DOI:
10.1039/C2RA20577G
(Paper)
RSC Adv., 2012,
2, 7492-7500
Extraction of Am(III) using novel solvent systems containing a tripodal diglycolamide ligand in room temperature ionic liquids: a ‘green’ approach for radioactive waste processing†
Received
29th March 2012
, Accepted 10th June 2012
First published on 13th June 2012
Abstract
Extraction of Am3+ from acidic feed solutions was investigated using novel solvent systems containing a tripodal diglycolamide (T-DGA) in three room temperature ionic liquids (RTIL), viz. [C4mim][NTf2], [C6mim][NTf2] and [C8mim][NTf2]. Compared to the results obtained with N,N,N′,N′-tetra-n-octyl diglycolamide (TODGA), T-DGA gave significantly higher distribution coefficients in these RTILs. The DAm values decreased with increasing carbon chain length in the RTILs, which was related to the solubility of the RTIL in the aqueous phase. The distribution studies included the effect of equilibration time, aqueous phase acid concentration variation and T-DGA concentration variation. In general, significantly higher equilibration times were observed for the extraction systems, which was partly due to the viscous RTIL phase and partly due to the slow conformational changes of the T-DGA ligand during complexation. Apart from Am3+, extraction of Pu4+, UO22+, Eu3+, Sr2+ and Cs+ was also investigated, since they have significant implications in radioactive waste processing. Stripping studies indicated >99% stripping in three stages using 0.5 M EDTA or DTPA in 1 M guanidine carbonate. Slope analysis indicated the extraction of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexed species of Am(III) with T-DGA. Time resolved laser fluorescence spectroscopy (TRLFS) studies showed a strong complexation with no inner-sphere water molecules in the Eu(III)–T-DGA complexes for [C4mim][NTf2] as the diluent. Radiolytic degradation studies of the solvent systems containing T-DGA in the three RTILs were also carried out and while the DAm values decreased marginally when the solvents were exposed to 500 kGy absorbed dose, a relatively sharp decrease (60%) was seen when the solvents were exposed further to 1000 kGy absorbed dose, suggesting the possibility of recycling. Extraction studies were also carried out at varying temperatures and the thermodynamic parameters were calculated.
:1 complexed species of Am(III) with T-DGA. Time resolved laser fluorescence spectroscopy (TRLFS) studies showed a strong complexation with no inner-sphere water molecules in the Eu(III)–T-DGA complexes for [C4mim][NTf2] as the diluent. Radiolytic degradation studies of the solvent systems containing T-DGA in the three RTILs were also carried out and while the DAm values decreased marginally when the solvents were exposed to 500 kGy absorbed dose, a relatively sharp decrease (60%) was seen when the solvents were exposed further to 1000 kGy absorbed dose, suggesting the possibility of recycling. Extraction studies were also carried out at varying temperatures and the thermodynamic parameters were calculated.
1. Introduction
Extraction of actinides is one of the key issues in the remediation of high level radioactive wastes emanating from the back end of the nuclear fuel cycle. Effective actinide extraction makes the waste benign and ready for disposal as vitrified waste blocks in deep geological repositories. However, conventional solvent extraction methods, though being routinely used for actinide separations, have several disadvantages, which include a large VOC (volatile organic compounds) inventory and generation of huge volumes of secondary wastes. Growing concern for the environment has led to increasing interest in room temperature ionic liquids (RTIL) as an alternative to molecular diluents in a myriad of applications including synthesis,1 catalysis,2 separation3 and electrochemistry.4 Out of these, the application of RTILs to separation science has increased enormously as can be seen from the rapid rise in the number of publications in this area in the last decade, due to their unique characteristics of high thermal stability and low volatility. Some of the thoroughly studied areas include the extraction of Cs+ and Sr2+ by crown ethers.5 Extraction of actinide ions, however, is hardly studied and only a handful of papers are available on this topic.
Some of the early work on actinide extraction using ionic liquids include extraction of UO22+ by tributyl phosphate (TBP) in [C4mim][PF6], [C6mim][PF6] or [C8mim][NTf2] (mim = 1-alkyl-3-methylimidazolium).6 Usually, the extraction mechanism with RTILs was found to be in variant to that reported with molecular diluents such as n-dodecane. However, the extraction mechanism and the mode of complexation have been surprisingly found to be identical for uranyl ion extraction using Cyanex 272 in n-dodecane or 1-decyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide as indicated by EXAFS studies when longer carbon chain RTILs were used.7 Task specific ionic liquids with a quaternary ammonium cation and bearing a phosphoryl group have been synthesized and used for the extraction of UO22+.8 CMPO (carbamoyl methyl phosphine oxide) grafted task specific ionic liquids have also been prepared and the solid phase extraction of Pu(IV) was investigated using PAN fibers and carbon nano tubes as the support.9 In another study, CMPO dissolved in RTILs has been used for the extraction of Am(III) from slightly acidic feed solutions and its separation from Eu(III) using a variety of complexing agents.10
Diglycolamide extractants, such as N,N,N′,N′-tetra-n-octyl diglycolamide (TODGA) have been found to be significantly more effective for minor actinide partitioning as compared to CMPO and several test counter-current runs have been carried out using ‘hot’ radioactive waste solutions.11 On the other hand, functionalized diglycolamides were found to be more efficient that TODGA and a tripodal diglycolamide (T-DGA) has shown unique actinide extraction properties.12–14 Usually, the dielectric constant of the diluent decides the number of extractant molecules being associated in the extracted species.15 With TODGA as the extractant, the number of extractant molecules in the extracted species vary from 2 for nitrobenzene (dielectric constant of 35.616) to 4 for n-dodecane (dielectric constant of 2.01216) for Am(III) bearing extracted species.15 The objective of using the tripodal diglycolamide extractant is to discount this diluent dependent metal ion extraction and to enable the formation of large organophilic complexes with high distribution ratio values in any diluent system. Further, in view of the improved extraction and separation behaviour of room temperature ionic liquids, it was of interest to carry out actinide extractions using T-DGA in ionic liquids.
The present work deals with the extraction of Am(III) from acidic feed solutions using a tripodal diglycolamide (T-DGA, Fig. 1(a)) in three commercially available room temperature ionic liquids, viz. [C4mim][NTf2], [C6mim][NTf2] and [C8mim][NTf2]. The extraction data are compared with those obtained with TODGA (Fig. 1(b)) which has been used in several process test runs. In addition to the effects of equilibration time and feed acidity, the stoichiometry of the complexes, the thermodynamic parameters, and the radiolytic stability are studied.
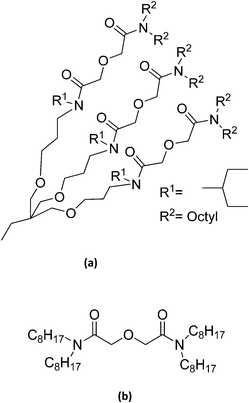 |
| | Fig. 1 Structural formulae of the extractants used. | |
2. Experimental
2.1 Materials
2.1.1 General.
N,N,N′,N′-tetra-n-octyl diglycolamide (TODGA) was obtained from Thermax Ltd, India and was characterized by NMR, HPLC, GC-MS as well as by distribution measurements (distribution data obtained at 3 M and 0.01 M HNO3 were compared with those reported previously17). 241Am, Pu (mainly 239Pu), and 233U tracers were purified prior to their use by ion-exchange methods, while 85,89Sr and 137Cs and 152,154Eu tracers were purchased from BRIT, Mumbai. Assaying of 241Am, 137Cs, 85,89Sr and 152,154Eu was done by gamma counting using a NaI(Tl) scintillation counter, while nuclides such as 239Pu and 233U were assayed by liquid scintillation counting.
2.1.2 Synthesis of T-DGA.
Recently, we reported the synthesis of the tripodal diglycolamide T-DGA12 by reaction of the tripodal amine 113 with diglycolic anhydride and subsequent reaction with di-n-octylamine. Meanwhile, we developed an improved procedure for T-DGA in which the tripodal amine 1 is reacted with p-nitrophenol activated DGA (2) to give the target compound in 74% yield (Scheme 1).
 |
| | Scheme 1 Synthesis of T-DGA. | |
A mixture of 1 (1.10 g, 2.1 mmol), p-nitrophenol activated DGA (2) (3.20 g, 6.6 mmol) and triethylamine (0.65 g, 6.5 mmol) in chloroform (70 mL) was refluxed for 2 d. The crude reaction mixture was successively washed with 2 M NaOH solution (3 × 50 mL), 1 M HCl (3 × 50 mL), and water (2 × 50 mL). The organic layer was concentrated under reduced pressure and the crude product was purified by column chromatography (SiO2, CH2Cl2:MeOH = 95:5 → 7:3) to afford T-DGA as an oil (2.42 g, 74%).
2.2 Methods
2.2.1 Distribution studies.
The distribution studies were carried out by mixing 1 mL of the ligand solution in a suitable room temperature ionic liquid with an equal volume of the aqueous phase containing the required radiotracers in a given concentration of HNO3. Studies with Am involved the use of 241Am tracer and the concentration of the metal ion in a typical distribution experiment was ∼10−7 M. The equilibration of the tubes was carried out in a thermostated water bath at 25 ± 0.1 °C for about 3 h, which was optimized after the studies with varying equilibration times. After centrifugation, the phases were separated and assayed radiometrically (usually 100 μL aliquots were taken for radiometric assay) as mentioned above. The valency of Pu to the +4 state was adjusted by using NaNO2 as the redox agent and ammonium metavanadate as the holding oxidant and its conversion was checked by the TTA extraction method.18 Radiometric assay of 233U and Pu was done using an alpha-liquid scintillation counting system (Hidex, Finland) using an Ultima Gold scintillator cocktail (Sisco Research Laboratory, Mumbai), while that of 85,89Sr, 137Cs and 239Np was done by gamma ray counting using a NaI(Tl) scintillation counter (Para Electronics, India) inter phased to a multi-channel analyzer (ECIL, India).
The distribution ratio, DM, was defined as the ratio of the activity per unit volume in the ionic liquid phase to that in the aqueous phase. In view of the uncertainties involved in sampling due to the high viscosity of the ionic liquid phase, the DM (distribution ratio of the metal ion) values were also calculated by obtaining the counts in the ionic liquid phase by the difference of the initial and final counts in the aqueous phase as follows:
where, Ci and Cf are the initial and final concentrations of the metal ion, respectively. The experiments were carried out in duplicate and the precision was within ±5%.
The radiolytic degradation studies were carried out using a 60Co irradiation source at a dose rate of 1.6 kGy h−1. Usually, about 5 mL of the solution containing T-DGA in RTIL were irradiated for the required period so as to correspond to absorbed doses of 500 and 1000 kGy. A thermostated water bath was used for carrying out the temperature variation studies wherein the two phases were equilibrated at varying temperatures in the range of 20–40 °C. Aliquots from both phases (usually ∼100 μL) were taken while keeping the equilibration tubes in the thermostat and the temperature changes were kept within ±0.2 °C of the set temperature.
Stripping studies were carried out by equilibrating the equal volumes of the loaded organic phase with aqueous phases containing either 0.05 M DTPA or EDTA in 1 M guanidine carbonate or a buffer mixture containing 0.1 M citric acid + 0.4 M formic acid + 0.4 M hydrazine hydrate which have been used for the stripping of Am(III) from CMPO loaded organic phases.19
2.2.2 Fluorescence studies.
Time resolved laser fluorescence spectroscopy (TRLFS) measurements were carried out using a luminescence spectrometer equipment supplied by Edinburgh Analytical Instruments, UK controlled by CD 920 controller and pumped by OPO lasers as excitation sources. The excitation wavelength was fixed at 230 nm, while emission spectrum was recorded in the range of 575–750 nm. The luminescence decay curves were fitted into the exponential function to obtain the lifetimes/decay rates of the excited states using inbuilt software GEM/3 (Edinburgh). The reproducibility of lifetimes of the excited states was within ±3 μs.
3. Results and discussion
Though no extraction of Am(III) was noticed with RTIL alone (DAm < 0.01), in the absence of the extractants TODGA or T-DGA, a sharp increase in the DM values was observed when a small amount (about 1.0 × 10−3 M) of the extractant was added to the ionic liquid phase. With comparable concentrations of the extractants, the DM values obtained with RTILs were significantly higher than those obtained with molecular diluents in the organic phase.14 This is in line with the observations made by Nakashima et al.,20 during their studies on lanthanide extraction using CMPO dissolved in RTILs. They have also reported that ionic liquids with NTf2− counter anions extracted the metal ions more efficiently as compared to those with PF6− counter anions. Similar observations were also made by us during our studies on Sr(II) extraction using di-tert-butylcyclohexano-18-crown-6 in both types of RTILs.21 The extraction data for Am(III) are listed in Table 1 with the three ionic liquids, viz. [C4mim][NTf2], [C6mim][NTf2] and [C8mim][NTf2] and a 1.0 × 10−3 M concentration of both the extractants. For comparison purposes, DAm values in molecular diluents, viz. n-dodecane and an n-dodecane/iso-decanol mixture (10:1) are also included in Table 1. As previously reported, T-DGA has limited solubility in n-dodecane alone and is fairly soluble in the diluent mixture, n-dodecane and iso-decanol (10:1).14 The observed trend of Am(III) extraction is [C4mim][NTf2] > [C6mim][NTf2] > [C8mim][NTf2], which is similar to that reported in several systems such as the extraction of Am3+ and UO22+ using CMPO and TODGA as the extractants, respectively.22 Apparently, the relatively higher aqueous solubility of the butyl form of the ionic liquid was responsible for the higher metal ion extraction as will be discussed below.
Table 1 Distribution data of Am(III) using 1.0 × 10−3 M TODGA and T-DGA in room temperature ionic liquids from aqueous nitric acid feed solutions
| [HNO3], M |
DAm in T-DGA in different RTILsa |
DAm in T-DGA in n-dodecane + iso-decanol |
DAm in TODGA in different RTILsa |
DAm in TODGA in n-dodecane |
| [C4mim][NTf2] |
[C6mim][NTf2] |
[C8mim][NTf2] |
[C4mim][NTf2] |
[C6mim][NTf2] |
[C8mim][NTf2] |
|
Note: the DAm values in the absence of the extractants were typically <0.01 in all the three ionic liquids.
|
| 3 |
0.6 |
0.2 |
0.17 |
30.1 |
0.25 |
0.13 |
0.12 |
0.003 |
| 0.5 |
91.1 |
32.0 |
3 |
0.05 |
0.76 |
0.29 |
0.22 |
0.002 |
| 1.0 × 10−2 |
1518 |
161 |
105 |
0.02 |
208 |
27.9 |
26.2 |
5.0 × 10−4 |
Though extraction kinetics is an important parameter in all liquid–liquid extraction studies, it requires great significance in studies involving ionic liquids as these diluents invariably display higher viscosities as compared to the molecular diluents. The extraction kinetics of Am(III) was studied from a feed solution containing 1.0 × 10−3 M T-DGA in [Cnmim][NTf2]. Fig. 2 represents the extraction kinetics data up to a period of 4 h, within which time complete attainment of the equilibrium was seen for all three RTILs. The rate of attainment of the equilibrium follows the trend: [C4mim][NTf2] > [C6mim][NTf2] ∼ [C8mim][NTf2], suggesting that while 1 h is sufficient for the butyl derivative, 2 h are required for both the n-hexyl and n-octyl derivatives of the ionic liquids. The observed slower kinetics in all cases as compared to the conventional diluents like n-dodecane can be attributed to the high viscosity of the ionic liquids.23 It is well known that the viscosity increases with the increase in the number of carbon atoms in the side chain.23 Therefore, the attainment of the equilibrium was the slowest with [C8mim][NTf2], while the extraction kinetics was much faster with [C4mim][NTf2]. Though the attainment of the equilibrium with [C6mim][NTf2] as the diluent should be in between those observed with [C4mim][NTf2] and [C8mim][NTf2], the results indicate an Am(III) extraction kinetics with [C6mim][NTf2] similar to that of [C8mim][NTf2].
![Extraction kinetics of Am from 0.5 M HNO3 feed into 1.0 × 10−3 M T-DGA in [Cnmim][NTf2] (n = 4, 6, 8).](/image/article/2012/RA/c2ra20577g/c2ra20577g-f2.gif) |
| | Fig. 2 Extraction kinetics of Am from 0.5 M HNO3 feed into 1.0 × 10−3 M T-DGA in [Cnmim][NTf2] (n = 4, 6, 8). | |
3.2 Relative extraction and separation behaviour
Apart from the Am3+ ion, the extraction of several actinide ions viz., UO22+ and Pu4+ was also investigated using 0.5 M HNO3 as the aqueous feed and 1.0 × 10−3 M T-DGA in the ionic liquids as the extractant; the results are listed in Table 2. Distribution ratio data for some important fission product elements were also determined under identical experimental conditions. The results are included in the table for comparison purposes, the extraction trend being Eu3+ > Am3+ > Pu4+ > UO22+ ∼ Sr2+ > Cs+. The extractability trend for the elements is similar to that reported with the T-DGA–n-dodecane–iso-decanol system.14 The relative extraction behaviour of Am3+vis-à-vis that of Sr2+ and UO22+ is interesting when compared to those observed with the analogous TODGA–n-dodecane system.24 The separation factor values are listed in Table 3. Though the Sr2+ extraction with TODGA–n-dodecane system was about 50%, the S.F. (separation factor is defined as DAm/DSr) value was as high as 302 (Table 3). On the other hand, the DSr (0.02) as well as the S.F. (1.08 × 104) values were reported to be favourable for Sr decontamination in T-DGA in n-dodecane–iso-decanol. It is clearly seen from Table 3 that U decontamination is significantly higher with T-DGA as the extractant, while ionic liquids are less effective than the molecular diluents (n-dodecane–iso-decanol mixture). On the other hand, a higher U decontamination was observed in T-DGA + ionic liquid solvent systems as compared to the TODGA–n-dodecane system. This indicates that the decontamination is due to T-DGA and not due to the ionic liquid, which is the opposite of the trend shown with a calix[4]arene-tetra-diglycolamide extractant.23 For Pu and Eu decontamination, the results are not very favourable and hence are not discussed here (Table 3).
Table 2 Comparative extraction data of actinides and fission product elements using 1.0 × 10−3 M T-DGA in different room temperature ionic liquids. Feed: 0.5 M HNO3
| Metal ions |
[C4mim][NTf2] |
[C6mim][NTf2] |
[C8mim][NTf2] |
| Am(III) |
91.1 |
32.6 |
3.1 |
| Pu(IV) |
30 |
16.9 |
1.4 |
| U(VI) |
0.24 |
0.23 |
0.06 |
| Cs(I) |
0.12 |
0.04 |
0.01 |
| Sr(II) |
0.27 |
0.12 |
0.02 |
| Eu(III) |
218.7 |
63.96 |
12.7 |
Table 3 Separation factors of Am(III) with respect to other actinides and fission products using T-DGA (1.0 × 10−3 M) in various room temperature ionic liquids. Feed: 0.5 M HNO3
| Extraction system |
Separation factors |
Reference |
| Am(III)/U(VI) |
Am(III)/Pu(IV) |
Am(III)/Eu(III) |
Am(III)/Sr(II) |
|
|
Note: data obtained with 3.0 M HNO3 as the feed.
0.1 M TODGA was used.
2.9 × 10−3 M T-DGA was used.
|
| T-DGA in [C4mim][NTf2] |
380 |
3.04 |
0.42 |
337 |
Present work |
| T-DGA in [C6mim][NTf2] |
142 |
1.93 |
0.51 |
272 |
Present work |
| T-DGA in [C8mim][NTf2] |
51.7 |
2.21 |
0.24 |
155 |
Present work |
| TODGA in n-dodecanea,b |
28.9 |
— |
1.04 |
302 |
Ref. 21
|
| T-DGA in n-dodecane + iso-decanola,c |
895 |
1.48 |
— |
1.08 × 104 |
Ref. 14
|
3.3 Effect of the feed acidity
Usually, for solvating type extractants, such as TBP, CMPO and TODGA, the extraction of the metal ion increases with increasing the feed acidity, which is based on an increase in the counter anion concentration which helps in the formation of neutral extractable species as per eqn (2):| | | Mn+ + nNO3− + mL(o) ↔ M(NO3)n·mL(o) | (2) |
where, species with the subscript ‘(o)’ indicate those present in the organic phase, while those without any subscript indicate species in the aqueous phase. Though this trend has invariably been observed in molecular diluents such as n-dodecane,12 an entirely different trend, i.e., decrease in the metal ion extraction with increasing aqueous feed acidity has been reported in ionic liquids as the diluent due to a ion-exchange mechanism,6,23 as indicated in eqn (3) (subscript “IL” means species in ionic liquid phase):
| | | Am3+ + nT-DGAIL + 3Cnmim+IL ↔ [Am(T-DGA)n] 3+IL + 3Cnmim+ | (3) |
As discussed above, eqn (3) suggests that a higher solubility of the [Cnmim]+ part of the ionic liquid will result in higher metal ion extraction. Fig. 3 shows the dependence of the distribution ratio of Am(III) with the change in the feed nitric acid concentration for the ionic liquid solvents containing 1.0 × 10−3 M T-DGA. It is clear that an increase in the feed nitric acid concentration led to a decrease in the DM values in the acidity range of 0.01–3 M, beyond which a slight increase was noticed. The decrease was less significant for [C6mim][NTf2] and [C8mim][NTf2] where the DAm values decreased by about 600–700 times as compared to about 2700 times when [C4mim][PF6] was used as the ionic liquid (Fig. 3). The extraction profiles obtained in the present study indicate that the extraction mechanism is not the same as that in the case of conventional molecular diluents such as n-dodecane is used. Apparently, an ion-exchange mechanism is responsible for the extraction of Am(III) to the ionic liquid phase. The ion-exchange mechanism also gets credence from the fact that the extraction of Am(III) increased with decreasing carbon chain length of the RTIL. As shown in Fig. 3, there is a marginal increase in the DAm values for all the three ionic liquids beyond 3 M HNO3 (about 3 times for the [C6mim][NTf2] and [C4mim][NTf2] and about 1.5 times for [C8mim][NTf2]). This can be attributed to a solvation mechanism with nitrate ion participation as described previously for a crown ether extraction system.25
![Extraction behavior of Am from different feed acidities into 1.0 × 10−3 M T-DGA in [Cnmim][NTf2] (n = 4, 6, 8) after 3 h equilibration.](/image/article/2012/RA/c2ra20577g/c2ra20577g-f3.gif) |
| | Fig. 3 Extraction behavior of Am from different feed acidities into 1.0 × 10−3 M T-DGA in [Cnmim][NTf2] (n = 4, 6, 8) after 3 h equilibration. | |
3.4 Effect of T-DGA concentration
In order to get an idea on the nature of the extracted species, the extraction of Am(III) with varying T-DGA concentrations was investigated using all the three RTILs from aqueous feed solutions of 0.1 M HNO3 and the time of equilibration was kept as 3 h. The results, presented in Fig. 4, show an increase in Am(III) extraction with increasing T-DGA concentration for all the three ionic liquids. The slope values of the plots are close to 1 (1.17 ± 0.05 for [C4mim][NTf2]; 1.13 ± 0.02 for [C6mim][NTf2]; and 1.05 ± 0.04 for [C8mim][NTf2]) suggesting that one T-DGA unit is present in the extracted species, which is in sharp contrast to the extracted species in the molecular diluent containing 95% n-dodecane + 5% iso-decanol mixture.14 It is well known that a lower number of ligands may be present in the extracted species with the increasing dielectric constant of the diluent.15 On the other hand, higher metal ion extraction has been reported with diluents of high dielectric constant even though lesser number of ligand molecules are associated in the extracted complexed species as compared to those with low dielectric constant diluents. In view of this, it is expected that the relatively high dielectric constant of the ionic liquids26 as compared to the molecular solvents used, would prevent the participation of a second T-DGA unit, which would otherwise lead to ‘stereochemical crowding’. A similar increase in the extraction of actinide ions with TODGA and C4DGA (a calix[4]arene containing four DGA moieties) was reported recently where [Cnmim][PF6] was used as the RTIL.23
![Dependence of Am(iii) extraction on varying T-DGA concentrations from 0.5 M HNO3 feed into [Cnmim][NTf2] (n = 4, 6, 8).](/image/article/2012/RA/c2ra20577g/c2ra20577g-f4.gif) |
| | Fig. 4 Dependence of Am(III) extraction on varying T-DGA concentrations from 0.5 M HNO3 feed into [Cnmim][NTf2] (n = 4, 6, 8). | |
3.5 Stripping studies
With room temperature ionic liquids, the stripping of the metal ion from the ionic liquid phase is one of the major challenges. Usually, with studies involving diglycolamide extractants such as TODGA, the extraction is carried out at higher acidity (3–6 M HNO3), while the stripping is done at lower acidity (pH 2.0). However, with ionic liquids as the diluent the D values at pH 2.0 are very high (vide supra), which do not decrease significantly with increasing the aqueous phase acidity, so that effective stripping is not possible under these conditions (Fig. 3). Nakashima et al.,20 have employed 0.05 M DTPA + 1 M guanidine carbonate, 0.05 M EDTA + 1 M guanidine carbonate and a buffer mixture comprising of 0.1 M citric acid + 0.4 M formic acid + 0.4 M hydrazine hydrate for the effective stripping of rare earth metal ions from ionic liquid extracts. These solutions were employed in the present study for the stripping of Am(III) from the T-DGA in [C4mim][NTf2] extracts. The results are shown in Fig. 5 which suggest that EDTA and DTPA solutions in 1 M guanidine carbonate are effective as strippant where close to 80% stripping of Am was observed in a single stage. On the other hand, the buffer mixture was not very effective as a strippant as only <10% stripping was obtained in a single step. The results are encouraging as >99% stripping of Am is possible using the complexing agents in three stages.†
![Stripping behaviour of Am(iii) from an extract made using 1.0 × 10−3 M T-DGA in [C4mim][NTf2].](/image/article/2012/RA/c2ra20577g/c2ra20577g-f5.gif) |
| | Fig. 5 Stripping behaviour of Am(III) from an extract made using 1.0 × 10−3 M T-DGA in [C4mim][NTf2]. | |
3.6 Laser induced fluorescence studies
Though the nature of the extracted species was determined by slope analysis from T-DGA concentration variation studies, further evidence was obtained by time resolved laser fluorescence spectroscopy (TRLFS) using the Eu3+–T-DGA complexes. In view of the similarities in the chemical properties of the trivalent actinides and lanthanides, Eu3+ was taken as the surrogate of Am3+ and the results of the Eu3+ fluorescence studies can be extended for the Am3+ system as well. Fluorescence spectroscopic investigations on the Eu3+ aqueous complex and Eu3+–T-DGA complex (both containing 1.0 × 10−3 M Eu3+) showed interesting behaviour as the intensity of the characteristics peaks at 617 nm (5D0→7F2 hyper sensitive transition, electric dipole), 592 nm (5D0→7F1 transition, magnetic dipole), and at 690 nm (5D0→7F4 transition, electric dipole, sensitive to Eu3+ environment) increased significantly (the former by more than 100 times) when T-DGA was added in an acetonitrile–water mixture as the solvating medium (Fig. 6). The acetonitrile–water mixture (5:1) contained dilute nitric acid to prevent hydrolysis of Eu3+. Further, an extract containing the Eu3+–T-DGA complex in [C4mim][NTf2] also showed a similar increase in the emission spectra intensities suggesting similarity between the complexes in both the acetonitrile–water mixture and the ionic liquid medium. The mode of complexation was studied by time resolved laser fluorescence spectroscopy (TRLFS), where the presence of inner-sphere water molecules or the lack of them would be reflected in the lifetime of the 5D0 emitting level of the Eu3+–T-DGA complex. It is well known that the luminescence lifetime depends on several radiation (independent of the environment) and non-radiation decay processes and that the number of inner-sphere water molecules is determined by the lifetime of the 5D0 emitting level of Eu3+.27 Uncomplexed Eu3+ has nine water molecules in its primary co-ordination sphere. In the presence of nitric acid in the aqueous phase, some nitrate complexation is expected, which can decrease the number of water molecules in the inner hydration sphere, which is also reflected by an increase of the emission lifetime. Further, with a decreasing number of water molecules in the primary hydration sphere, which results in the addition of the T-DGA ligand, the lifetime should show an increasing trend. The fluorescence decay profiles of Eu3+ and Eu3+–T-DGA both in the acetonitrile–water mixture and the extract containing the Eu3+–T-DGA complex in [C4mim][NTf2] point to the presence of a single complexed species with 1:1 stoichiometry in all cases; the lifetime data† are presented in Table 4. The number of water molecules was calculated from the lifetime (τ) using eqn (4):28| | | NH2O = (1.06/τ) − 0.19 | (4) |
![Emission spectra of Eu3+ (1 mM) in the absence and presence of T-DGA in acetonitrile : water (5 : 1) and those of the Eu3+–T-DGA extract in [C4mim][NTf2] obtained from different feed acidities.](/image/article/2012/RA/c2ra20577g/c2ra20577g-f6.gif) |
| | Fig. 6 Emission spectra of Eu3+ (1 mM) in the absence and presence of T-DGA in acetonitrile:water (5:1) and those of the Eu3+–T-DGA extract in [C4mim][NTf2] obtained from different feed acidities. | |
| Sample |
Lifetime (ms) |
Inference |
| Eu in ACN-3 M HNO3 |
0.155 |
6 water; 3 nitrate |
| Eu–T-DGA complex in ACN–3 M HNO3 |
2.285 |
No inner-sphere water |
| Eu–T-DGA complex in [C4mim][NTf2], feed acidity 0.5 M HNO3 |
2.251 |
No inner-sphere water |
| Eu–T-DGA complex in [C4mim][NTf2], feed acidity 0.01 M HNO3 |
2.249 |
No inner-sphere water |
The lifetime of Eu3+ in the absence of T-DGA was found to be 155 μs, which increased to about 2.5 ms in the presence of the complexing extractant. It has been reported that the Eu3+ aqueous ion has a fluorescence lifetime of 114 μs, which increases in the presence of nitric acid, apparently due to the replacement of some inner-sphere water molecules by nitrate ions.29 The appearance of an additional peak at 617 nm indicates a strong interaction with the DGA moieties of T-DGA similar to that observed with a calix[4]arene containing four DGA units.30 In case of the extract containing the Eu3+–T-DGA complex in [C4mim][NTf2], a similar pattern was observed with a lifetime of 2.5 ms. However, the intensity of the split lines at 617 nm changed significantly. These results indicate very strong complex formation with the DGA moieties of T-DGA with practically no inner-sphere water molecules. Comparable lifetimes for extracts made from both 0.01 M and 0.5 M HNO3 suggested extraction of similar ‘inclusion’ complex species, in which all the water molecules are replaced by the coordinating sites of the DGA groups.
3.7 Radiolytic degradation
A recycling option in process applications for radioactive waste treatment requires that the solvent system should have an adequate radiation stability. Fig. 7 shows the changes in the DAm values as a function of absorbed radiation dose (gamma ray) up to a maximum of 1000 kGy. Interestingly, the decrease in the DAm values was insignificant when the solvents were exposed to a 500 kGy dose for all the three ionic liquids. However, an appreciable decrease in the DAm values was seen when the solvents were exposed to a 1000 kGy radiation dose. It is interesting to note that the DAm value of 91.2 with 1.0 × 10−3 M T-DGA in [C4mim][NTf2] decreased to 37 when the solvent was irradiated up to a 1000 kGy dose. This decrease is little over 60%, which means that T-DGA is far more stable than a Cs-selective calix-crown extractant containing the same ionic liquid ([C4mim][NTf2]) where the decrease was >95% even for an absorbed dose of 550 kGy.31 Though the very poor irradiation stability of the Cs-selective solvent system was attributed to the degradation of the ionic liquid leading to acidic products, a similar effect might have affected the DAm values in the present system as well, which is shown by the sharp dependence of the Am(III) extraction on the aqueous phase acidity. On the other hand, Allen et al.,32 have reported that the radiation stability of the ionic liquids is higher than that of diluents like n-dodecane, due to the presence of aromatic groups such as the imidazolium group. The radiation stability of solutions of T-DGA in ionic liquids is far higher compared to that of TODGA in a molecular solvent, where the degradation was >95% for an absorbed dose of 1000 kGy.33 The amazingly high stability of the solvent system containing T-DGA [C4mim][NTf2] may be attributed to the high viscosity of the ionic liquid system (leading to recombination of the free radicals) as compared to the TODGA in n-dodecane solvent system. Similar high radiolytic stability of T2EDGA (a branched diglycolamide extractant) was seen when undiluted extractant was irradiated, which can also be attributed to the recombination of the radicals in a viscous medium.34 These results, together with the very high extraction and effective stripping, suggest that the solvent containing T-DGA in [C4mim][NTf2] may be used as a possible ‘green’ alternative for actinide partitioning instead of TODGA–n-dodecane based solvents.
![Effect of absorbed dose on the extraction of Am(iii) from 0.5 M HNO3 using 1.0 × 10−3 M T-DGA solutions in [Cnmim][NTf2] (n = 4, 6, 8).](/image/article/2012/RA/c2ra20577g/c2ra20577g-f7.gif) |
| | Fig. 7 Effect of absorbed dose on the extraction of Am(III) from 0.5 M HNO3 using 1.0 × 10−3 M T-DGA solutions in [Cnmim][NTf2] (n = 4, 6, 8). | |
3.8 Determination of thermodynamic parameters
With T-DGA as the extractant, significant stereochemical ordering is required during the complexation reaction. The thermodynamic parameters were determined to throw light on the nature of the complexation. Temperature variation studies on Am(III) extraction were carried out from 0.5 M HNO3 using 1.0 × 10−3 M T-DGA in the three RTILs being studied. The Van't Hoff plots for the distribution ratio data as a function of temperature† are presented in Fig. 8. The change in enthalpy (ΔH) during the complexation was calculated by using the Van't Hoff eqn (5):| | | ΔH = −2.303 R Δlog D/Δ(1/T) | (5) |
![Variation in distribution ratio of Am(iii) at different temperatures from 0.5 M HNO3 feed using 1.0 × 10−3 M T-DGA in [Cnmim][NTf2] (n = 4, 6, 8) after 3 h equilibration.](/image/article/2012/RA/c2ra20577g/c2ra20577g-f8.gif) |
| | Fig. 8 Variation in distribution ratio of Am(III) at different temperatures from 0.5 M HNO3 feed using 1.0 × 10−3 M T-DGA in [Cnmim][NTf2] (n = 4, 6, 8) after 3 h equilibration. | |
The slope (−ΔH/2.303 R) values of the log D vs. 1/T plots are listed in Table 5. The Gibb's free energy (ΔG) and the entropy change (ΔS) at a particular temperature were calculated from eqn (6) and (7):
| Extractant |
Ionic liquid |
DAm |
Log Kex |
Slope |
Intercept |
ΔG (kJ mol−1) |
ΔH (kJ mol−1) |
ΔS (J K−1 mol−1) |
| T-DGA |
[C4mim][NTf2] |
91 |
5.56 |
32.9 |
19.1 |
−31.937 |
−0.630 |
552.3 |
| [C6mim][NTf2] |
32 |
5.27 |
28.83 |
16.5 |
−30.271 |
−0.551 |
450 |
| [C8mim][NTf2] |
3 |
4.66 |
27.53 |
14.8 |
−26.767 |
−0.532 |
443 |
The thermodynamic parameters are also listed in Table 5. The ΔH values are similar in all the three ionic liquids and are very small; the extraction reaction was exothermic. This is possibly due to the large amount of heat required to dehydrate the hydrated metal ion, which is nearly compensated by the strong complexation with the polydentate T-DGA ligand. The complete removal of the inner-sphere water molecules is understood from the very high positive entropy values. Overall, the reactions are spontaneous with large ΔG values. It is quite clear that larger distribution coefficient values in the RTIL system as compared to the molecular diluents resulted in higher extraction constants (log Kex, Table 5) which are responsible for the large negative ΔG values.
4. Conclusions
The solvent extraction studies of Am(III) using T-DGA in various room temperature ionic liquids clearly demonstrate that the ionic liquid medium extracts the metal ions more efficiently as compared to molecular diluents such as n-dodecane. Furthermore, the tripodal diglycolamide T-DGA is a far superior extractant as compared to TODGA. It extracts Am(III) from acidic feed solutions with the butyl derivative being the most efficient ionic liquid. The extracted species follow an ion-exchange mechanism at lower acidity and possibly a solvation mechanism at higher acidities. Though the extraction of the tetravalent actinide ion was much lower as compared to the trivalent lanthanide and actinide ions (both Eu(III) and Am(III) were highly extracted), the extraction of the hexavalent actinyl ion was insignificant. The extracted species was a 1:1 complex formed between Am(III) and T-DGA with no inner-sphere water molecules, which was confirmed by the TRLFS studies using the analogous Eu(III) complexes. The stripping results were very encouraging with the possibility of >99% stripping after three stages when a complexing agent such as EDTA or DTPA was used in guanidine carbonate. The thermodynamics of the extraction indicated a highly entropy driven reaction, suggesting the removal of a large number of water molecules from the inner-coordination sphere. Radiolytic stability studies showed a significantly higher stability of the ionic liquid solvent systems as compared to those reported previously in the literature, which may lead to possible long term recycling options. This also makes the proposed solvent system a viable ‘green’ alternative to the TODGA based solvent systems with molecular diluents.
Acknowledgements
The authors (A.S., S.V.G. and P.K.M.) thank Dr A. Goswami, Head, Radiochemistry Division, BARC for his keen interest in this work.
References
-
(a) C. J. Adam, M. J. Earle and K. R. Sneddon, Green Chem., 2000, 2, 21–23 RSC;
(b) G. Toma, B. Gotov and E. Solkaniova, Green Chem., 2000, 2, 149–151 RSC;
(c) C. E. Song and E. J. Roh, Chem. Commun., 2000, 837–838 RSC;
(d) P. J. Dyson, D. J. Ellis, D. C. Parker and T. Welton, Chem. Commun., 1999, 25–26 RSC.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS.
-
(a) J. G. Huddleston, H. D. Willauer, R. P. Swatloski, A. N. Visser and R. D. Rogers, Chem. Commun., 1998, 1765–1766 RSC;
(b) L. A. Blanchard, D. Hancut, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28–29 CrossRef;
(c) A. G. Fadeev and M. M. Meagher, Chem. Commun., 2001, 295–296 RSC;
(d) V. A. Cocalia, M. P. Jensen, J. D. Holbrey, S. K. Spear, D. C. Stepinski and R. D. Rogers, Dalton Trans., 2005, 1966–1971 RSC;
(e) A. E. Visser and R. D. Rogers, J. Solid State Chem., 2003, 171, 109–113 CrossRef CAS.
-
(a) I. W. Sun and C. L. Hussey, Inorg. Chem., 1989, 28, 2731–2737 CrossRef CAS;
(b)
C. L. Hussey, inChemistry of nonaqueous solvents: current progress, ed. G. Mamantov and A. I. Popov, VCH, New York, 1994 Search PubMed.
-
(a) A. N. Turanov, V. K. Karandashev and V. E. Baulin, Solvent Extr. Ion Exch., 2010, 28, 367–387 CrossRef CAS;
(b) S. Dai, Y. H. Yu and C. E. Barnes, J. Chem. Soc., Dalton Trans., 1999, 1201–1202 RSC;
(c) A. E. Visser, R. P. Swatloski, W. M. Reichert, S. T. Griffin and R. D. Rogers, Ind. Eng. Chem. Res., 2000, 39, 3596–3604 CrossRef CAS;
(d) H. Luo, S. Dai, P. V. Bonnesen, A. C. Buchanan III, J. D. Holbrey, N. J. Bridges and R. D. Rogers, Anal. Chem., 2004, 76, 3078–3083 CrossRef CAS;
(e) S. A. Ansari, P. K. Mohapatra, D. R. Raut and V. K. Manchanda, Radiochim. Acta, 2011, 99, 713–717 CrossRef CAS.
- P. Giridhar, K. A. Venkatesan, T. G. Srinivasan and P. R. V. Rao, J. Nucl. Radiochem. Sci., 2004, 5, 21–26 CAS.
- V. A. Cocalia, M. P. Jensen, J. D. Holbrey, S. K. Spear, D. C. Stepinski and R. D. Rogers, Dalton Trans., 2005, 1966–1971 RSC.
- A. Ouadi, O. Klimchuk, C. Gaillarda and I. Billard, Green Chem., 2007, 9, 1160–1162 RSC.
- I. L. Odinets, E. V. Sharova, O. I. Artyshin, K. A. Lyssenko, Y. V. Nelyubina, G. V. Myasoedova, N. P. Molochnikova and E. A. Zakharchenro, Dalton Trans., 2010, 39, 4170–4178 RSC.
- A. Rout, S. Karmakar, K. A. Venkatesan, T. G. Srinivasan and P. R. Vasudeva Rao, Sep. Purif. Technol., 2011, 81, 109–115 CrossRef CAS.
-
(a)
R. B. Gujar, S. A. Ansari, D. R. Prabhu, P. K. Mohapatra, P. N. Pathak, A. Sengupta, S. K. Thulasidas and V. K. Manchanda, Solv. Extr. Ion Exch., 2012, in press Search PubMed;
(b) G. Modolo, A. Hanna, C. Schreinemachers and H. Vijgen, Solvent Extr. Ion Exch., 2007, 25, 703–729 CrossRef CAS.
- D. Jańczewski, D. N. Reinhoudt, W. Verboom, C. Hill, C. Allignol and M. T. Duchesne, New J. Chem., 2008, 32, 490–495 RSC.
- D. Jańczewski, D. N. Reinhoudt, W. Verboom, E. Malinowska, M. Pietrzak, C. Hill and C. Allignole, New J. Chem., 2007, 31, 109–120 RSC.
- P. K. Mohapatra, M. Iqbal, D. R. Raut, W. Verboom, J. Huskens and V. K. Manchanda, J. Membr. Sci., 2011, 375, 141–149 CrossRef CAS.
-
(a) Y. Sasaki, Y. Sugo, S. Suzuki and S. Tachimori, Solvent Extr. Ion Exch., 2001, 19, 91–103 CrossRef CAS;
(b) S. Panja, P. K. Mohapatra, S. C. Tripathi, P. M. Gandhi and P. Janardan, J. Membr. Sci., 2012, 403–404, 71–77 CrossRef CAS.
-
W. M. Haynes, Handbook of chemistry and physics, CRC Press, Florida, 1992 Search PubMed.
- R. B. Gujar, S. A. Ansari, M. S. Murali, P. K. Mohapatra and V. K. Manchanda, J. Radioanal. Nucl. Chem., 2010, 284, 377–385 CrossRef CAS.
- M. S. Sajun, V. V. Ramakrishna and S. K. Patil, J. Radioanal. Chem., 1981, 63, 57–63 CrossRef CAS.
- A. K. Dinkar, P. K. Mohapatra, A. Dakshinamoorthy and V. K. Manchanda, Desalin. Water Treat., 2009, 12, 73–78 CrossRef CAS.
- K. Nakashima, F. Kubota, T. Maruyama and M. Goto, Ind. Eng. Chem. Res., 2005, 44, 4368–4372 CrossRef CAS.
- A. Sengupta and P. K. Mohapatra, Supramol. Chem Search PubMed , accepted for publication.
-
(a) A. Rout, K. A. Venkatesan, T. G. Srinivasan and P. R. Vasudeva Rao, J. Radioanal. Nucl. Chem., 2011, 290, 215–219 CrossRef CAS;
(b) Y. Shen, X. Tan, L. Wang and W. Wu, Sep. Purif. Technol., 2011, 78, 298–302 CrossRef CAS.
- A. Sengupta, P. K. Mohapatra, M. Iqbal, W. Verboom and J. Huskens, Dalton Trans., 2012, 41, 6970 RSC.
- S. A. Ansari, P. K. Mohapatra, D. R. Prabhu and V. K. Manchanda, J. Membr. Sci., 2007, 298, 169–174 CrossRef CAS.
- M. L. Dietz and D. C. Stepinski, Green Chem., 2005, 7, 747–750 RSC.
- T. Singh and A. Kumar, J. Phys. Chem. B, 2008, 112, 12968–72 CrossRef CAS.
-
(a) W. D. Horrocks and D. R. Sudnick, J. Am. Chem. Soc., 1979, 101, 334–340 CrossRef CAS;
(b) W. D. Horrocks and D. R. Sudnick, Acc. Chem. Res., 1981, 14, 384–392 CrossRef CAS.
- S. Colette, B. Amekraz, C. Madic, L. Berthon, G. Cote and C. Moulin, Inorg. Chem., 2004, 43, 6745–6751 CrossRef CAS.
- P. N. Pathak, S. A. Ansari, S. V. Godbole, A. R. Dhobale and V. K. Manchanda, Spectrochim. Acta, Part A, 2009, 73, 348–352 CrossRef CAS.
- P. K. Mohapatra, M. Iqbal, D. R. Raut, W. Verboom, J. Huskens and S. V. Godbole, Dalton Trans., 2012, 41, 360–363 RSC.
- C. Xu, L. Yuan, X. Shen and M. Zhai, Dalton Trans., 2010, 39, 3897–3902 RSC.
- D. Allen, G. Baston, A. E. Bradley, T. Gorman, A. Haile, I. Hamblett, J. E. Hatter, M. J. F. Healey, B. Hodgson, R. Lewin, K. V. Lovell, B. Newton, W. R. Pitner, D. W. Rooney, D. Sanders, K. R. Seddon, H. E. Sims and R. C. Thied, Green Chem., 2002, 4, 152–158 RSC.
- R. B. Gujar, S. A. Ansari, A. Bhattacharyya, A. S. Kanekar, P. N. Pathak, P. K. Mohapatra and V. K. Manchanda, Solv. Extr. Ion Exch., 2012, 30, 278–290 CrossRef CAS.
- P. Deepika, K. N. Sabharwal, T. G. Srinivasan and P. R. Vasudeva Rao, Solvent Extr. Ion Exch., 2011, 29, 230–246 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra20577g |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.