The first bromide ion directed double helicate and its role in catalysis†‡
P. M.
Selvakumar
,
P. Yowan
Jebaraj
,
Jashobanta
Sahoo
,
E.
Suresh
,
K. Jeya
Prathap
,
R. I.
Kureshy
* and
P. S.
Subramanian
*
CSIR-Central Salt and Marine Chemicals Research Institute (Council of Scientific & Industrial Research), Bhavnagar–364 002, Gujarat, India. E-mail: siva@csmcri.org; rkureshy@csmcri.org; Fax: +91-278-25676970; Tel: +91-2782567760
First published on 15th June 2012
Abstract
A bromide ion directed double stranded helicate is synthesised and its structure is reported for the first time. Synthesis of a series of β-aminocarbonyls using this water soluble helicate as a catalyst for a three-component amionalkylation reaction is also explored by adopting solvent-free reaction conditions.
Studies on recognition, detection and extraction of anions1 have electrified interest among researchers. Motivation behind such anion recognition studies are the immense implications in biology,2 medicine,3 catalysis,4 molecular assembly and host–guest chemistry,5etc. In this direction, supramolecular chemistry with various construction components6 has provided varieties of brilliant helical architectures in nature, e.g., α-helical protein (single helix), DNA (double helix), collagen (triple helix), etc. The coordination chemistry of anions as witnessed from the rich literature7 has attracted immense interest among supramolecular chemists; anion helicates are rare in the literature when compared to metal ion directed helicates. De Mandoza8 reported the first sulphate-surrounded tetraquanidinium double helix solely supported by ROESY NMR. Subsequently, Hannon,9 Yoshida10 and Kruger11 had reported varieties of metallohelicates using a series of Schiff base ligands. Kruger11 protonated the same ligand and demonstrated the first structurally characterized chloride based double stranded helicate, diammonium–bis-pyridinium salt [(H4L2Cl)2]·6Cl·H2O. Recently Gale and co-workers12 have reported the first fluoride-based double helix using an isophthalamide ligand. Though the first fluoride- and chloride-directed double helicates have already been reported, bromide ion directed double helicates have not been reported until recently. Although anion encapsulated compounds covering derivatives of various macrocycles,13a amides,13b urea,13c pyrrole,13d ammonium,13e guanidinium13f and pyridinium moieties13g are abundant in the literature, the studies on such anion salts are restricted to recognition, detection and their stereochemistry. In this series, the present paper reports the first structurally derived bromide ion based double stranded helicate and demonstrates that such anion salts can be further used as a catalyst in many organic reactions, opening up a new avenue for their catalytic potentiality. In general, Lewis or Brønsted acids are used as catalysts in various reactions. Aminoalkylation (Mannich) is a classical organic reaction, and the conventional acids used as catalysts14a,b are proline,14c,d acetic acid,14ep-dodecyl-benzenesulfonic acid,14f metal complexes14g, etc.
Bis-imine bispyridine ligand 1 was obtained by mixing a methanolic solution of pyridine-2-carbaxaldehyde and 4,4′-methylenedianiline in 2:1 ratio. Following the standard sodium borohydride reduction, the respective protonated ligand 1a, H4L was obtained, for which the crystal structure is already available (CSD ref code: ICIBOL). The ligand 1a was treated with HBr (48%) until it reached pH 1 and the resultant precipitate was isolated. While the corresponding chlorocomplex as reported by Kruger was pale yellow, the respective bromo complex was isolated as a white powder. The bromide complex 1b crystallized upon slow evaporation from aqueous solution at room temperature.
An ORTEP diagram showing the side view and top view of the double helical arrangement and the interaction of Br2 anions with the intertwined H4L ligand moiety is shown in Fig. 1. Tetrabromo diammonium-bis-pyridinium salt H4L·Br4 crystallized in orthorhombic space group Pban with the tetraprotonated cationic ligand moiety possessing a two-fold axis passing through the methylene carbon C13 along with two of the bromide ions (Br2 and Br3) occupying special positions (lying on another two-fold axis) and the third one (Br1) in a general position. The molecular structure of this organic salt can be described as “two symmetrically disposed ligand moieties that are flexibly intertwined and wrapped around the bromide ions (Br2) and generating a double helical arrangement”. The double helical arrangement of the intertwined strands generates channels in which bromide ions (Br2) are encapsulated via N–H⋯Br coordination involving pyridinium hydrogen H1C (with N1⋯Br2 distance 3.161(5)Å ) as depicted in Fig. 1a. Fig. 2 represents the intermolecular hydrogen bonding interactions of the various bromide ions. The adjacent intertwined cationic ligands are interconnected in a double helical arrangement via C–H⋯Br and N–H⋯Br hydrogen bonds involving Br1 and the aromatic hydrogen H1; ammonium hydrogen H2B with C1⋯Br1 and N2⋯Br1 distances 3.595(7) and 3.171(5) Å respectively. The Br2⋯Br2 separation distance between symmetrically disposed anions residing within the cleft generated by the hydrogen bonded intertwined ligand strand is 4.56 Å. The double stranded hydrogen bonded helical nets oriented in the ac-plane are cross linked via N–H⋯Br from the ammonium hydrogen H2A and Br3, generating a two dimensional hydrogen bonded network as depicted in Fig. 2, where Br3 is occupied between the exteriors of the helical nets.
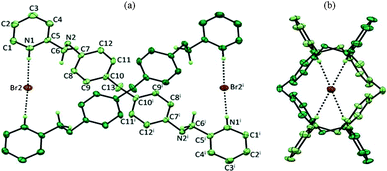 | ||
| Fig. 1 ORTEP diagram depicting the interaction of Br2 anions with the intertwined H4L ligand moiety (symmetry transformations used to generate equivalent atoms ‘i’ is; −x + 1/2, y, z; 30% probability factor for the thermal ellipsoids) (a) side view (b) top view down a-axis. | ||
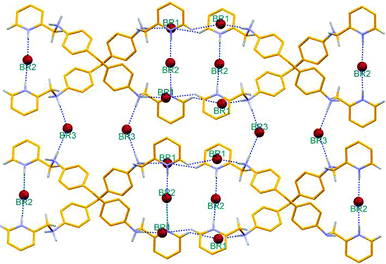 | ||
| Fig. 2 Mercury diagram depicting the helical network generated by the cationic part of the organic ligand and the various hydrogen bonding interactions between the anion and cation in generating the two dimensional network in the organic salt. | ||
Details of the pertinent hydrogen bonding interactions with symmetry code are given in Table 1. The templating effect of the bromide anion upon coordination with the protonated ligand and the conformational flexibility of the cationic ligand concomitantly favors the formation of a helical network, enabling the anions to reside inside the shielded intertwined cationic cavity of organic strands. An almost similar structure, with the same cationic ligand in which a chloride anion is within the helical cavity and one water molecule acts as a solvent of crystallization, reported by Keegan et al.11a is noteworthy.
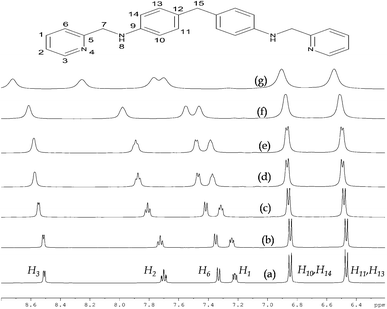
While the crystal structure establishes the helical architecture in the solid, the corresponding 1H-NMR spectra illustrates solution state behaviour. The series of 1H-NMR spectra of 1a titrated against HBr are shown in Fig. 3(a)–(g). [(a) 1:0, (b) 1:1, (c) 1:2, (d) 1:3, (e) 1:4, (f) 1:6 and (g) 1:20 equivalent ratio of 1a against HBr]. The spectra (a) represent the neat 1a, (b)–(g) illustrate the responses of 1a upon adding HBr. The 1H-NMR signals are assigned to the appropriate hydrogens of 1a. A fine triplet appeared at 6.15δ attributable to the NH proton disappeared immediately upon HBr addition. While the hydrogens attached to pyridyl domains showed a larger downfield shift, the hydrogens H10, H14 and H11, H13 of diphenyl spacer exhibit comparatively less shift and confirm the respective strength of their interaction with the bromide ions.
The binding of the bromide ion with a terminal pyridyl moiety affects the proximal hydrogens H6, H4, H3 and H5 of the pyridyl group significantly. However, since the diphenyl spacer is away from the bromide environment, the respective hydrogens show less or no shift in δ. The CH2 hydrogens residing away from the bromide environment show no change in the chemical shift with respect to the free ligand. Since the complex 1b contains eight HBr anions, the pH of the isolated complex measured as 3.5 is justifiable.
Brønsted acids are routinely used as catalysts in many organic reactions. Researchers study a variety of catalysts and work assiduously on modifying the catalyst, solvents and reaction conditions, etc., for various organic reactions. Although there are rich anionic compounds available in the literature, no attempt has been made to explore their catalytic role before this report, despite their high acidic nature. With this understanding, we were inspired to explore the catalytic capability of the HBr complex of 1a. Since the compound 1b is composed of HBr, its acidic and water soluble nature is considered to be advantageous. Further, in the current interest of developing new catalysts to work under aqueous media15 and solvent-free16 conditions, this study gains more importance. Hence a three-component C–C bond coupling (Mannich) reaction17 is known to have significant importance towards the preparation of various secondary and tertiary amine derivatives, such as β-amino carbonyls used as intermediates in pharmaceuticals, and natural products which are investigated in this present analysis.
Initially, in order to find out the optimal catalyst loading, the three-component aminoalkylation reaction (Scheme 1) was carried out using benzaldehyde (1 mol), aniline (1 mol) and cyclohexanone (2 mol) as representative reactants (Entries 1–5; Table 2, S3) in solvent-free conditions at RT, following the general catalysis protocol described in S2 of the ESI.† In the absence of a catalyst, no product had been formed after 48 h (entry 1). The compound 1b, with 2 mol%, provided 90% yield after 70 min (entry 2). Upon increasing the amount of 1b to 5 mol%, the amount of yield increased to 94% with a reduction in the reaction time to 45 min (Entry 3). However, a further increase to 10 mol% of 1b showed no change in the yield and reaction time, thus the optimal amount of 1b as catalyst was fixed at 5 mol%. Since the best result in terms of yield was achieved within 45 min using 5 mol% catalyst, three-component reactions adopting cyclohexanone, aniline and substituted aldehydes (as shown in entries 5–13) were conducted under these conditions (catalyst 5 mol%, 45 min, RT) and the respective data are depicted in Table 2 along with syn/anti ratio calculated using the 1H-NMR spectra. Based on the product formation, entries 5, 8, 9, 11 and 12 provided yields of 90–94%, entries 6, 7, 10 and 13 provided 76–80%, which suggests that the substitutional effect is a key factor. Compared to the electron withdrawing group, the electron donating substitution enhances the formation of the product. Further confirmation was achieved by obtaining the crystal structure of the product in entry 5.
| Entry | Substratesa | % yieldb | [Cat] mol% | syn/anti c | Time | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: catalyst 1b, substrate aniline and cyclohexanone, NR = no reaction, substituted aldehydes as substrates. b Reaction conditions: catalyst 1b, substrate aniline and cyclohexanone, NR = no reaction, isolated yield. c Reaction conditions: catalyst 1b, substrate aniline and cyclohexanone, NR = no reaction, syn/anti ratio was calculated from the 1H-NMR spectra. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Benzaldehyde | NR | Nila | NR | 48 h | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Benzaldehyde | 90 | 2a | — | 70 min | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Benzaldehyde | 94 | 5a | — | 45 min | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Benzaldehyde | 94 | 10a | — | 45 min | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Benzaldehyde | 94 | 5b | 24:76 | 45 min | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 2-F-benzaldehyde | 79 | 5b | 33:67 | 45 min | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 4-Cl-benzaldehyde | 76 | 5b | 39:61 | 45 min | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 2-CH3-benzaldehyde | 93 | 5b | 30:63 | 45 min | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 3-CH3-benzaldehyde | 90 | 5b | 32:68 | 45 min | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 4-NO2-benzaldehyde | 77 | 5b | 34:66 | 45 min | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 2-OCH3-benzaldehyde | 90 | 5b | 24:76 | 45 min | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 4-OCH3-benzaldehyde | 92 | 5b | 35:65 | 45 min | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | 2-OC6H5-benzaldehyde | 80 | 5b | 34:66 | 45 min | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
As mentioned above, the literature survey indicates that varieties of catalysts such as Brønsted acids and Lewis acids are applied for the reaction. Among the Lewis acids, H3PMo12O40 was reported recently18 with a yield of 84% and with a 63:37 syn/anti ratio. Similarly, as an example Brønsted acid, trifluoroacetic acid was used recently by Joseph and coworkers.19 In the case of trifluoroacetic acid, the reaction time was 4 h and the yield was reported as 57%. A similar anionic compound, bromodimethylsulfonium bromide, was used as a catalyst20 for a three-component aminoalkylation reaction, the yield being 86% and the reaction time had been tremendously reduced to 15 min. A comparative analysis with these catalysts certainly reveals that the present catalyst 1b is superior in view of the yield.
At this point, in addition to the catalysis, it is considered as important to establish the reaction path, since a three-component system is generally known to proceed either through aldol or imine intermediates as indicated in Scheme 2. Accordingly, an experiment conducted by mixing benzaldehyde and cyclohexanone in the presence of 1b for 6 h failed to give any aldol. However, for a similar experiment performed with benzaldehyde and aniline, the imine product was allowed to react with cyclohexanone in the presence of 1b at RT, which resulted in the formation of the product β-amino ketone. Based on these observations, it is concluded that among these two pathways, path-B is found to be operative.
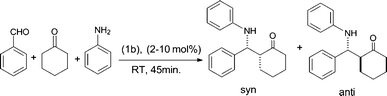 | ||
| Scheme 1 Catalytic studies on the Mannich reaction using complex 1b as a catalyst. | ||
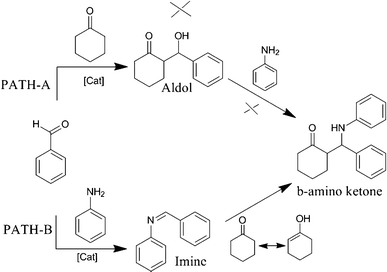 | ||
| Scheme 2 Mechanism for following a three-component Mannich reaction. | ||
With the catalyst 1b being a novel reagent, our systematic approach demonstrated above clearly indicates that 1b catalyses the reaction along with its simultaneous transformation of 1b into L1 and HBr. The nucleophilic addition of enolizable ketones into these imines in the presence of HBr followed by hydrolysing, β-aminocarbonyl compounds was clearly established.
To understand the stability of the catalyst, and the catalytic reaction up to four cycles, the 1H-NMR spectra (S6) recorded for 1b before and the after the catalytic recycle clearly indicate that the compound is highly stable.
In summary, this work presents the first structurally characterized bromide ion directed double stranded anion helicate. Secondly, by demonstrating the catalytic activity using the double stranded anion helicate, this work opens up a new avenue that allow such rich anionic compounds reported earlier to also be used as catalysts in addition to the routine recognition, detection and extraction studies. Furthermore, this reaction illustrates a solvent-free three-component catalytic system, and the catalyst is considered to be environmentally friendly. The catalytic reaction demonstrates the first experiment using this type of anionic compound as a catalyst, and further planning to synthesize some chiral double helicates is under progress. Our experiments clearly suggest that the catalyst 1b is a highly stable and active catalyst.
Preparation of ligands: 1 and 1a were prepared as per the literature.11b
Complex 1b. The ligand 1a (0.361 g, 0.001 mmol) was dissolved in methanol and mixed with 48% HBr and was continuously stirred in cold conditions, which resulted in the formation of a precipitate. The precipitate was isolated and dissolved in methanol and produced colorless crystals upon slow evaporation at RT in 48 h. Yield 85%, C50H58Br8N8O: calc (found) C 42.64 (42.89), H 4.01 (4.40), N 7.96 (7.87); 1H NMR (DMSO-D6, TMS, 500 MHz): δ = 11.0 (sbr, 2H, NH), 8.92, 8.90 (d, 2H, J = 10 Hz), 8.59, 8.57, 8.55 (t, 2H, J = 10 Hz), 8.07, 8.03 (d, 2H, J = 20 Hz), 8.02, 8.00, 7.98 (t, 2H, J = 10 Hz), 6.98, 6.96 (d, 4H, J = 10 Hz), 6.67, 6.64, 6.63, 6.60 (dd, 4H, J = 15 Hz), 4.84 (s), 4.72 (s, 2H, –CH2–).
Acknowledgements
Author PSS acknowledges the DST New Delhi for funding and PMS acknowledges CSIR for a SRF Grant. We thank Ms. Prathiba for her help in catalysis. We acknowledge both the reviewers for their useful comments.References
- (a) Topics in Heterocyclic Chemistry, ed. P. A. Gale, W. Dehaen, Springer, New York, 2010, vol. 24, pp. 145–176 Search PubMed; (b) S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3665 RSC; (c) B. A. Moyer and R. P. Singh, Fundamentals and applications of anion separation, Kluwer academic Plenum Publishers, New York, 2003 Search PubMed.
- K. L. Kirk, Biochemistry of the halogens and inorganic halides, Plenum Press, New York, 1991 Search PubMed.
- (a) P. A. Gale, Acc. Chem. Res., 2011, 44, 216–226 CAS; (b) F. Okamoto, H. Kajiya, K. Toh, S. Uchida, M. Yoshikawa, S. Sasaki, M. A. Kido, T. Tanaka and K. Olabe, Am. J. Physiol., 2008, 294, C693–701 CrossRef CAS; (c) T. Agou, M. Sekine, J. Kobayashi and T. Kawashima, Chem.–Eur. J., 2009, 15, 5056–5062 CrossRef CAS.
- K. Li, T. He, C. Li, X.-W. Feng and X.-Q. Yu, Green Chem., 2009, 11, 777–779 RSC.
- T. Yamaguchi, S. Tashiro, M. Tominaga, M. Kawano, T. Ozeki and M. Fujita, Chem.–Asian J., 2007, 2, 468–476 CrossRef CAS.
- J. W. Steed, D. R. Turner and K. Wallace, Core Concepts in Supramolecular Chemistry and Nano Chemistry, Wiley, 2007 Search PubMed.
- (a) P. A. Gale, Coord. Chem. Rev., 2003, 240, 191 CrossRef CAS; (b) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486 CrossRef CAS; (c) A. Bianchi, K. Bowman-James and E. Garcia-Espana, Supramolecular Chemistry of AnionsWiley-VCH, New York, 1997 Search PubMed; (d) V. McKee, J. Nelson and R. M. Town, Chem. Soc. Rev., 2003, 32, 309 RSC.
- J. Sanchez-Quesada, C. Seel, P. Prados, J. de Mendoza, I. Dalcol and E. Giralt, J. Am. Chem. Soc., 1996, 118, 277–278 CrossRef CAS.
- (a) M. J. Hannon, C. L. Painting and W. Errington, Chem. Commun., 1997, 307 RSC; (b) M. J. Hannon, C. L. Painting, A. Jackson, J. Hamblin and W. Errington, Chem. Commun., 1997, 1807 RSC; (c) M. J. Hannon, C. L. Painting and N. W. Alcock, Chem. Commun., 1999, 2023 RSC.
- (a) N. Yoshida, K. Ichikawa and M. Shiro, J. Chem. Soc., Perkin Trans. 2, 2000, 17 RSC; (b) N. Yoshida, H. Oshio and T. Ito, Chem. Commun., 1998, 63–64 RSC.
- (a) J. Keegan, P. E. Kruger, M. Nieuwenhuyzen, J. O'Brien and N. Martin, Chem. Commun., 2001, 2192–2193 RSC; (b) P. E. Kruger, N. Martin and M. Nieuwenhuyzen, J. Chem. Soc., Dalton Trans., 2001, 1966–1970 RSC.
- S. J. Coles, J. G. Frey, P. A. Gale, M. B. Hursthouse, M. E. Light, K. Navakhan and G. L. Thomas, Chem. Commun., 2003, 568–569 RSC.
- (a) A. Szumna and J. Jurczak, Eur. J. Org. Chem., 2001, 4031–4039 CrossRef CAS; (b) M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC; (c) J. S. Mondy, M. A. Saeed, F. R. Fronczek, D. R. Powell and Md. A. Hossain, Inorg. Chem., 2010, 49, 7223–7225 CrossRef; (d) I. V. Korendovych, M. Cho, P. L. Butler, R. J. Staples and E. V. Rybak-Akimova, Org. Lett., 2006, 8(15), 3171–3174 CrossRef CAS; (e) S. Li, C. Jia, B. Wu, Q. Luo, X.- Huang, Z. Yang, Q.-S. Li and X.-J. Yang, Angew. Chem., Int. Ed., 2011, 50, 5721–5724 CrossRef CAS; (f) Y. Zhang, Z. Yin, Z. Li, J. Hea and J.-P. Chenga, Tetrahedron, 2007, 63, 7560–7564 CrossRef CAS; (g) J. M. Llinares, D. Powell and K. Bowman-James, Coord. Chem. Rev., 2003, 240, 57–75 CrossRef CAS; (h) P. Bose, B. N. Ahamed and P. Ghosh, Org. Biomol. Chem., 2011, 9, 1972–1979 RSC.
- (a) P. Desai, K. Schildknegt, K. A. Agrios, C. Mossman, G. L. Milligan and J. Aube, J. Am. Chem. Soc., 2000, 122, 7226–7232 CrossRef CAS; (b) A. Cordova, Acc. Chem. Res., 2004, 37, 102–112 CrossRef CAS; (c) R. I. Kureshy, S. Agarawal, S. Saravanan, N. H. Khan, A. K. Shah, S. H. R. Abdi, H. C. Bajaj and E. Suresh, Tetrahedron Lett., 2010, 51, 489–494 CrossRef CAS; (d) R. O. Duthaler, Angew. Chem., Int. Ed., 2003, 42, 975–978 CrossRef CAS; (e) S. Saito, T. Tsubogo and S. Kobayashi, Chem. Commun., 2007, 1236–1237 RSC; (f) K. Manabe, Y. Mori and S. Kobayashi, Tetrahedron, 2001, 57, 2537–2544 CrossRef CAS; (g) M. Rueping, C. Vila, R. R. M. Koenigs, K. Poscharny and D. C. Fabry, Chem. Commun., 2011, 47, 2360–2362 RSC.
- (a) T. Ollevier, E. Nadeau and A.-A.-G. Bégin, Tetrahedron Lett., 2006, 47, 8351 CrossRef CAS; (b) C. T. Chang, B.-S. Liao and S.-T. Liu, Tetrahedron Lett., 2006, 47, 9257 CrossRef CAS; (c) Y.-S. Wu, J. Cai, Z. Y. Hu and G.-X. Lin, Tetrahedron Lett., 2004, 45, 8949–8952 CrossRef CAS.
- N. H. Khan, S. Agrawal, R. I. Kureshy, S. H. R. Abdi, S. Singh and R. V. Jasra, J. Organomet. Chem., 2007, 692, 4361–4366 CrossRef CAS.
- J. M. M. Verkade, L. J. C. V. Hemert, P. J. L. Quaedflieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2008, 37, 29–41 RSC.
- N. Aziz, L. Torkiyan and M. R. Saidi, Org. Lett., 2006, 8, 2079–2082 CrossRef.
- S. Sahoo, T. Joseph and S. B. Halliguidi, J. Mol. Catal. A: Chem., 2006, 244, 179–182 CrossRef CAS.
- A. T. Khan, T. Parvin and L. H. Choudhury, Eur. J. Org. Chem., 2008, 834–839 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 867091. For ESI and crystallographic data in CIF or other electronic format see 10.1039/c2ra20507f |
| ‡ Crystal data for 1b: C25H28Br4N4, M = 704.15, orthorhombic, Pban, a = 17.826(5); b = 12.506(3); c = 12.588(3) Å; α = β = γ = 90°, V = 2806.4(12) Å3, Z = 4, ρcalc = 1.667 mg m−3, F(000) = 1384, absorption coefficient = 5.756 mm−1, crystal size 0.70 × 0.65 × 0.20 mm3, μ(Mo-Kα) = 0.71073 Å, T = 293(2) K. 15615 reflections collected, 3322 independent reflections, [Rint = 0.0718], GOF = 1.049, final R indices = [I ≥ 2σ(I)], R1 = 0.0614, wR2 = 0.1740. |
| This journal is © The Royal Society of Chemistry 2012 |