromatic moieties. These aromatic moieties lead, however, also to rigidity of the molecular chain and difficult intermolecular interactions, which result in problems in dissolving or melting polyimides before their decomposition temperature. The commonly used Kapton@ type polyimide can be processed only in its precursor form, polyamic acid (PAA). This, however, also causes problems as the polyamic acid is easily decomposes on storage, which leads to a decrease in molecular weight. Therefore, it must be stored at low temperature. The imidization process has to be carried out at high temperatures (250–300 °C). These high temperatures can cause damage to the substrate dramatically affecting the final material properties. Furthermore, a large amount of water is generated during the imidization process, which reduces the chain packing and affects final polymer properties. To improve the processability of polyimides, many attempts have been made to prepare polyimides which are either soluble or thermoplastic.Despite their processing difficulties, aromatic polyimides received great attention due to their outstanding thermal, mechanical and electrical properties.1–7 Polyimides have been in mass production since 1955. The polyimide materials are lightweight, flexible and resistant to heat and chemicals. Polyimides have found many applications in microelectronics, aircraft and the space industry, and new applications in the field of separation techniques are currently under development.8–12 These polymers are further used in the electronics industry for flexible cables, as an insulating film on magnetic wire and for medical tubing. For example, in a laptop computer, the cable that connects the main logic board to the display is often polyimide based with copper conductors. Examples of commercial polyimide films include Apical®, Kapton®, UPILEX®, VTEC PI®, Norton TH® and Kaptrex®. In the semiconductor industry polyimides are used as a high-temperature adhesives; it is also used as a mechanical stress buffer. Some polyimides can be used as photoresists; both “positive” and “negative” types of photoresist-like polyimide exist on the market. However, their applications have been limited in many fields because aromatic polyimides are normally insoluble in common organic solvents and have extremely high glass transition or melting temperatures, which preclude melt processability by injection moulding or extrusion. Hence, one of the drawbacks in using aromatic polyimides is their poor processability. Acordingly, great effort has been made to improve the processing characteristics of these intractable polyimides.13,14 Numerous methods to obtain polyimides with chemically modified chain structures have been introduced for balancing the thermal stability and the processability. The most important methods include incorporation of hinge atoms, kink units, or flexible spacer units either into the dianhydride or diamine fragment or both. Accordingly, many attempts have been undertaken to design and synthesize new diamines15–19 and dianhydrides,20–22 therefore producing a great variety of soluble and processable polyimides for various purposes and applications. Mainly, there are three methods to improve the processability of polyimides. One is the improvement of their solubility, as demonstrated, by Harris and Lanier,23 and other scientists.24 They successfully produced several organo-soluble polyimides25–27 by using aromatic monomers with bulky side-groups. The second is the improvement of solubility and thermoplasticity that was first shown by Takekoshi and co-workers of the General Electric Co. (4-1, Table 4).27 Later, they successfully produced and industrialised polyetherimides named as ULTEM® with an excellent processability. The last method is focused only on improvement of thermoplasticity. The National Aeronautics and Space Administration (NASA) and Mitsui Toatsu Chemicals, Inc. were involved in improving the melt flowability of polyimide in order to improve the thermoplasticity and the output was the melt flowable polyimide, LARC#1500 (4-2, Table 4).28,29 This review will focus on polyimide synthesis, difficulties encountered in processing aromatic polyimides, and finally methods and approaches employed so far to overcome the difficulties leading to processable polyimides.
2. Polyimides
2.1 Synthesis
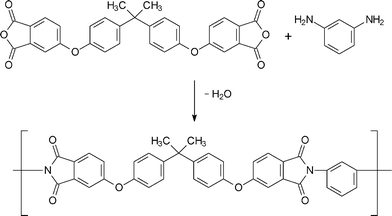
Polyimides are condensation polymers, generally derived from organic diamines and organic tetracarboxylic acids (or derivatives). Synthesis of polyimides is generally done by a two-step method that consists of the formation of the poly(amic acid) precursor followed by cyclodehydration (Scheme 1). Mainly, the synthetic techniques for polyimide formation involve thermal imidization, solution imidization and chemical imidization. A single stage homogeneous solution polymerization technique is also known for the polyimides, which are soluble in organic solvents at polymerization temperature. One of the examples of this type of polymer is the condensation product of 4,4′-(4,4-isopropylidenediphenoxy)bis(phthalic anhydride) (BPADA) and m-phenylene diamine (mPD) (Scheme 2). In this method of polymerization, completely cyclized polyimides are obtained directly from their corresponding stoichiometric mixture of tetracarboxylic acid dianhydride and diamine.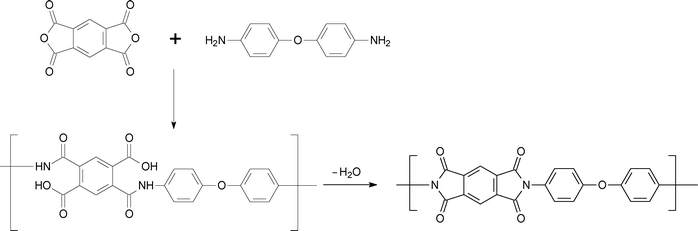 | ||
| Scheme 1 General synthesis of polyimide by a two-step method. | ||
 | ||
| Scheme 2 Synthesis of polyimide by a single-step method. | ||
2.2 Concepts to overcome solubility restrictions in aromatic polyimides
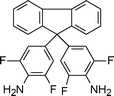
The difficulties in processing conventional aromatic polyimides are due to the inherent molecular features of aromatic polyimides. Molecular stiffness, high polarity and high intermolecular association forces make these polymers virtually insoluble in any organic medium, and raise the glass transition temperatures and melting to well above the decomposition temperature, which causes difficulties in both synthesis and processing. Therefore, the processing of the aromatic polyimides is usually carried out with soluble poly(amic acid) precursors, which are cast onto glass plates and converted to thin polyimide films by a rigorous thermal treatment. This process has been observed to have certain limitations, including the emission of volatile byproducts during curing and the storage instability of the poly(amic acid) intermediate.39 Thus, strategies towards processable aromatic polyimides arose which focused on chemical modifications, mainly by preparing new monomers that provide less molecular order, torsional mobility and lower intermolecular bonding. In the various alternatives to design new processable polyimides, some general approaches universally adopted are: introduction of aliphatic or another kind of flexible segments which reduce chain stiffness, introduction of bulky side substituents which help for separation of polymer chains and hinder molecular packing and crystallization, use of enlarged monomers containing angular bonds which suppress coplanar structures, use of 1,3-substituted instead of 1,4-substituted monomers, and/or use of asymmetric monomers, which lower regularity and molecular ordering, use of fluorinated diamines and dianhydrides and preparation of co-polyimides from two or more dianhydrides or diamines and introduction of branching. However, factors leading to better solubility or lower glass transition temperature (Tg) or melting temperature (Tm) in a polymer often conflict with other important requirements, such as high mechanical properties, thermal resistance or chemical resistance. Therefore, an adjusted degree of modification should be applied to balance the properties. For example, incorporation of siloxane oligomeric segments into a polyimide backbone yields soluble processable poly(imide siloxane) copolymers with a variety of good property characteristics. Although some thermal and thermooxidative stability is compromised through introduction of the siloxane segment, a number of improvements are also observed in processability, toughness, flexibility and in electrical properties.They reported that polyimides prepared from 6FDA (apart from when m = 12) could be dissolved in a wide range of strong polar solvents namely DMAc, DMF, NMP, m-cresol, as well as in conventional polar solvents such as THF and chloroform. On the other hand polyimides prepared from BTDA and ODPA could not be dissolved in all the above mentioned solvents. One can obviously infer that the introduction of flexible chains in the polymer backbone could not solely increase the solubility. Other factors, such as structural regularity of the flexible chain as well as the functional groups of the dianhydride units can hinder polymer solubility. The polyimide containing the hexaisopropylidene groups showed complete solubility for m = 2 to 10 where the bulky hexaisopropylidene groups increased the fractional free volume resulting in loose chain packing. However, when m = 12, structural regularity of the long flexible chain dominates over the loose chain packing coming from the hexafluoroisopropylidene units, leading to insolubility. Polyimides with BTDA and ODPA as a dianhydride unit, despite having long flexible –CH2– linkages, were insoluble in the above mentioned solvents due to the high regularity of the flexible chains. The carbonyl group present in BTDA also promotes insolubility.
Marek et al.44 reported the preparation of new poly(pyromellitimide)s based on 4,4′-(alkylenediyldioxy)dianiline (DA) where m = 4, 6, 10. The representative diamine structure is shown in Scheme 3.
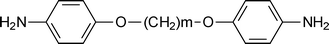 | ||
| Scheme 3 Structure of 4,4′-(alkylenediyldioxy)dianiline. | ||
The presence of dioxyalkylene structure in the diamine part of the polypyromellitimide repeat unit, however, does not lead to enhanced polymer solubility. Polyimides, and even polyisoimides, based on PMDA/DA-n are not soluble in conventional solvents (dimethylformamide, dimethylacetamide, N-methylpyrrolidone, chlorinated hydrocarbons, ethers, cresol); they can only be dissolved in sulphuric acid. This fact indicates that even dilution of the pyromellitimide structures and reduction of local rigidity by dioxyalkylene chains cannot improve the poor solubility of polypyromellitimides. Later, Marek et al.45 changed the dianhydride system to 4,4′-(hexafluoroisopropylidene) bis(phthalic anhydride) (6FDA) since polyimides prepared from monomers containing hexafluoroisopropylidene groups are attractive materials because of their high transparency, low dielectric constant, low moisture absorption, resistance to photochemical degradation, good solubility and gas permeation properties.46–50 Moreover, 6FDA was less reactive in the acylation of amines than pyromellitic dianhydride (PMDA) and gave polyimides with lower molecular weights,51 with the latter being strongly dependent on the diamine monomer that is used. All the resulting polyimides with 6FDA as a dianhydride unit were soluble in tetrahydrofuran and polar aprotic solvents except the polyimide with DA = 10, (6FDA/DA-10) whereas dimethyl sulfoxide at room temperature was not a good solvent for these polymers.
 | ||
| Scheme 4 Structure of polyimide taken from Ref. 52. | ||





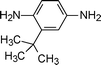

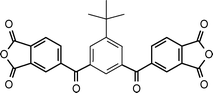

Flexible links such as –O–, –SO2–, or –CH2–, or bulky groups (hexafluoroisopropylidene, isopropylidene, etc.) are commonly employed as solubilizing moieties,72–74 but they often bring about a loss of thermal stability and chain stiffness of polyimides. Alternative approach towards tractable aromatic polyimides is to use nonplanar monomers where the nonplanarity provides the conformational freedom to lower chain regularity and rigidity.72,75 With the aim of improving the tractability, polyimides derived from m-terphenyl dianhydride have been synthesized, yet they still showed limited solubility in organic solvents.76 Furthermore, it has been observed that the incorporation of a tert-butyl pendent group in the 5′ position of the m-terphenyl moiety improves their solubility. Therefore, polyimides from 5′-tert-butyl-m-terphenyl-3,4,3′′,4′′-tetracarboxylic acid dianhydride (BTPDA) (1-7, Table 1) were soluble in m-cresol, N-methylpyrrolidinone (NMP), and N,N-dimethylacetamide (DMAc).77 They also observed that the presence of tert-butyl side groups showed minimum detrimental effect to the thermal stability of the polyimides compared to the polyimides without tert-butyl groups and showed a thermal decomposition temperature above 500 °C. This finding is somewhat contrary to that observed by Huang et al.62 indicating that the decrease in interchain interactions brought about by the pendent tert-butyl groups can be overcome by the effect of structural rigidity imparted by the tert-butyl substituents. These soluble m-terphenyl polyimides are promising materials in different fields, including gas separation applications, because these polymers exhibit a combination of desirable properties such as high thermal stability, high glass-transition temperatures, and good mechanical properties. In addition, the effective chain separation effect caused by the presence of the bulky tert-butyl pendant group, results in a hindered molecular packing affording high free volume, and hence, they have an excellent permeability to small molecules.78 Calle et al.79 incorporated a pivaloyl substituent in position 2′ of dianhydride (1-9, Table 1) which showed noteworthy improvement in polymer chain rigidity with better gas separation capability and improved solubility relative to those polyimides prepared from BTPDA.
Augl et al.83 have improved the solubility of aromatic polyimides without reducing their thermal stability by introducing an ordered sequence of phenylated quinoxaline moieties into the polymer chain by using appropriate diamines (2-1, Table 2). The imidequinoxalines were not soluble in chloroform, nitrobenzene, or dichlorobenzene. They were, however, soluble in phenolic solvents such as m-cresol, m-methoxyphenol, hexafluoroacetone, hexafluoroisopropanol and are moderately soluble in tetrachloroethane. Feld et al.84 reported an improvement in solubility by the introduction of pendent phenyl substituents. The resulting polymers were soluble in chlorinated hydrocarbon solvents such as chloroform and sym-tetrachloroethane and in aromatic solvents such as benzene.
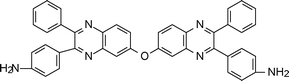

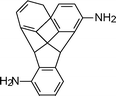
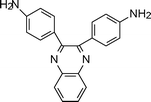







Akutsu et al.85 reported the improvement of solubility of the aromatic polyimides by introduction of rigid and zigzag structures through the 2,3-quinoxalinediyl diamine monomer (2-2, Table 2). Further studies by Akutsu et al. showed improvement of solubility by introduction of a 2-phenyl-4,5-imidazolediyl86 and rigid 2,7-triptycenediyl structure (2-3, Table 2)87 into the polyimide backbone without lowering their high thermal stabilities. Introduction of a 2,7-triptycenediyl structure87 into the polymer backbone makes the main chain three-dimensionally zigzag thereby improving the solubility. In general, introduction of 1,2-heteroarylene units leads to a zigzag main chain structure which increases the bulkiness and reduces the symmetry of the polymer chain.
A series of new polyimides containing triptycene moieties were prepared by Zhang et al.88 from a synthesized dianhydride monomer namely 1,4-bis[4-(3,4-dicarboxylphenoxy)]triptycene dianhydride (2-4, Table 2) and various diamines like 4,4′-oxydianiline (ODA), 1,3-phenylenediamine (MPD) 4,4′-methylenedianiline (MDA), 3,3′-dimethyl-4,4′-methylenedianiline (DMMDA) and 2,2′-dimethyl-4,4′-biphenylenediamine (DMB). The triptycene-containing polyimides, showed improved solubility both in polar solvents, such as DMF, DMAc, DMSO, NMP and m-cresol, and in less polar solvents, such as THF, CH2Cl2, chloroform and TCE at room temperature and solubility varied depending upon the diamine used. The improved solubility of these polyimides could be attributed to the presence of flexible aryl ether linkages and bulky side substituents, together with the incorporation of bulky alicyclic units of the triptycene structure.
Kasashima et al.89 showed that polyimides prepared from diamines containing ortho-terphenyldiyl structures (2-5, Table 2) provide thermally stable and soluble polyimides. Introduction of bulky, propeller-shaped packing disruptive triphenylamine (TPA) groups (2-6, Table 2)90 into the polymer backbone increases the solubility of polyimides without sacrificing thermal stability. The polyimides showed a good film forming ability.
The approach to improve polymer solubility by incorporation of soluble tetra-substituted perylene dianhydride containing phenoxy and 4-tert-butylphenoxy groups (2-7, Table 2) was studied by Dotcheva et al.91 The improved results obtained are caused by the increased solubility of the 4-tert-butylphenoxy-substituted monomers and oligomers during the polycondensation. Thus, it is evident that tert-butyl groups, which lead to the improved solubility of the materials, inhibit the kind of side-chain crystallization which is usually observed for longer n-alkyl-substituted side chains.
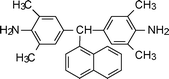
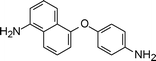
Yang et al.92,93 have reported a 2,3-naphthalene moiety and 1,5-diamino naphthalene-based polyimides from an appropriate diamine monomer (2-8, Table 2). Wang et al.94 synthesized diamine monomers with pendant naphthalene groups and flexible ether linkages to produce a series of polyimides having easy processability. The introduction of a bulky naphthalene side group and flexible ether segments into the polyimides resulted in excellent solubility.
Soluble aromatic polyimides have also been prepared by introducing bulky substituents, aryl or heterocyclic rings. Rusanov et al.95 reported soluble polyimides with pendent phenyl groups. The phenyl group does not affect the thermal stability, but introduces molecular irregularity and separation of chains, that causes an increase in free volume and lowering of the cohesive energy density.96–99 Fluorene diamines and the so-called “cardo” monomers also present valuable alternatives for the preparation of processable polyimides.80,100 Moreover, the presence of the bulky side substituents in polyimides or in any other linear polymer causes a lowering of the chain's torsional mobility and generally an increment of the glass transition temperature.101,102
Ghaemy et al.103 prepared soluble polyimides from a diacetamido–diamine monomer, N′-[7-(acetyl-4-aminoanilino)-9,9-dioctylfluoren-2-yl]-N′-4-aminophenyl)acetamide (ADOAc) (2-9, Table 2), with a fluorene-based structure. The excellent solubility of polyimides based on ADOAc was a result of structural modification through the incorporation of two octyl groups at the 9-position of the fluorene ring and acetamide (–N(CO(CH3))–) moieties in the repeating unit of the polymer chain. These groups disturb the planarity of aromatic units, close packing of polymer chains and crystallinity, increase fractional free volume that increases chain rotational and flexural mobility, and thus lead to the increase in solubility. Mathews et al.104 reported carbazole containing diamines, namely 4′′-carbazole-9-yl-[1,1′,2′,1′′] terphenyl-4-4′-diamine(CzTPDA) and 2-(6-carbazol-9-yl-hexyl)-biphenyl-4,4′-diamine (CzHBPDA) (2-10 and 2-11, Table 2). Polyimides prepared from PMDA and 6FDA with these monomers showed improved solubility. They inferred that the presence of aliphatic moieties imparted more flexibility than the aromatic groups, while the side chain units loosen the close chain packing of polymer chains leading to enhanced solubility.
Behniafar et al.105 reported naphthalene-based and imide ring-containing dicarboxylic acids bearing a flexible ether linkage namely 5-(4-trimellitimidophenoxy)-1-trimellitimido naphthalene (2-12, Table 2), and used those to prepare aromatic poly(amide ether imide)s. The polymers were easily dissolved in highly polar solvents and even in moderately polar solvents such as tetrahydrofuran (THF). In fact, the corresponding diamines had no significant effect on the solubility because the polarity extent of the structures is determined by the presence of the imide ring-containing aryl ether moieties. On the other hand, the orientation of the polar linkages attached to the central naphthalene ring into the polymer backbone interrupted a good chain packing thus leading to high solubility of the polymers in highly polar solvents. Table 2 shows a few examples of monomers with rigid and zigzag structures.
One of the most successful approaches to attaining solubility without sacrificing backbone rigidity and linearity has involved the polymerization of 4,4′-diamino 2,2′-disubstituted biphenyls with aromatic dianhydrides.106,107 The steric repulsion of the substituents in the 2- and 2′-positions of the biphenyl moieties twists the rings dramatically out-of-plane.108 The resulting twisted conformation inhibits chain packing and crystallization. The severe twist also breaks up the conjugation along the backbone and hinders the formation of intermolecular charge-transfer complexes. Both of these effects contribute to a dramatic reduction in color.
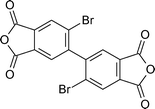
The introduction of twisted biphenyl structures to polyimide backbones is reported by using 2,2′-dibromo-4,4′-5,5′-biphenyltetracarboxylic dianhydride (DBBPDA) and 2,2′-diphenyl-4,4′-5,5′-biphenyltetracarboxylic dianhydride (DPBPDA) (2-13 and 2-14, Table 2) and their polymerization with 4,4′-diamino 2,2′-disubstituted biphenyls in refluxed m-cresol containing isoquinoline.109 Due to the groups in the 2- and 2′-positions, the solubility of the resulting polyimides was improved. One of the polymers was soluble in acetone, and several were soluble in tetrahydrofuran (THF). The acetone-soluble polymer was obtained from DBBPDA and the fluorinated diamine 4,4′-diamino-2,2′-bis(trifluoromethyl)biphenyl (PFMB). PFMB had also been previously polymerized with the fluorinated dianhydride 2,2′-hexafluoroisopropylidene bis(phthalic anhydride) (6FDA) to afford a polyimide that was soluble in acetone.
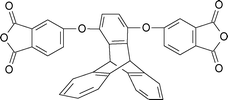
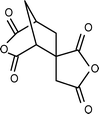
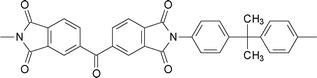
The introduction of spiro-annulated segment through diamines (3-1 and 3-2, Table 3) preserved the rigidity of the polymer backbone, but the spiro structure in polyimides reduces their crystallization tendency and enhances their solubility, glass-transition temperature (Tg), and thermal stability.115–120 Hsaio et al.121 reported soluble thermoplastic polyimides derived from spiro-linked diamine (3-3, Table 3) structures in an effort to address these aspects. More importantly, the fluorene-containing polymers have been proven to exhibit high refractive indices due to their high aromatic contents, so some of the fluorene-containing polymers have been successfully utilized to make optical lenses.122 On the other hand, the incorporation of bulky fluorene groups, which sterically hinder the intermolecular interaction of the PI chains, reduces the packing density, thus increasing the transparency of the polyimides.123,124 Recently, Zhang et al.125 reported a diamine monomer (3-4, Table 3) by combining spiro-fluorene and xanthene moieties to afford good solubility together with a host of other properties. All the polyimides were soluble in dipolar amide-type solvents, such as NMP, DMAc, and DMF, at room temperature or upon heating. Some of them also were soluble in DMSO and phenol solvents like m-cresol and ortho-chlorophenol, as well as chlorinated solvents like chloroform. The polyimides derived from less stiff dianhydride components were also soluble in low boiling point organic solvents such as THF and 1,4-dioxane. In general, the presence of kinked spirofluorene units, with flexible aryl ether linkages along the polymer backbone, increased the disorder in the chains and hindered dense chain packing, thus, reducing the interchain interactions to enhance the solubility. The polyimides prepared by chemical imidization exhibited higher solubility than those by thermal curing. The lower solubility of the latter is possibly due to the presence of partial interchain crosslinking or denser aggregation of the polymer chains during the thermal imidization process. Jiang et al.126 incorporated spirobifluorene units in the diamine (3-5, Table 3) and prepared polyimides with improved solubility. Shu et al. also developed highly soluble and thermally stable polyimides by inclusion of spiro-fused orthogonal bifluorene segments along with two extra ether linkages in both dianhyride and diamine structures (3-6 and 3-7, Table 3).127,128 Kudo et al. and Matsomoto et al. prepared polyimide based unsymmetric tricyclic dianhydrides (3-8 and 3-9, Table 3) that showed good solubility towards dipolar aprotic solvents.129,130 Inclusion of a spiro system into the polyimides to increase the free volume in the polymer has also been successfully reported by using the appropriate diamine (3-10)131,132 and dianhydride monomers (3-11, Table 3).131,132 The increase in free volume as well as rigidity of the polyimides after incorporation of this rigid spiro fused bi-indane moiety render higher gas permeability along with good permselectivity through the polymeric membrane. Table 3 shows selected monomers with spiro skeleton linkage.









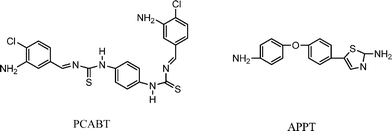 | ||
| Scheme 5 Structures of PCABT and APPT. | ||
A foremost drawback of azomethine polymers is their limited solubility in most organic solvents owing to their rigid chain structure. The overall superior solubility of polymers containing additional phenylthiourea units relative to other azomethine polymers is attributable to the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S that reduces macromolecular hydrogen bonding and has a positive effect on the solubility. On the other hand, the chloro-substituted structure in poly(phenylthiourea azomethine imides)s might direct a lower chain ordering of polyimides to diminish chain-to-chain interaction, thereby, also enhancing their solubility.
S that reduces macromolecular hydrogen bonding and has a positive effect on the solubility. On the other hand, the chloro-substituted structure in poly(phenylthiourea azomethine imides)s might direct a lower chain ordering of polyimides to diminish chain-to-chain interaction, thereby, also enhancing their solubility.
Introducing an unsymmetrical structure into the polyimide main chain in the form of heteroaromatic rings can lead to improvement of solubility and melt processability. The thiazole ring with heteroaromatic structure has an excellent chemical, thermal and thermooxidative stability derived from its molecular symmetry and aromaticity. In addition, the thiazole rings in the polyimide backbones which can interact with metal, enhance the adhesion of the polymer chains to metal. Zhao et al. (Scheme 5, APPT)137 reported the synthesis of a series of polyimides from a thiazole ring containing the unsymmetrical diamine monomer, 2-amino-5-[4-(4′-aminophenoxy)phenyl]-thiazole (APPT) by polycondensation with various aromatic dianhydrides via a one-step process. The good solubility of these polymers was attributed to the flexible ether group and the noncoplanar and unsymmetrical structure in the polyimide main chain. Scheme 5 shows the structure of 1,4-phenylene bis((E)-1-(4-chloro-3-aminobenzylidene)thiourea) (PCABT) and 2-amino-5-[4-(4′-aminophenoxy)phenyl]-thiazole (APPT) taken from Ref. 133 and 137 respectively.
O, –SO2–, –C(CH3)2–, –CH2– and –C(CF3)2 (4-1 to 4-6, Table 4).142–146 This table gives an idea how the structural manipulation in polyimides through bridging functional groups affects the glass transition temperature and the processability. The “kink” linkages between aromatic rings cause a breakdown of the planarity and an increase of the torsional mobility. Furthermore, the additional bonds lead to an enlargement of the repeating unit and, hence a separation of the imide rings take place whose relative density is actually responsible for the polymer tractability. The suppression of the coplanar structure is maximal when voluminous groups are introduced in the main chain, for instance sulfonyl or hexafluoroisopropylydene groups, or when the monomers are enlarged by more than one flexible linkage. Hexafluoropropylidene, carbonyl and sulfonyl are the groups most advantageously incorporated concerning processability. This is due to the relatively large volume of these groups, and to the conformational characteristics imparted by them to the polymer chain. The poly(ether imide), marketed under the trade name Ultem® is an amorphous, soluble polymer that shows Tg values around 217 °C (4-1, Table 4). Ultem® can be processed from the melt by conventional means, and offers a price-performance balance that enables it to compete successfully in the market of engineering thermoplastics.





Cho et al.149 synthesized PI from ethylene glycol bis(anhydro trimellitate) and 1,3-bis(4-aminophenoxy) benzene (Scheme 6). The two monomers consist of highly flexible ether and ester linkages within their chemical structure, which contributed to improving the processability. Together with the processability, the thermal stability and mechanical properties of the PIs must be also taken into consideration in electronic applications because the packaging process is done at a high temperature. Further, Leu et al.150 reported on the synthesis of a series of polyimides containing the bisphenol unit and aryl–ether linkage to understand the relationship between the structure and properties of polyimides.
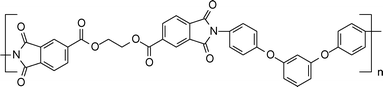 | ||
| Scheme 6 Structure of polyimide taken from Ref. 149. | ||
Polyimides with aromatic asymmetrical ether diamine moieties (5-1, Table 5)151,152 showed enhanced solublility caused by molecular chain nonlinearity. Butt et al.153 synthesized 2-methyl-1,3-bis(4-aminophenyloxy) benzene (MAPB) (5-2, Table 5) with the view to influence the chain flexibility and enhance the solubility of the polyimides.




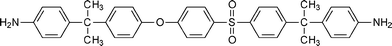






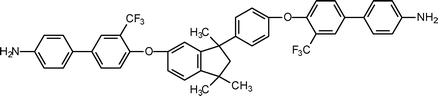

Liaw et al.154 reported good solubility by structural modification of the polyimide structure through incorporation of flexible isopropylidene and methyl substituted arylene ether groups. These results could be explained by the presence of the flexible isopropylidene and ether groups, which reduced the rigidity of the polymer chain which in turn reduced the crystallinity and improved the solubility of polymer. In addition, methyl groups on the phenylene also inhibited close packing of the polymer chains.155 Liaw et al.156 synthesized a flexible diamine (5-3, Table 5) containing flexible ether and isopropylidene units and used those to prepare a series of polyimides. The improved solubility of these polymers were attributed to the presence of the flexible moieties on the polyimide backbone that considerably decrease the rigidity of the polymer chain. Most of the polyimides showed excellent solubility in a variety of solvents such as N-methyl-2-pyrrolidinone, DMAc, N,N-dimethylformamide, dimethyl sulfoxide, pyridine, cyclohexanone and tetrahydrofuran. Such polyimides had better solubility than those containing only one isopropylidene unit or a hexafluoroisopropylidene linkages in the repeating units of the polyimide backbone.
Sava et al.157 synthesized polyimides with a diamine containing flexible methylene bridge namely, 3,3′-dimethyl-4,4′-diaminodiphenylmethane and compared the properites with those of polyimides synthesized from commercially available diamine, dimethyl-4,4′-diaminodiphenylmethane with the same dianhydrides. They reported that most of the polymers, were easily soluble in DMAc, N-methylpyrrolidone (NMP), N,N′-dimethylformamide (DMF), and dimethylsulfoxide (DMSO). Their good solubility may be explained by the presence of methyl groups or isopropylidene or hexafluoroisopropylidene units,158 which increase the free volume allowing for the small solvent molecules to penetrate more easily among the polymer chains. The incorporation of the two methyl groups on the diamine moiety leads to an increase in the free volume and a resultant decrease in the molecular packing. The steric hindrance from the methyl groups might also lead to a distortion of the packing of the polyimide backbones. For these reasons, the polyimides based on 3,3′-dimethyl-4,4′-diaminodiphenylmethane have a better solubility compared with polymers obtained from 4,4′-diaminodiphenylmethane.159,160
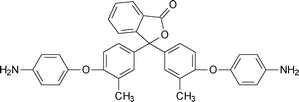
Banihashemi et al. synthesized polymers containing a specific structure namely benzofuro-benzofuran.161–166 A representative monomer167 benzofuro [2,3-b]benzofuran-2,3,8,9-tetracarboxylic dianhydride (BBTDA) (5-4, Table 5) was used for the synthesis of a series of polyimides. The synthesized polyimides were soluble in phenolic solvents, and also polar aprotic solvents. Polyimides containing sulfone, methane, ether, and carbonyl groups, respectively were more soluble, and a shorter time was needed for their complete dissolution in solvents.
Amutha et al.168 incorporated a pyridyl moiety with ether linkages to improve the solubility of polyimides. Beniafar et al.169 synthesized a diamine monomer containing two phenoxy phenylene lateral groups. The polyimides showed excellent organosolubility in common polar solvents. The phenylene lateral groups attached to the macromolecular backbones had no substantial retreating effect on the thermal stability of these polyimides.
A series of polyimides were synthesized, from aromatic oxadiazole-diamines and a dianhydride containing a siloxane bridge (–R2Si–O–SiR2–), by Rusanov et al.170 The resulting polymers exhibited good solubility in certain organic solvents and high thermal stability with decomposition above 440 °C and relatively low glass transition temperatures in the range 160–190 °C. These polymers showed strong photoluminescence in the blue spectral region. The introduction of oxadiazole rings together with siloxane groups into the chains of aromatic polyimides gives highly thermostable polymers with remarkable solubility, film-forming abilities and that emit blue light, being attractive for applications in micro- and nanoelectronics and other related advanced fields.
Hsiao et al.171 incorporated flexible ether and isopropylidene in the polyimide backbone together with sulfone groups through a diamine monomer (5-5, Table 5) into the macromolecular chain which generally results in improved solubility, together with interesting properties such as increased Tg and high thermal stability.172–176 The presence of methyl substituents at the ortho positions to the ether linkage substantially increases their processability due to difficulty in the chain rotation177,178 The thermal stability of these segmented polyimides is dependent on their chemical structure and mainly on the flexible chain. Polyimides containing polyethylene glycol sequences are thermally unstable, whereas polysiloxane,179,180 perfluoroalkanes,181 and even alkanes45,182 provide a reasonable thermal stability. The combination of conventional and synthesized monomers is providing an expansion of the scope to synthesize processable polyimides containing flexible spacers.183,184 Perfluoroalkylenes are acceptable groups to be introduced as flexibilizing moieties as they provide a substantial lowering of the Tg without greatly impairing the thermal, mechanical and chemical properties. Soluble and meltable aromatic polyimides containing flexible linkages have been developed to the growing demand of specific materials for advanced technologies. All of them are soluble in some specific organic media, showing glass transition temperatures in the range 250–380 °C.
Recently two aromatic diamine monomers, bis-4,4′[(4-aminophenyl-2,2′-isopropylidene phenyloxy)] diphenylsulfone and bis-4,4′[(4-aminophenyl-2,2′-isopropylidene phenyloxy)] benzophenone (5-6 and 5-7, Table 5) were synthesized.185 The resulting polyimides were soluble in polar aprotic solvents but an increment in solubility in common organic solvents was observed due to the presence of flexible groups and methyl groups in the chain backbone.
Incorporation of an arylene ether linkage into a polymer macrochain is known to impart processability to the polymer with little reduction in thermal stability.186,187 Yang et al.188–190 reported thermostable, organo-soluble polyimides containing the arylene ether cyclic group. Polyimides based on the diamine, 3,3-bis[4-(4-aminophenoxy)-3-methylphenyl] phthalide (BAMP) (5-8, Table 5)191 and various aromatic dianhydrides were prepared through a two-stage procedure that included ring opening, followed by thermal or chemical conversion to polyimides. Most of the polyimides obtained through the chemical cyclodehydration procedure were found to be soluble in DMAc, NMP, m-cresol, and ortho-chlorophenol. Hence, combining aryl ether and cardo groups of the polymer chain may increase the solubility of polymers without severe loss of thermal stability.
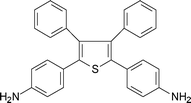
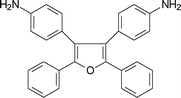
One of the successful approaches to increase solubility and processability of polyimides without sacrificing their high thermal stability is the introduction of pendant phenyl groups into the polymer backbone. Some organo-soluble aromatic polyimides have been synthesised using highly phenylated tetracarboxylic dianhydrides.82,192 Studies also revealed that highly phenylated aromatic diamines such as 2,5-bis-(4-aminophenyl)-3,4-diphenylthiophene (5-9, Table 5),82,192,193 3,4-bis(4-aminophenyl)-2,5-diphenylfuran (5-10, Table 5),194 3,4-bis (4-aminophenyl)-2,5-diphenylpyrrole (5-11, Table 5),195 and 1,1-bis(4-aminophenyl)-2,2-diphenylethylene (5-12, Table 5)196 were used for the preparation of organo-soluble high temperature aromatic polyimides. Oishi et al.197 synthesized soluble aromatic polyimides using triphenylamine units containing diamine, 4,4′-diaminotriphenylamine (5-13, Table 5).
Bacosca et al.198 synthesized aromatic polyimides by solution polycondensation reaction of an aromatic diamine having cyano group and two ether units (5-14, Table 5) with different aromatic dianhydrides incorporating flexible units, such as ether, carbonyl or hexafluoroisopropylidene. Most of the prepared polyimides were soluble in polar organic solvents such as N-methylpyrrolidone, dimethylsulfoxide or dimethylformamide, except the polymer based on pyromellitic dianhydride having no flexible groups, which precipitated during the cyclization reaction. The polyimide containing 6F group was also soluble in less polar solvents like chloroform and tetrahydrofuran. The good solubility of the polymers was explained by the structural modification, and the flexible bridges such as ether, carbonyl, hexafluoroisopropylidene, cyano, disturbing the packing. The disturbed packing of macromolecular chains facilitates the diffusion of small molecules of solvents between the polymer chains which leads to better solubility. The good solubility of these polymers allowed a one-pot solution imidization.
In an effort to prepare new polyimides with high thermal stability and improved solubility, Yeganeh et al.199 synthesized a diisocyanate containing aliphatic oxyethylene moieties (5-15, Table 5). One of the main objectives of this study was to produce modified polyimides with improved solubility. The solubility of the polymers studied at a concentration of 5 g dL−1 and at ambient temperature in different solvents were found to be soluble in common aprotic solvents such as N-methyl-2-pyrrolidone, DMAc, N,N-dimethylformamide, and dimethyl sulfoxide.
Banerjee et al. reported a series of organosoluble polyimides with high thermal stability with good mechanical properties. The polyimide series based on an indane moiety (5-16, Table 5)200 and phthalimidine containing fluorinated diamine (5-17, Table 5)201 in combination with various aromatic dianhydrides including pyromellitic dianhydride and benzenetetracarboxylic acid dianhydrides. The polyimides showed excellent organo-solubility in various common organic solvents like less polar dichloromethane, tetrahydrofuran, chloroform and dipolar aprotic dimethylsulfoxide, N-methyl pyrrolidone, pyridine etc. The key factors for the organo-solubility of those polyimides are inclusion of cardo groups in addition to flexible ether linkages and pendant trimethyl group into the polymer backbone. Table 5 shows some representative structures with flexible ether/isopropylidene/benzofuran/pyridyl/isocyanate moieties.
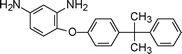
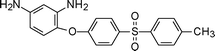


Studies by Yang et al.203 reported polyimides from mono-phenyl containing 2,5-bis(4-aminophenoxy)-biphenyl (6-3, Table 6) with higher thermal stability and better solubility than the tert-butyl containing one. Incorporation of this unsymmetrical and hindered moiety disturbed the coplanarity of the aromatic unit to the reduce packing efficiency and crystallinity hence enhancing solubility and maintaining high glass transition temperatures.
Ghaemy et al.204 prepared polyimides by reaction with commercially available dianhydrides such as PMDA, BTDA, and bicyclo[2.2.2]-oct-7-ene-2,3,5,6-tetracarboxylic dianhydride (BCDA) (6-4, Table 6) and from a synthesized unsymmetrical diamine monomer, 2,4-diaminophenyl [4′-(2′′,6′′-diphenyl-4′′-pyridyl)phenyl] ether (DAP). The prepared PIs were readily soluble in polar aprotic solvents such as NMP, DMAc, DMF, DMSO, and even in less polar solvents like pyridine and m-cresol at room temperature but showed poor solubility in dioxan and THF. The good solubility should be the result of formation of some intermolecular spacing due to introduction of the bulky pendent group in the polymer backbone. The unsymmetrical structure of the diamine monomer interrupts the regular packing of polymer chains, and polarizability resulting from the nitrogen atom in the pyridine ring. Table 6 shows structures of phenylene diamines taken from Ref. 202, 203 and 204 respectively.




Kwak et al.214 synthesized a phosphorus containing diamine monomer bis(3-aminophenyl)-4-(1-adamantyl)phenoxyphenyl phosphine oxide (mDAATPPO) (7-3, Table 7) and prepared polyimides from commercially available dianhydrides, like 6FDA, BTDA, ODPA and PMDA. The polymers exhibited good solubility in NMP, DMAc, and TCE depending on the rigidity of the dianhydrides. Adamantyl moieties are known to impart high glass transition temperatures, excellent thermal stability, and good dielectric properties. The improvement of solubility and thermal properties of adamantyl containing polymers results from the bulkiness and the rigidity of the adamantyl moiety, which greatly reduces the chain mobility and inhibits chain packing.
Kung et al.215 synthesized an adamantoxytriphenylamine-containing diamine monomer namely 4-(1-adamantoxy)-4′, 4′′-diaminotriphenylamine (7-4, Table 7). The monomer was reacted with various aromatic dianhydrides to form polyimides. The polyimides exhibited good solubility in polar solvents such as NMP, DMAc, DMF, DMSO, and m-cresol at room temperature or upon heating at 70 °C, except for the polyimides derived from dianhydrides such as PMDA and BPDA. In general, these polymers revealed an enhanced solubility in respect to conventional aromatic polyimides. Their high solubility and amorphous properties could be attributed to the incorporation of the bulky, three-dimensional adamantoxy-tripenylamine moiety along the polymer backbone, which results in a high steric hindrance for close packing, and thus reduces their crystallization tendency and interchain interactions. Table 7 shows the structures of adamantane containing diamines taken from Ref. 211, 212, 214 and 215 respectively.
















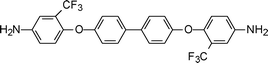


The presence of trifluoromethyl groups and, in general, the substitution of fluorine for hydrogen, causes a dramatic change in properties. The combination of electronic and steric effects reduces the ability for interchain interactions and, particularly, hinders the formation of charge transfer complexes, which is a major factor of molecular packing and intractability in aromatic polyimides. Furthermore, the C–F bond is a high energy bond, so the polyimides containing fluorine generally possess high Tg and excellent thermal properties, comparable to those of the conventional aromatic polyimides. Fluorinated polyimides showed low dielectric constants, high optical transparency, excellent mechanical properties, low moisture absorption, increased solubility, low optical loss and low refractive indices. This excellent balance of properties has made fluorinated polyimides very attractive for some applications in advanced technologies, such as in high performance structural resins, thermally stable coatings and films, polymeric membranes for gas separation, polymeric waveguides, and other electronic and optoelectronic applications.
It is well known that the incorporation of fluorinated substituents into polymers decreases the dielectric constant due to the small dipole and low polarizability of the C–F bond. Thus, fluorinated polyimides were developed in order to decrease the dielectric constant. Furthermore, the free volume can be increased by replacement of methyl groups by trifluoromethyl groups thus enhancing solubility and hence many fluorinated polyimides have been prepared from several fluorinated dianhdrides (8-3, Table 8) and diamines (8-4, 8-5, 8-6, Table 8).220–224 This increase in solubility might be attributed to the molecular asymmetry and the presence of bulky trifluoromethyl groups, which increase the disorder in the chains and hinder the dense chain stacking, thereby reducing the interchain interactions and thus enhancing solubility. Ge et al.225 synthesized a fluorinated aromatic diamine (8-7, Table 8) which was then employed to prepare a series of fluorinated polyimides with commercial aromatic dianhydrides, such as PMDA, 6FDA, BTDA and ODPA. The polyimides exhibited good solubility in strong dipolar solvents such as NMP, DMAc, DMF and m-cresol as well as in some of low boiling point organic solvents as THF and CHCl3.
Recent studies have demonstrated that polyimides derived from diamines with ether bridge and –CF3 substituents are soluble in most organic solvents and have good thermal stability and low moisture uptake. Banerjee et al.226–232 have incorporated trifluoromethyl groups (–CF3) as well as ether linkages in the polyimide backbone by designing several diamine monomers. A few representative diamine structures are shown in Table 8 (8-8 to 8-13, Table 8). The presence of the ether linkage broke the planarity and improved the mobility which ultimately led to an improvement in solubility in not only aprotic solvents but also to some extent in protic solvents. Furthermore a reduced dielectric constant and low moisture uptake, high gas permeability and high optical transparency was also observed in this type of fluorinated poly(ether imide)s.
Shao et al.233 synthesized an unsymmetrical ether diamine with a trifluoromethyl pendent group, 1,4-(2′-trifluoromethyl-4′,4′′-diaminodiphenoxy)benzene (8-14, Table 8), and polymerized it with various aromatic tetracarboxylic acid dianhydrides, including ODPA, BTDA, 6FDA, and PMDA, via both the conventional two-step thermal and the chemical imidization methods. The polymers exhibited very good solubility behavior in polar solvents such as DMSO, DMF, N,N-dimethyl acetamide (DMAc), and NMP and they could also be dissolved in common solvents such as chloroform, tetrahydrofuran (THF), and toluene upon heating. The polyimides prepared by thermal curing had much poorer solubility than those prepared by chemical imidization, possibly due to the presence of partial intermolecular crosslinking during the thermal imidization stage.
Li et al.234 synthesized polynaphthalimides (PNI) from asymmetrical fluorinated naphthalene substituted monomers (8-15, Table 8). All PNIs exhibited high solubility in common organic solvents, such as NMP, DMAc, TCE, and CHCl3 at room temperature. The solubility of PNIs was found to be comparable to ULTEM poly(ether imide), which is soluble in polar aprotic solvents and chlorinated solvents. The high solubility behavior of these PNIs was attributed to their regioirregular microstructures within the PNI backbone.
A fluorinated diamine monomer with a tert-butyl group, 4-tert-butyl-[1,2-bis(4-amino-2-trifluoromethylphenoxy)phenyl]benzene (8-16, Table 8), was prepared by Yang et al.235 and was reacted with various aromatic dianhydrides by thermal imidization to prepare polyimides. The prepared polyimides were soluble in polar aprotic solvents such as NMP, DMAc, DMF and DMSO and were partially soluble in less polar solvents like m-cresol, pyridine, dioxane, THF, CH2Cl2 and acetone. This is attributed to the molecular chain containing the tert-butyl group and the –CF3 group leading to increased solubility.
Wang et al.236 synthesized fluorinated diamines, 4,4′-bis(3-amino-5-trifluoromethylphenoxy)biphenyl (m-6FBAB) and 4,4′-bis(4-amino-5-trifluoromethylphenoxy) biphenyl (p-6FBAB) (8-17, 8-18, Table 8) and prepared polyimides from these monomers on reaction with various aromatic dianhydrides PMDA, ODPA, BTDA and 6FDA in the presence of phthalic anhydride (PA) as a molecular weight-controlling and end capping agent. Polyimides derived from 6FDA and ODPA showed better solubility than those derived from PMDA and BPDA. Polyimides derived from 6FDA showed good solubility not only in strong aprotic solvents such as NMP and DMAc but also in common organic solvents such as THF and chloroform.
Diamine momomers containing phosphine oxide and fluorine, bis(3-aminophenyl) 3,5-bis(trifluoroethyl)phenyl phosphine oxide (mDA6FPPO) (8-19, Table 8), were prepared by Jeong et al.237 This diamine monomer was utilized to prepare polyimides on reaction with different dianhydrides. The solubility of the polyimides was greatly enhanced by the phosphine oxide moiety and further by trifluoromethyl phenyl group. The excellent solublity of the mDA6FPPO based polyimides could be attributed to the presence of bulky –CF3 groups, leading to increased free volume and also to the phosphine oxide segments providing strong intermolecular forces with solvent molecules.238 Satoh et al.239 studied the effect of the trifluoromethyl groups at the 2,2′-positions in terms of solubility and other properties in comparison to the previously prepared polyimides, which had a phenyl group at the 2,2′-position of the diphenyl ether. Table 8 shows some examples of fluorinated monomers.
Efforts at the NASA Langley Research Center were made by St. Clair et al. in the early 1980s244,245 and by McGrath et al.246–250 on block copolymers comprising of polydimethyl siloxane and polyimides. PDMS containing polyimide copolymers yielded segmented or multiblock copolymers. The architecture influences the elastomeric behaviour, melt rheology and toughness in rigid materials. Two types of poly(imide siloxane) copolymers were generally prepared, namely randomly segmented copolymers and perfectly alternating segmented copolymers. Block copolymers are composed of two segments, namely hard polymer segments and soft polymer segments, which are arranged alternately. The distribution of small domains of the soft segment in the hard phase behave as toughened glassy polymers251 while the distribution of the hard segments in the continuous soft segment give rise to thermoplastic elastomers.252 It has been found from the literature that perfectly alternating segmented copolymers have a higher structural regularity and give better microphase separation with higher glass transition temperature and improved tensile strength compared to the randomly segmented copolymers.253
The most common method followed for the preparation of randomly segmented poly(imide siloxane) copolymers involves the reaction of preformed amino propyl terminated polydimethylsiloxane with a dianhydride and diamine monomer.254,255 The resulting amic acid intermediate is cyclodehydrated either by thermal or high temperature solution imidization techniques. Perfectly alternating segmented copolymers were synthesized by polymerizing two different oligomers via reaction of their end groups by a two pot synthesis. In this method the average block length of each segment is known before copolymerization. Jwo et al.256 studied the dependence of poly(imide siloxane)s on the solubility parameter of unmodified polyimides on the molecular weight and on the content of α,ω-bis(3-aminopropyl) polydimethylsiloxane.
Recently, Babanzadeh et al.257 synthesized a group of siloxane diamines containing bulky aromatic groups (Scheme 7) namely, bis(p-aminophenoxy)diphenylsilane (BPS), bis(m-aminophenoxy)diphenylsilane (BMS) and bis(5-amino-1-naphthoxy)diphenylsilane (BAS), the related polyimides were prepared from these diamines and different aromatic dianhydrides. Incorporation of bulky groups and low-polarizing siloxane units into the polymer backbones effectively improved the solubility of the polyimides. The diamines were designed in such a way to induce flexibility into the structure of the final polyimides due to the presence of flexible ether and siloxane units.
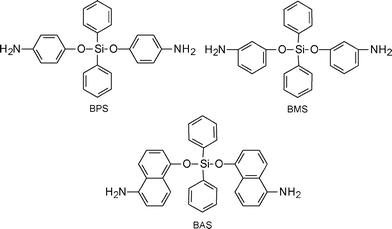 | ||
| Scheme 7 Siloxane diamines taken from Ref. 257 | ||
There are several reports on random fluorinated copoly(imide siloxane)s so far.258–264 The exceptionally high solubility of the poly(imide siloxane)s could be ascribed to the presence of flexible aminopropyl linkages in the polymer backbone which imparts ease of penetration of solvent molecules in between the polymer chains and the presence of bulky –CF3 groups reduces the chain packing density and hence enhances the solubility to a great extent. Thermal stability showed a decrease for the siloxane containing polymers compared to the respective homopolymers without siloxane units because of the weak aminopropyl linkages present in the polymer backbone. One of the most interesting compromises between processability and thermal stability is given by imide-siloxane block copolymers.265
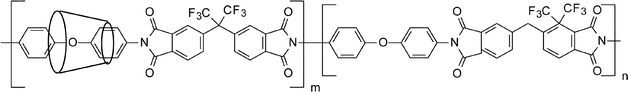
Block copoly(imide siloxane)s were prepared by our research group by varying the hard and soft block length and their thermal and mechanical properties were compared with random copoly(imide siloxane) analogues.259,263 Anhydride terminated soft blocks and amine terminated hard blocks of different chain lengths were prepared by tailoring the stoichiometric imbalance in two separate reaction pots and finally block copolyimides were obtained on their reaction. Both DSC and DMA tan δ results indicated that as the block length increases the hard block glass transition temperature of the respective polymers increases. Therefore, the formation of a block structure with excellent solubility in common organic solvents also showed an increase in glass transition temperature slowly approaching the range of the homopolyimide without siloxane moiety, with an increase in hard block lengths. Thus, with perfectly alternating copolymers of various block lengths, it is possible to control the Tg and modulus of the block copoly(imide siloxane)s. Scheme 8 shows fluorinated random copoly(imide siloxane) and fluorinated block copoly(imide siloxane) with alternating hard and soft block segments where K and L are degrees of polymerization for the hard and soft block, respectively, taken from Ref. 260 and 263.
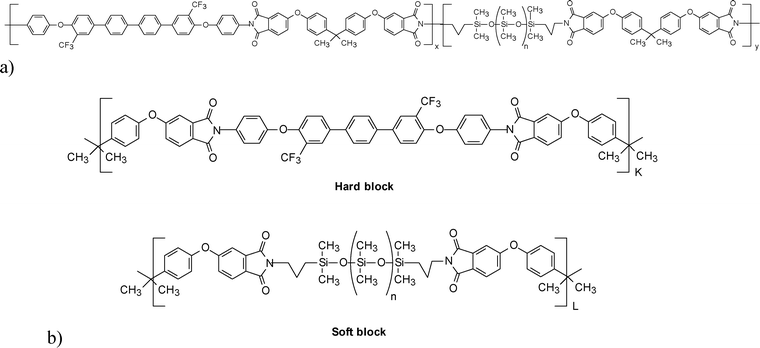 | ||
| Scheme 8 (a) Fluorinated random copoly(imide siloxane) (b) fluorinated block copoly(imide siloxane) with alternating hard and soft block segments where K and L are hard and soft block degrees of polymerization, taken from Ref. 260 and 263. | ||
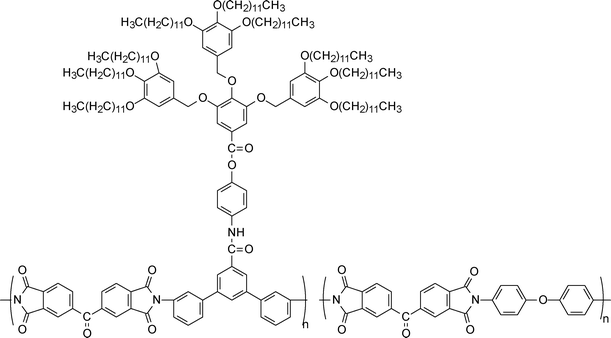 | ||
| Scheme 9 Structure of dendronized polyimide taken from Ref. 266 | ||
Hyperbranched polyimides, which have a highly branched structure and a large number of terminal groups, have attracted increasing attention in recent years because they are expected to have unique properties when compared to their linear analogues.267,268 Generally, hyperbranched polyimides show good solubility due to lack of chain entanglement caused by the highly branched structure and terminal groups also influence the solubility.269–274 Predominantly, the A2 + B3 approach has been adapted in order to achieve highly branched soluble polyimides: Cheng et al.273 synthesized hyperbranched polyimides of high molecular weight from 2,6,12-triaminotriptycene, with high molecular weight modified with different terminal functional groups from a triamine monomer (9-1, Table 9). The prepared hyperbranched polyimides showed good solubility in CHCl3, DMF and THF. The polymers did not show any detectable glass transition temperatures up to 330 °C and thermal stability was as high as 455 °C at 5% weight loss. Chen et al.274 synthesized a triamine monomer, 1,3,5-tris(4-aminophenoxy)benzene (TAPOB) (9-2, Table 9), and polymerized it to hyperbranched polyimides reacting it with commercially available dianhydrides namely 6FDA, ODPA and BTDA in different monomer molar ratios to yield hyperbranched polyimides. The 6FDA-based hyperbranched polyimides exhibited good solubility even in common low-boiling solvents such as chloroform, tetrahydrofuran, and acetone. Gao et al.275 designed and synthesized a kind of –CF3 terminated hyperbranched polyimide (CF3-HQDPA) using a triamine monomer, 1,3,5-tris(2-trifluoromethyl-4-aminophenoxy)benzene (TFAPOB) (A3) (9-3, Table 9) , as a ‘core’ molecule, 1,4-bis(3,4-dicarboxyphenoxy)-benzene dianhydride (HQDPA) as a B2 monomer and 3,5-ditrifuoromethylaniline as an endcapping reagent. The obtained polyimide films were soluble in a variety of organic solvents such as NMP, DMAc, DMF, and cyclohexanone. Hence, polyimides with hyperbranched structures based on new monomer structures like ABx and BB2268,276 have also been investigated as a possible route to improve processability and solubility by disrupting the molecular symmetry and regularity. Chen et al.277 synthesized a new BB′2-type triamine (9-4, Table 9) and prepared series of amine- and anhydride-terminated triphenylpyridine-containing hyperbranched polyimides under microwave irradiation. The synthesized polyimides were soluble in a number of solvents namely m-cresol, NMP, DMSO, DMAc and DMF at room temperature. The polymers showed glass transition temperatures more than 300 °C and 5% weight loss as high as above 480 °C. A new AB2 monomer (9-5, Table 9) was designed by Hao et al.278 by multistep synthesis. Hyperbranched polyimides, prepared from the new AB2 monomer were observed to be soluble in high boiling point solvents, namely DMF, DMAc, DMSO and NMP. An AB2 type monomer (9-6, Table 9), was synthesized by Yamanaka et al.279 starting from 3,5-dimethoxyphenol. Hyperbranched aromatic polyimides were prepared by chemical imidization in the presence of acetic anhydride and pyridine. The synthesized polyimides were soluble in polar aprotic solvents like NMP, DMF and DMSO. Table 9 shows a few examples of monomers synthesized for the preparation of hyperbranched polyimides.
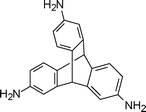




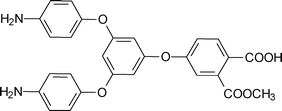
3. Summary
The difficulties in processing conventional aromatic polyimides are due to the inherent molecular features of aromatic polyimides. Molecular stiffness, high polarity and high intermolecular association forces make these polymers virtually insoluble in any organic medium, and shift up the transition temperatures to well above the decomposition temperatures. Thus, the overall goal of ongoing research activities in this field of research is to modify the structure of aromatic polyimides to attain solubility in common organic solvents without forfeiting the thermal and mechanical properties of this class of polymers that arise from the rigidity of their backbone. Solubility is sought after to allow processing in the imide form and, thus, avoid many problems associated with handling poly(amic acid) (PAA) precursors. Thus, the strategies to produce novel processable aromatic polyimides have been focused on chemical modifications, mainly by preparing new monomers that provide less molecular order, torsional mobility and lower intermolecular bonding. Of the various alternatives to design novel processable polyimides, some general approaches universally adopted are: introduction of aliphatic or another kind of flexible segments which reduce chain stiffness, introduction of bulky side substituents which help for separation of polymer chains and hinder molecular packing and crystallization, use of enlarged monomers containing angular bonds which suppress coplanar structures, use of 1,3-substituted instead of 1,4-substituted monomers, and/or asymmetric monomers, which lower regularity and molecular ordering, and preparation of copolyimides from two or more dianhydrides or diamines. The introduction of free volume in a polymer decreases the number of polarizable groups per unit volume. The addition of pendant groups, flexible bridging units, and bulky groups which limit chain packing density have all been used to enhance free volume in polyimides. However, factors leading to better solubility or lower Tg or Tm in a polymer often conflict with other important requirements, such as mechanical properties, thermal resistance or chemical resistance. With perfectly alternating copoly(imide siloxane)s of various block lengths, it is possible to control the Tg and modulus of the block copoly(imide siloxane)s together with excellent solubility. Branched polymer systems such as hyperbranched polymers due to higher fractional free volume also show significantly improved solubility and processability while still demonstrating high thermal properties but low toughness. Therefore, an adjusted degree of modification should be applied to optimize the balance of the properties.Acknowledgements
The author, A.G. would like to thank the Alexander von Humboldt Foundation for a postdoctoral research fellowship and S.B. acknowledges the financial support from the Alexander von Humboldt Foundation through a follow-up program.References
- K. L. Mittal, in Polyimides and other high temperature polymers, VSP Brill, Leiden, 2005, vol. 1 Search PubMed.
- K. L. Mittal, in Polyimides and other high temperature polymers, VSP Brill, Leiden, 2007, vol. 4 Search PubMed.
- K. L. Mittal, in Polyimides and other high temperature polymers, VSP Brill,Leiden, 2009, vol. 5 Search PubMed.
- D. M. Stoakley, A. K. St. Clair and C. I. Croall, J. Appl. Polym. Sci., 1994, 51, 1479 CrossRef CAS.
- J. P. Gao and Z. Y. Wang, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1627 CrossRef CAS.
- D. Wilson, H. D. Stenzenberger and P. M. Hergenrother, Polyimides, Blackie & Son Ltd., Glasgow and London, 1990 Search PubMed.
- A. Kausar, S. Zulfiqar, Z. Ahmed and M. I. Sarwar, Polym. Degrad. Stab., 2010, 95, 1826 CrossRef CAS.
- N. Yoda and H. Hiramoto, J. Macromol. Sci., Chem., 1984, 21, 1641 CrossRef.
- T. H. Chiang, S. L. Liu, S. Y. Lee and T. E. Hsieh, Eur. Polym. J., 2008, 44, 3482 CrossRef CAS.
- C. L. Hendrics and S. G. Hill, in Adhesion chemistry, ed. L. H. Lee, Plenum, New York, 1984, p. 617 Search PubMed.
- L. Thnaka, H. Kita, M. Okano and K. Okamoto, Polymer, 1992, 33, 58 Search PubMed.
- H. Hachisuka, Y. Tsujita, A. Takizawa and T. Kinoshita, Polym. J., 1989, 21, 681 CrossRef CAS.
- Y. Imai, High Perform. Polym., 1995, 7, 337 CAS.
- Y. Imai, React. Funct. Polym., 1996, 30, 3 CrossRef CAS.
- S. Mehdipour-Ataei and H. Heidari, Macromol. Symp., 2003, 193, 159 CrossRef CAS.
- S. Mehdipour-Ataei and H. Heidari, J. Appl. Polym. Sci., 2004, 91, 22 CrossRef CAS.
- W. Zhang, H. J. Xu, J. Yin, X. X. Guo, Y. F. Ye and J. H. Fang, J. Appl. Polym. Sci., 2001, 81, 2814 CrossRef CAS.
- M. T. Reetz, K. M. Kuhling, A. Deege, H. Hinrichs and D. Belder, Angew. Chem., Int. Ed., 2000, 39, 3891 CrossRef CAS.
- M. Avalos, R. Babiano, P. Cintas, M. M. Chavero, F. J. Higes and J. L. Jimenez, J. Org. Chem., 2000, 65, 8882 CrossRef CAS.
- W. Guo, W. T. Leu and S. H. Hsiao, J. Polym. Res., 2007, 14, 359 CrossRef CAS.
- S. Liu, C. Tang, B. Ho, M. Ankersen, C. E. Stidsen and A. M. Crider, J. Med. Chem., 1998, 41, 4693 CrossRef CAS.
- A. Hari and B. L. Miller, Org. Lett., 2000, 2, 3667 CrossRef CAS.
- F. W. Harris and L. H. Lanier, in Structure–solubility relationships in polymers, ed. F. W. Harris and R. B. Seymour, Academic Press, New York, 1997, p.183 Search PubMed.
- D. J. Kumar, Polym. Sci.: Polym. Chem., 1984, 20, 3439 Search PubMed.
- F. W. Harris, S. L. C. Hsu and C. C. Tso, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1990, 31, 324 Search PubMed.
- I. K. Spiliopoulos and J. A. Mikroyaunidis, Polymer, 1996, 37, 3331 CrossRef CAS.
- T. Takekoshi, J. L. Webb, P. P. Anderson and C. E. Olsen, Proceedings of IUPAC 32nd International Symposium on Macromolecules, Kyoto, 1988, p. 464 Search PubMed.
- D. J. Proger, T. L. St Clair, H. Burks, C. Gautreavx, A. Yamaguchi and M. Ohta, Int. SAMPE, Tech. Conf. Series, 1989, 21, 544 Search PubMed.
- M. Ohta, S. Tamai, T. W. Towell, N. J. Johnston and T. L. St Clair, Int. SAMPE Symp., 1990, 35, 1030 CAS.
- L. W. Frost and J. Kesse, J. Appl. Polym. Sci., 1964, 8, 1039 CrossRef CAS.
- C. Feger and M. M. Khojasteh, in Polyimides: materials, chemistry, and characterization, ed. J. E. McGrath, Elsevier, Amsterdam, 1989 Search PubMed.
- R. W. Synder, B. Thomson, B. Bartges, D. Czeriwski and P. C. Painter, Macromolecules, 1989, 22, 4166 CrossRef.
- C. E. Sroog, in Encyclopedia of polymer science and technology, ed. N. M. Bikales, Interscience, New York, 1969, ch. 11, p. 247 Search PubMed.
- P. C. Cassidy and N. C. Fawcett, in Encyclopedia of chemical technology, New York, Wiley, 1982, p. 704 Search PubMed.
- R. J. Angelo, R. C. Golike, W. E. Tatum and J. A. Kreuz, in Advances in polyimide science and technology, ed. W. D. Weber and M. R. Gupta, Brookfield, CT, 1985, p. 67 Search PubMed.
- ed. D. Wilson, H. D. Stenzenberger and P. M. Hergenrother in Polyimides, Blackie & Son Ltd., Glasgow and London, 1990 Search PubMed.
- Y. J. Kim, T. E. Glass, G. D. Lyle and J. E. McGrath, Macromolecules, 1993, 26, 1344 CrossRef CAS.
- M. H. Kailani, C. S. P. Sung and S. Haung, Macromolecules, 1992, 25, 3751 CrossRef CAS.
- C. E. Sroog, J. Polym. Sci., Macromol. Rev., 1976, 11, 161 CrossRef CAS.
- P. M. Heregenrother and S. J. Havens, Macromolecules, 1994, 27, 4659 CrossRef.
- M. S. Butt, Z. Akhtar, M. Zafar-Uz-Zaman and A. Munir, Eur. Polym. J., 2005, 41, 1638 CrossRef CAS.
- J. Yin, Y. F. Ye and Z. G. Wang, Eur. Polym. J., 1998, 34, 1839 CrossRef CAS.
- C. Koning, L. Teuwen, E. W. Meijer and J. Moonen, Polymer, 1994, 35, 4889 CrossRef CAS.
- M. Marek, Jr., J. Labský, B. Schneider and J. Stokr, Eur. Polym. J., 1991, 27, 487 CrossRef.
- M. Marek, Jr., D. Dosko Cilová, P. Schmidt, B. Schneider, J. Kôiÿ, J. Labský and R. Puffr, Polymer, 1994, 35, 4881 CrossRef.
- T. Matsuura, M. Ishisawa, Y. Hasuda and S. Nishi, Macromolecules, 1991, 25, 3540 CrossRef.
- T. Omote, K. Koseki and T. Yamaoka, Polym. Eng. Sci., 1989, 29, 945 CAS.
- T. Omote, T. Yamaoka and K. Koseki, J. Appl. Polym. Sci., 1989, 38, 389 CrossRef CAS.
- A. N. K. Lau and J. P. Vo, Macromolecules, 1992, 25, 7294 CrossRef CAS.
- P. J. Cassidy, T. M. Aminathavi and J. M. Farley, J. Macromol. Sci., Part C, 1989, 29, 365 CrossRef.
- G. Hougham, G. Tesoro and J. Shaw, in Polyimides: materials, chemistry and characterization, ed. C. Feger, M. M. Khojasteh and J. E. McGrath, Elsevier, Amsterdam, 1989, p. 465 Search PubMed.
- J. Y. Yang, B. T. Jung and D. H. Suh, Polymer, 2001, 42, 8349 CrossRef CAS.
- A. Harada and M. Kamachi, Macromolecules, 1990, 23, 2821 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Macromolecules, 1995, 28, 8406 CrossRef CAS.
- G. Wenz and B. Keller, Angew. Chem., Int. Ed. Engl., 1992, 31, 197 CrossRef.
- N. Ogata, K. Sanui and J. Wada, J. Polym. Sci., Polym. Lett. Ed., 1976, 14, 459 CrossRef CAS.
- M. B. Steinbrunn and G. Wenz, Angew. Chem., Int. Ed. Engl., 1996, 35, 2139 CrossRef CAS.
- G. Wenz, M. B. Steinbrunn and K. Landfester, Tetrahedron, 1997, 53, 15575 CrossRef CAS.
- B. Casu, M. Reggiani, G. G. Gallo and A. Vigevani, Tetrahedron, 1968, 24, 803 CrossRef.
- J. Szejtli, A. Liptak, I. Jodal, P. Fugedi, P. Nanasi and A. Neszmelyi, Starch/Staerke, 1980, 32, 165 CrossRef CAS.
- J. P. Casey, P. A. Lucas and M. S. Vratsanos, US Pat., 5 066 764, 1991 Search PubMed.
- W. Huang, D. Yan, Q. Lu and P. Tao, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 229 CrossRef CAS.
- H. Yagci and L. J. Mathias, Polymer, 1998, 39, 3779 CrossRef CAS.
- S. H. Hsiao, C. P. Yang and S. H. Chen, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1551 CrossRef CAS.
- D. J. Liaw and B. Y. Liaw, Polym. J., 1996, 28, 970 CrossRef CAS.
- M. Langsam and W. F. Burgoyne, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 909 CrossRef CAS.
- S. K. Ahn, Y. H. Kim, D. C. Shin and S. K. Kwon, Bull. Korean Chem. Soc., 2000, 21, 377 CAS.
- Y. H. Kim, H. S. Kim, S. K. Ahn, S. O. Jung and S. K. Kwon, Bull. Korean Chem. Soc., 2002, 23, 933 CrossRef CAS.
- K. H. Becker and H. W. Schmidt, Macromolecules, 1992, 25, 6784 CrossRef CAS.
- C. García, Ph.D. Thesis, Autonoma University, Madrid, 1996.
- D. Ayala, A. E. Lozano, J. G. de la Campa and J. de Abajo, Polym. Prep., 1997, 38, 359 CAS.
- J. de Abajo and J. G. de la Campa, Adv. Polym. Sci., 1999, 140, 23 CrossRef CAS.
- D. J. Liaw, B. Y. Liaw and C. W. Yu, Polymer, 2001, 42, 5175 CrossRef CAS.
- D. J. Liaw and F. C. Chang, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5766 CrossRef CAS.
- H. S. Kim, Y. H. Kim, S. K. Ahn and S. K. Kwon, Macromolecules, 2003, 36, 2327 CrossRef CAS.
- J. G. de la Campa, C. Tauler and J. de Abajo, Macromol. Rapid Commun., 1994, 15, 417 CrossRef CAS.
- C. Garcı'a, A. E. Lozano, J. G. de la Campa and J. de Abajo, Macromol. Rapid Commun., 2003, 24, 686 CrossRef.
- C. Garcı'a, J. Espeso, A. E. Lozano, J. G. de la Campa and J. de Abajo, Macromol. Symp., 2003, 19, 293 Search PubMed.
- M. Calle, A. E. Lozano, J. G. de La Campa and J. de Abajo, Macromolecules, 2010, 43, 2268 CrossRef CAS.
- V. Korshak, S. V. Vinogradova and Y. S. Vygodskii, J. Macromol. Sci., Part C, 1974, C11, 45 CrossRef.
- F. W. Harris, W. A. Feld and L. H. Lanier, J. Appl. Polym. Sci.: Appl. Polym. Symp., 1975, 26, 421 CAS.
- Y. Imai, N. N. Malder and M. Kakimoto, J. Polym. Sci., Polym. Chem. Ed., 1984, 22, 2189 CrossRef CAS.
- J. M. Augl, J. Polym. Sci., Part A-1, 1970, 8, 3145 CrossRef CAS.
- W. A. Feld, L. Serico, B. G. Genez and L. L. Friar, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 883 CrossRef CAS.
- F. Akutsu, S. Kuze, K. Matsuo, K. Naruchi and M. Miura, Makromol. Chem. Rapid Commun., 1990, 11, 673 CrossRef CAS.
- F. Akutsu, T. Kataoka, H. Shimizu, K. Naruchi and M. Miura, Macromol. Rapid Commun., 1994, 15, 441 CrossRef.
- F. Akutsu, G. Saito, M. Miyamoto, Y. Kasashirna, M. Inoki and K. Naruchi, Macromol. Chem. Phys., 1996, 197, 2239 CrossRef CAS.
- Q. Zhang, S. Li, W. Li and S. Zhang, Polymer, 2007, 48, 6246 CrossRef CAS.
- Y. Kasashima, H. Kumada, K. Yamamoto, F. Akutsu, K. Naruchi and M. Miura, Polymer, 1995, 36, 645 CrossRef CAS.
- C. W. Chang, H. J. Yen, K. Y. Huang, J. M. Yeh and G. S. Liou, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7937 CrossRef CAS.
- D. Dotcheva, M. Klapper and K. Miillen, Macromol. Chem. Phys., 1994, 195, 1905 CrossRef CAS.
- C. P. Yang and W. T. Chen, Macromolecules, 1993, 26, 4865 CrossRef CAS.
- C. P. Yang and W. T. Chen, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2799 CrossRef CAS.
- C. S. Wang and T. S. Leu, Polymer, 2000, 41, 3581 CrossRef CAS.
- A. L. Rusanov and Z. B. Shifrina, High Perform. Polym., 1993, 5, 107 CAS.
- F. W. Harris and S. O. Norris, J. Polym. Sci., Polym. Chem. Ed., 1973, 11, 2143 CrossRef CAS.
- R. Giesa, U. Keller, P. Eiselt and H. W. Schmidt, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 141 CrossRef CAS.
- F. Akutsu, T. Kataoka, H. Shimizu, K. Naruchi and M. Miura, Macromol. Rapid Commun., 1994, 15, 411 CrossRef CAS.
- I. K. Spiliopoulos and J. A. Mikroyannidis, Macromolecules, 1996, 29, 5313 CrossRef CAS.
- V. V. Korshak, S. V. Vinogradova, Y. S. Vygodskii, Z. M. Nagiev, Y. G. Urman, S. G. Alekseeva and I. Y. Slonium, Makromol. Chem., 1983, 184, 235 CrossRef CAS.
- A. E. Lozano, J. G. de la Campa, J. de Abajo and J. Preston, Polymer, 1994, 35, 873 Search PubMed.
- H. R. Kricheldorf, G. Schwarz and E. Volker, Makromol. Chem. Rapid Commun., 1989, 10, 243 CrossRef CAS.
- M. Ghaemy and M. Barghamadi, J. Appl. Polym. Sci., 2009, 112, 815 CrossRef CAS.
- A. S. Mathews, D. Kim, Y. Kim, I. L. Kim and C. S. Ha, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 8117 CrossRef CAS.
- H. Behniafar and M. Ghorbani, Polym. Degrad. Stab., 2008, 93, 608 CrossRef CAS.
- F. W. Harris and S. L. C. Hsu, High Perform. Polym., 1989, 1, 3 CAS.
- F. W. Harris, S. L. C. Hsu and C. C. Tso, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1990, 31, 342 CAS.
- H. G. Rogers, R. A. Gaudiana, W. C. Hollinsed, P. S. Kalyamaraman, J. S. Manello, C. McGowan, R. A. Minns and R. Sahatjian, Macromolecules, 1985, 18, 1058 CrossRef CAS.
- F. W. Harris, S. H. Lin, F. Li and S. Z. D. Cheng, Polymer, 1996, 37, 5049 CrossRef CAS.
- J. Salbeck, N. Yu, J. Bauer, F. Weissöortel and H. Bestgen, Synth. Met., 1997, 91, 209 CrossRef CAS.
- N. Johansson, J. Salbeck, J. Bauer, F. Weissoörtel, P. Broöms, A. Andersson and W. R. Salaneck, Adv. Mater., 1998, 10, 1136 CrossRef CAS.
- F. Steuber, J. Staudigel, M. Stoössel, J. Simmerer, A. Winnacker, H. Spreitzer and J. Wei, Adv. Mater., 2000, 12, 130 CrossRef CAS.
- J. H. Weisburger, E. K. Weisburger and F. E. Ray, J. Am. Chem. Soc., 1950, 72, 4250 CrossRef CAS.
- R. Wu, J. S. Schumm, D. L. Pearson and J. M. Tour, J. Org. Chem., 1996, 61, 6906 CrossRef CAS.
- F. I. Wu, R. Dodda, D. S. Reddy and C. F. Shu, J. Mater. Chem., 2002, 12, 2893 RSC.
- C. L. Chiang and C. F. Shu, Chem. Mater., 2002, 14, 682 CrossRef CAS.
- S. H. Hsiao and C. T. Li, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1403 CrossRef CAS.
- C. H. Chou, D. S. Reddy and C. F. Shu, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3615 CrossRef CAS.
- C. P. Yang, Y. Y. Su, S. J. Wen and S. H. Hsiao, Polymer, 2006, 47, 7021 CrossRef CAS.
- C. Y. Wang, X. Y. Zhao, G. Li and J. M. Jiang, Polym. Degrad. Stab., 2009, 94, 1746 CrossRef CAS.
- S. H. Hsiao, C. P. Yang and C. Y. Yang, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1487 CrossRef CAS.
- S. Yoshida, T. Fujimori and N. Kato, Jpn Pat., 57915, 2007 Search PubMed.
- R. Mercado, Y. Wang, T. Flaim, W. DiMenna and U. Senapati, Proc. SPIE–Int. Soc. Opt. Eng., 2004, 5351, 276 CAS.
- C. A. Terraza, J. G. Liu, Y. Nakamura, Y. Shibasaki, S. Ando and M. Ueda, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1510 CrossRef CAS.
- S. J. Zhang, Y. F. Li, T. Ma, J. J. Zhao, X. Y. Xu, F. C. Yang and X. Y. Xiang, Polym. Chem., 2010, 1, 485 RSC.
- G. M. Jiang, X. Jiang, Y. F. Zhu, D. Huang, X. H. Jing and W. D. Gao, Polym. Int., 2010, 59, 896 CrossRef CAS.
- D. S. Reddy, C. H. Chou, C. F. Shu and G. H. Lee, Polymer, 2003, 44, 557 CrossRef.
- D. S. Reddy, C. F. Shu and F. I. Wu, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 262 CrossRef CAS.
- J. Li, K. Kudo and S. Shiraishi, Macromol. Chem. Phys., 2000, 201, 2289 CrossRef CAS.
- T. Matsumoto, J. Photopolym. Sci. Technol., 2001, 14, 695 CrossRef CAS.
- S. K. Sen and S. Banerjee, J. Membr. Sci., 2010, 365, 329 CrossRef CAS.
- B. S. Ghanem, N. B. McKeown, P. M. Budd, N. M. Al-Harbi, D. Fritsch, K. Heinrich, L. Starannikova, A. Tokarev and Y. Yampolskii, Macromolecules, 2009, 42, 7881 CrossRef CAS.
- A. Kausar, S. Zulfiqar, Z. Ahmad and M. I. Sarwar, Polym. Degrad. Stab., 2010, 95, 2603 CrossRef CAS.
- Z. M. Michalska, B. Ostaszewski and K. Strzelec, J. Organomet. Chem., 1995, 496, 19 CrossRef CAS.
- Y. P. Yen and K. W. Ho, Tetrahedron Lett., 2006, 47, 7357 CrossRef CAS.
- S. I. Kondo and M. Sato, Tetrahedron, 2006, 62, 4844 CrossRef CAS.
- X. Zhao, Y. F. Li, S. J. Zhang, Y. Shao and X. L. Wang, Polymer, 2007, 48, 5241 CrossRef CAS.
- H. R. Kricheldrof and V. Linzer, Polymer, 1995, 36, 1893 CrossRef.
- M. Acevedo and F. W. Harris, Polymer, 1994, 35, 4456 CrossRef CAS.
- W. Huang, Y. T. Tong, J. Xu and M. Ding, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 143 CrossRef CAS.
- G. Hougham, in Fluoropolymers, New York, Kluvert, 1999, vol. 2, ch. 14 Search PubMed.
- N. Adrova, M. Bessonov, L. A. Laius and A. P. Rudakov, in Polyimides: anew class of heatresistant polymers, IPST Press, Jerusalem, 1969 Search PubMed.
- R. A. Dine-Hart and W. W. Wright, Makromol. Chem., 1972, 153, 237 CrossRef CAS.
- V. L. Bell, B. L. Stump and H. Gager, J. Polym Sci. Part A: Polym. Chem., 1976, 14, 2275 CAS.
- P. E. Cassidy, T. M. Aminabhavi and J. M. Farley, J. Macromol. Sci., Part C, 1989, C29, 365 CrossRef CAS.
- T. Lin, K. W. Stickney, M. Rogers, J. S. Riffle, J. E. McGrath, H. Marand, T. H. Yu and R. M. Davis, Polymer, 1993, 34, 772 CrossRef CAS.
- D. M. White, T. Takehoshi, F. J. Williams, H. M. Relles, P. F. Donahue, H. J. Klopfer, G. R. Loucks, J. S. Manello, R. O. Mathews and R. W. Schluens, J. Polym. Sci. Part A: Polym. Chem., 1981, 19, 1635 CAS.
- R. O. Johnson and H. S. Burlhis, J. Polym. Sci., Polym. Symp., 1983, 70, 129 CrossRef CAS.
- Y. H. Cho, J. M. Park and Y. H. Park, Macromol. Res., 2004, 12, 38 CrossRef CAS.
- T. S. Leu and C. S. Wang, J. Appl. Polym. Sci., 2003, 87, 945 CrossRef CAS.
- Y. Shao, Y. F. LI, X. L. Wang, S. J. Zhang, X. Zhao and T. Ma, Chin. Chem. Lett., 2006, 17, 635 CAS.
- S. H. Hsiao and K. H. Lin, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 331 CrossRef CAS.
- M. S. Butt, Z. Akhter, M. Bolte, H. M. Siddiqi, H. Nawaz and M. Zafar-uz-Zaman, J. Appl. Polym. Sci., 2009, 114, 2101 CrossRef CAS.
- D. J. Liaw and B. Y. Liaw, Macromol. Chem. Phys., 1998, 199, 1473 CrossRef CAS.
- D. J. Liaw and B. Y. Liaw, Eur. Polym. J., 1997, 33, 1423 CrossRef CAS.
- D. J. Liaw, B. Y. Liaw and C. W. Yu, Polymer, 2001, 42, 5175 CrossRef CAS.
- I. Sava, S. Chisca, M. Bruma and G. Lisa, Polym. Bull., 2010, 65, 363 CrossRef CAS.
- M. Bruma, J. Fitch and P. Cassidy, J. Macromol. Sci., Part C, 1996, C36, 119 CrossRef CAS.
- Q. H. Lu, J. Yin, H. J. Xu, J. M. Zhang, L. M. Sun, Z. K. Zhu and Z. G. Wang, J. Appl. Polym. Sci., 1999, 72, 1299 CrossRef CAS.
- X. Y. Shang, Z. K. Zhu, J. Yin and X. D. Ma, Chem. Mater., 2002, 14, 71 CrossRef CAS.
- A. Banihashemi and B. Pourabbas, Eur. Polym. J., 2000, 36, 2031 CrossRef CAS.
- A. Banihashemi and B. Akhlaghinia, Macromol. Chem. Phys., 1999, 200, 2284 CrossRef CAS.
- A. Banihashemi and F. Atabaki, Eur. Polym. J., 2002, 38, 2119 CrossRef CAS.
- A. Banihashemi and S. H. Firoozifar, Iran Polym. J., 2000, 92, 111 Search PubMed.
- A. Banihashemi, A. Abdolmaleki, Proceedings of the Fifth Seminar on Polymer Science and Technology, Tehran, 2000 Search PubMed.
- B. Pourabbas and A. Banihashemi, Polym. Int., 2002, 51, 1086 CrossRef.
- A. Banihashemi and A. Abdolmaleki, Eur. Polym. J., 2004, 40, 1629 CrossRef CAS.
- N. Amutha and M. Sarojadevi, J. Macromol. Sci., Part A: Pure Appl. Chem., 2007, 44, 1013 CrossRef CAS.
- H. Behniafar and P. Boland, J. Polym. Res., 2010, 17, 511 CrossRef CAS.
- M. D. Damaceanu, I. Bacosca, M. Bruma, J. Robison and A. Rusanov, Polym. Int., 2009, 58, 1041 CrossRef CAS.
- S. H. Hsiao, C. P. Yang and T. K. Lo, J. Polym. Res., 1998, 5, 193 CrossRef CAS.
- G. L. Brode, J. H. Kawakami, G. T. Kwiatkowski and A. W. Bedwin, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 575 CrossRef.
- G. T. Kwiatkowski, G. L. Brode, J. H. Kawakami and A. W. Bedwin, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 589 CrossRef CAS.
- C. Chiriac and J. K. Stille, Macromolecules, 1974, 10, 712 CrossRef.
- J. Adduci, L. L. Chapoy, G. Jonsson, J. Kops and B. M. Shinde, J. Appl. Polym. Sci., 1983, 28, 2069 CrossRef CAS.
- S. B. Idage, B. B. Idage, B. M. Shinde and S. P. Vernekar, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 583 CrossRef CAS.
- Y. Delaviz, A. Gungor, J. E. McGrath and H. W. Gibson, Polymer, 1993, 34, 210 CrossRef CAS.
- F. Keitou, M. Kakimoto and Y. Imai, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 317 CrossRef.
- V. M. Svetlichny, V. M. Denisov, V. V. Kudryatsev, G. A. Polotskaya and Y. P. Kuznetsov, in Polyimides and other high-temperature polymers, ed. M. J. M. Abadie and B. Sillion, Elsevier, Amsterdam, 1991, p. 525 Search PubMed.
- G. N. Buba, in Polyimides: synthesis, characterization and applications, ed. K. L. Mittal, Plenum, New York, 51, 1985 Search PubMed.
- J. P. Critchley and M. A. White, J. Polym. Sci., Part A-1, 1972, 10, 1809 CrossRef CAS.
- H. R. Kricheldorf and V. Linzer, Polymer, 1995, 36, 1893 CrossRef CAS.
- M. E. Rogers, T. E. Glass, S. J. Mecham, D. Rodrigues, G. L. Wilkes and J. E. McGrath, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2663 CrossRef CAS.
- Y. D. Moon and Y. M. Lee, J. Appl. Polym. Sci., 1993, 50, 1461 CrossRef CAS.
- P. Thiruvasagam and D. Venkatesan, High Perform. Polym., 2010, 22, 682 CrossRef CAS.
- W. F. Hale and A. G. Famham, J. Polym. Sci., Part A-1, 1967, 5, 2399 CrossRef CAS.
- B. F. Malichenko and V. V. Sherikova, Vysokomol Soedin, Ser B, 1972, 14, 423 CAS.
- C. P. Yang and J. H. Lin, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2153 CrossRef CAS.
- C. P. Yang and J. H. Lin, J. Polym. Sci., Part A: Polym. Chem., 1993, 32, 369 CrossRef.
- C. P. Yang and J. H. Lin, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 423 CrossRef CAS.
- C. P. Yang and S. Y. Tang, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 455 CrossRef CAS.
- Y. Imai and M. Kakimoto, Polym.-Plast. Technol. Eng., 1989, 28, 371 CrossRef CAS.
- Y. Oishi, M. Kakimoto and Y. Imai, in Polyimides: Materials, chemistry and characterization, ed. C. Feger, M. M. Khojasteh, and J. E. McGrath, Elsevier, New York, 1989, p. 139 Search PubMed.
- H. J. Jeong, Y. Oishi, M. Kakimoto and Y. Imai, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 39 CrossRef CAS.
- H. J. Jeong, M. Kakimoto and Y. Imai, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 1691 CrossRef CAS.
- M. Xie, Y. Oishi, M. Kakimoto and Y. Imai, Polym. Prepr. Jpn., 1988, 37, 2966 Search PubMed.
- Y. Oishi, M. Ishida, M. A. Kakimoto, Y. Imai and T. Kurosaki, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 1027 CrossRef CAS.
- I. Bacosca, E. Hamciuc, M. Bruma and M. Szesztay, Revue Roumaine de Chimie, 2009, 54, 1023 CAS.
- H. Yeganeh and S. Mehdipour-Ataei, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1528 CrossRef CAS.
- B. Dasgupta, S. K. Sen and S. Banerjee, J. Membr. Sci., 2009, 345, 249 CrossRef CAS.
- S. K. Sen and S. Banerjee, J. Membr. Sci., 2009, 350, 53 CrossRef.
- R. R. Pal, P. S. Patil, R. T. Dere, M. M. Salunkhe, N. N. Maldar and P. P. Wadgaonkar, J. Appl. Polym. Sci., 2005, 97, 1377 CrossRef CAS.
- C.P. Yang and R. S. Chen, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 429 CrossRef CAS.
- M. Ghaemy, R. Alizadeh and F. H. Nasr, J. Appl. Polym. Sci., 2010, 118, 3407 CrossRef CAS.
- C. E. Sroog, Prog. Polym. Sci., 1991, 16, 561 CrossRef CAS.
- R. C. Fort and P. V. R. Schleyer, Jr., Chem. Rev., 1964, 64, 277 CrossRef CAS.
- Y. T. Chern and W. H. Chung, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 117 CrossRef CAS.
- Y. T. Chern and H. C. Shiue, Macromolecules, 1997, 30, 4646 CrossRef CAS.
- Y. T. Chern, K. S. Lin and S. C. Kao, J. Appl. Polym. Sci., 1998, 68, 315 CrossRef CAS.
- Y. T. Chern, H. C. Shiue and S. C. Kao, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 785 CrossRef CAS.
- Y. T. Chern and H. C. Shiue, Macromol. Chem. Phys., 1998, 199, 963 CrossRef CAS.
- S. H. Hsiao and C. T. Li, Macromolecules, 1998, 31, 7213 CrossRef CAS.
- S. H. Hsiao and C. T. Li, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1435 CrossRef CAS.
- S. M. Kwak, J. H. Yeon and T. H. Yoon, J. Polym. Sci. Part A: Polym. Chem., 2006, 44, 2567 CrossRef CAS.
- Y. C. Kung, G. S. Liou and S. H. Hsiao, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1740 CrossRef CAS.
- Z. Qiu, S. Zhang, W. Li, Z. Qiu and W. Li, J. Appl. Polym. Sci., 2007, 104, 2395 CrossRef CAS.
- R. Hariharan and M. Sarojadevi, Polym. Int., 2007, 56, 22 CrossRef CAS.
- X. J. Zhao, J. G. Liu, J. M. Rui, L. Fan and S. Y. Yang, J. Appl. Polym. Sci., 2007, 103, 1442 CrossRef CAS.
- B. Y. Myung, J. S. Kim, J. J. Kim and T. H. Yoon, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3361 CrossRef CAS.
- B. Y. Myung, C. J. Ahn and T. H. Yoon, Polymer, 2004, 45, 3185 CrossRef CAS.
- Z. Li, J. Liu, Z. Gao, Z. Yin, L. Fan and S. Yang, Eur. Polym. J., 2009, 45, 1139 CrossRef CAS.
- C. L. Chung, W. F. Lee, C. H. Lin and S. H. Hsiao, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1756 CrossRef CAS.
- K. Xie, J. G. Liu, H. W. Zhou and S. Y. Yang, Polymer, 2001, 42, 7267 CrossRef CAS.
- H. Choi, S. Y. Kim, I. S. Chung, K. Hong and C. E. Park, Polymer, 2008, 49, 2644 CrossRef CAS.
- Z. Ge, L. Fan and S. Yang, Eur. Polym. J., 2008, 44, 1252 CrossRef CAS.
- S. Banerjee, M. K. Madhra, A. K. Salunke and G. Maier, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1016 CrossRef CAS.
- M. K. Madhra, A. K. Salunke, S. Banerjee and S. Prabha, Macromol. Chem. Phys., 2002, 203, 1238 CrossRef CAS.
- S. Banerjee, M. K. Madhra, A. K. Salunke and D. K. Jaiswal, Polymer, 2003, 44, 613 CrossRef CAS.
- V. Kute and S. Banerjee, Macromol. Chem. Phys., 2003, 204, 2105 CrossRef CAS.
- A. Ghosh, S. Banerjee and B. Voit, High Perform. Polym., 2009, 21, 173 CrossRef CAS.
- V. Kute and S. Banerjee, J. Appl. Polym. Sci., 2007, 103, 3025 CrossRef CAS.
- A. Ghosh and S. Banerjee, J. Macromol. Sci., Part A: Pure Appl. Chem., 2008, 45, 578 CrossRef CAS.
- Y. Shao, Y.-F. Li, X. Zhao, X.-L. Wang, T. Ma and F.-C. Yang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6836 CrossRef CAS.
- W. Li, S. Zhang, G. Chen and Q. Zhang, Polymer, 2007, 48, 3082 CrossRef CAS.
- C. P. Yang, Y. Y. Su and H. C. Chiang, React. Funct. Polym., 2006, 66, 689 CrossRef CAS.
- K. Wang, L. Fan, J. G. Liu, M. S. Zhan and S. Y. Yang, J. Appl. Polym. Sci., 2008, 107, 2126 CrossRef CAS.
- K. U. Jeong, J. J. Kim and T. H. Yoon, Korea Polym. J., 2008, 8, 215 Search PubMed.
- S. Wang, Q. Ji, C. N. Tchatchoua, A. R. Shultz and J. E. McGrath, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 1849 CrossRef CAS.
- A. Satoh and A. Morikawa, High Perform. Polym., 2010, 22, 412 CrossRef CAS.
- W. C. Liaw and K. P. Chen, J. Polym. Res., 2007, 14, 5 CrossRef CAS.
- X. Jiang, J. Gu, Y. Shen, S. Wang and X. Tian, Desalination, 2011, 265, 74 CrossRef CAS.
- I. Novák, P. Sysel, J. Zemek, M. Špírková, D. Velič, M. Aranyosiová, Š. Florián, V. Pollák, A. Kleinová, F. Lednický and I. Janigová, Eur. Polym. J., 2009, 45, 57 CrossRef.
- I. Novák, P. Sysel, I. Chodák, M. Špírková and I. Janigová, Chemické Listy., 2009, 103, 76 Search PubMed.
- S. Maudgal and T. L. St Clair, Int. J. Adhes. Adhes., 1984, 4, 87 CrossRef CAS.
- S. Maudgal and T. L. St Clair, SAMPE Quarterly, 1984, 16, 6 CAS.
- I. Yilgor, E. Yilgor, B. J Johnson, J. Eberle, G. L. Wilkes and J. E. McGrath, Polym. Prep., 1983, 24, 78 CAS.
- I. Yilgor, E. Yilgor, J. Eberle, W. Jr Steckle, B. C. Johnson, D. Tyagi, G. L. Wilkes and J. E. McGrath, Polym. Prep., 1983, 24, 167 CAS.
- B. C. Johnson. PhD thesis. Materials engineering science. High-performance polyimide copolymers: synthesis and characteristics.Virginia Polytechnic Institute and State University. 1984 Search PubMed.
- B. C. Johnson, I. Yilgor and J. E. McGrath, Polym Prep., 1984, 25, 54 CAS.
- J. E. McGrath, in American Chemical Society, ACS Symposium Series, ed. J. E. McGrath, Washington DC, 1985, 286, 1 Search PubMed.
- R. Z. Zelinski and C. W. Childers, Rubber Chem. Technol., 1968, 41, 161 CrossRef CAS.
- N. R. Legge, G. Holden and H. E. Schroeder, Thermoplastic elastomers: a comprehensive review, New York, Hamser Publishers, 1987 Search PubMed.
- M. Kajiyama, M.-A. Kakimoto and Y. Imai, Macromolecules, 1990, 23, 1244 CrossRef CAS.
- F. L. Keohan and J. E. Hallgren, Adv. Chem. Series, 1990, 224, 165 CrossRef.
- S. Nakata, M. Kawata, M. A. Kakimoto and Y. Imai, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 3425 CrossRef CAS.
- S. L. Jwo, W. T. Whang and W. C. Liaw, J. Appl. Polym. Sci., 1999, 74, 2832 CrossRef CAS.
- S. Babanzadeh, A. R. Mahjoub and S. Mehdipour-Ataei, Polym. Degrad. Stab., 2010, 95, 2492 CrossRef CAS.
- A. Ghosh and S. Banerjee, J. Appl. Polym. Sci., 2008, 107, 1831 CrossRef CAS.
- A. Ghosh and S. Banerjee, J. Appl. Polym. Sci., 2008, 109, 2329 CrossRef CAS.
- A. Ghosh and S. Banerjee, Polym. Adv. Technol., 2008, 19, 1486 CAS.
- A. Ghosh, S. Banerjee and B. Voit, High Perform. Polym., 2010, 22, 28 CrossRef CAS.
- K. Pareek, A. Ghosh, S. K. Sen and S. Banerjee, Des. Monomers Polym., 2010, 13, 221 CrossRef CAS.
- A. Soules, C. P. Vázquez, B. Améduri, C. J. Duhamel, M. Essahli and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3214 CrossRef CAS.
- A. Ghosh, S. Banerjee, H. Komber, K. Schneider, L. Häuβler and B. Voit, Eur. Polym. J., 2009, 45, 1561 CrossRef CAS.
- A. Ghosh, S. Banerjee, L. Häuβler and B. Voit, J. Macromol. Sci., Part A: Pure Appl. Chem., 2010, 47, 671 CrossRef CAS.
- Y. Tsuda, J. M. OH and R. Kuwahara, Int. J. Mol. Sci., 2009, 10, 5031 CrossRef CAS.
- J. Fang, H. Kita and K. Okamoto, Macromolecules, 2000, 33, 4639 CrossRef CAS.
- M. Jikei and M. Kakimoto, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1293 CrossRef CAS.
- N. B. McKeown, P. M. Budd and D. Book, Macromol. Rapid Commun., 2007, 2, 995 CrossRef.
- B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2505 CrossRef CAS.
- M. Jikei and M. Kakimoto, Prog. Polym. Sci., 2001, 26, 1233 CrossRef CAS.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183 CrossRef CAS.
- L. Cheng, Z. Xu, X. Q. Xiong, J. X. Wang and B. Jing, Chin. J. Polym. Sci., 2010, 28, 69 CrossRef CAS.
- H. Chen and J. Yin, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3804 CrossRef CAS.
- H. Gao, D. Wang, W. Jiang, S. Guan and Z. Jiang, J. Appl. Polym. Sci., 2008, 109, 2341 CrossRef CAS.
- Q. Zhang, J. He, Y. Q Fang and Y. H. Wang, Liq. Cryst., 2008, 35, 385 CrossRef CAS.
- W. Chen, W. Yan, S. Wu, Z. Xu, K. W. K. Yeung and C. Yi, Macromol. Chem. Phys., 2010, 211, 1803 CrossRef CAS.
- J. Hao, M. Jikei and M. Kakimoto, Macromolecules, 2003, 36, 3519 CrossRef CAS.
- K. Yamanaka, M. Jikei and M. Kakimoto, Macromolecules, 2000, 33, 6937 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |