Biodegradable soft elastomers: synthesis/properties of materials and fabrication of scaffolds
Yuan
Li
a,
George A
Thouas
b and
Qi-Zhi
Chen
*a
aDepartment of Materials Engineering, Monash University, Clayton, Victoria 3800, Australia.. E-mail: qizhi.chen@monash.edu; qzchen202@gmail.com; Fax: 61 (3) 99054940; Tel: 61 (3) 9053599
bDepartment of Zoology, The University of Melbourne, Parkville, Victoria 3010, Australia
First published on 12th July 2012
Abstract
Recently there have been increasing research efforts in the development of elastomeric biomaterials with desirable biocompatibility, degradation profiles and mechanical properties for use in soft tissue engineering. This review provides an update on the progresses of developing biodegradable, soft elastomeric biomaterials and their tissue engineering scaffolding techniques. Following a brief review on traditional thermoplastic elastomers, including polyurethane (PU), polyhydroxyalkanoates (PHA) and aliphatic copolyesters, detailed review is devoted to the synthesis, properties and scaffold fabrication of recently developed soft, biodegradable elastomers, including poly(polyol sebacate) (PPS), PPS-based elastomers, and citric-acid based elastomers. Although biodegradable, soft elastomeric biomaterials have advantages (compliant and biodegradable), this review also identified a number of issues associated with these elastomers, including cytotoxicity, rapid in vivo degradation rates, and poor reproducibility. Future research directions are discussed to address these issues.
 Qi-Zhi Chen | Qi-Zhi Chen received a PhD degree in Biomaterials from Imperial College London. She is currently an academic in the Department of Materials Engineering at Monash University. Previously she was also employed by the National Heart and Lung Institute London and the University of Cambridge. She has produced more than 100 peer-reviewed journal articles and book chapters. Her research interests broadly cover polymeric, ceramic, metallic and composite biomaterials for applications in biomedical engineering. |
1. Introduction
A common strategy in tissue engineering is to use a three-dimensional (3D) scaffold for proper cell growth and differentiation.1 Existing evidence suggests that the mechanical conditioning regimens of elastomeric scaffolds promote improved tissue formation and allow gradual stress transfer from the degrading synthetic matrix to the newly formed tissue.2,3 However, the biodegradable thermoplastics currently dominantly being used in tissue engineering, e.g. polyglycolide (PGA), polylactide (PLA) and their copolymers, do not provide satisfactory performance in soft tissue engineering due to their high stiffness, plastic deformation and mechanical failure when exposed to long-term cyclic strain. In the last decade, synthetic biodegradable elastomers have increasingly become a popular option for soft tissue engineering, and researchers have explored a wide range of scaffolds having mechanical properties resembling those of natural tissues.4,5,6–10 This review aims to provide an update on biodegradable elastomers, with a focus on relatively new soft elastomers, in particular poly(polyol sebacate) (PPS), which have tuneable mechanical properties to closely resemble those of soft tissues, and thus are promising for wide applications in tissue engineering of soft types.2. Overview of elastomeric biomaterials
An elastomer is a polymer with a glass transition temperature (Tg) below room temperature and that has the ability to undergo elastic deformation; that is, to stretch and return to its original shape in a reversible way.11 In order to work with mechanically dynamic tissues such as muscle, an elastomer must have the following characteristics: good biocompatibility with the host tissue and a glass transition temperature (Tg) lower than body temperature (i.e., 37 °C) so as to recover from various deformations at the body temperature.Biomedical elastomers can be classified into two categories according to the polymer sources: naturally occurring biological elastomers and chemically synthesised elastomers. Synthetic elastomers can further be divided into thermoplastic elastomers and cross-linked elastomers based on the type of “cross-link” used to join their chains. Table 1 provides important biomedical elastomers reported up to now, and more detailed reviews on their biomedical applications are available in the literature.5,8,9,12–15
| Elastomers | Abbreviations | Ref |
|---|---|---|
| 1. Synthetic elastomer | ||
| Thermoplastic elastomers | ||
| Poly(hydroxyalkanoate) | PHA | 19 |
| Poly(4-hydroxybutyrate) | P4HB | 20 |
| Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) | PHBV | 19 |
| Polyurethane | PU | 9 |
| Poly(phosphazene) | PPHOS | 21 |
| Poly(ether/ester) | PEE | 21 |
| Poly(ethyleneglycol)/poly(butylene terephthalate) | PEG/PBT | 21 |
| Poly(ε-caprolactone)copolymer | 21 | |
| Poly(glycolide-co-caprolactone) | PGCL | 21 |
| Poly(caprolactone-co-lactide) | PCLA | 21 |
| Poly(p-dioxanone-co-ε-caprolactone) | PDSCL | 22 |
| Poly(1,3-trimethylene carbonate) | PTMC | 21 |
| cross-linked elastomers | ||
| Poly(polyolsebacate) | PPS | 23 |
| Poly(glycerol sebacate) | PGS | 4 |
| Poly(xylitol sebacate) | PXS | 24 |
| Poly(glycerol sebacate acrylate) | PGSA | 25 |
| Poly(diol citrate) | PDC | 6 |
| Poly(1,8-octanediol-co-citric acid) | POC | 6 |
| Poly(1,8-octanediol-co-citric acid)-cysteine | POC-Cys | 26 |
| Poly(1,10-decanediol-co-citric acid) | PDC | 6 |
| Poly((1,2-propanediol-sebacate)-citrate) | PPSC | 27 |
| Poly(octamethylene maleate (anhydride) citrate) | POMaC | 28 |
| Poly(ethylene glycol) maleate citrate | PEGMC | 29 |
| Star-poly(esters) copolymer | SCP | |
| Poly(ε-caprolactone-D,L-lactide-glycerol-co-2,2-bis(ε-CL-4-yl)-propane | SCP-co-BCP | 30 |
| Poly(ε-caprolactone-D,L-lactide-glycerol-acrylate) | ASCP | 31 |
| Poly(ester amide) | ||
| Poly(1,3-diamino-2-hydroxypropane-co-polyolsebacate) | APS | 32 |
| 2. Natural degradable polymers | ||
| Polysaccharides | 33 | |
| Alginate | 34,35 | |
| Elastic Proteins | ||
| Collagen | 35 | |
| Gelatin | 36 | |
| Fibrin | 35 | |
| Elastin | 37 | |
| Elastin-like polypeptides | ELPs | 38 |
| Silk–elastin like protein polymers | SELPs | 39 |
The use of elastomers for medical devices can be traced back to as early as the mid 1890s, when the rubber industry began to expand.16 Vulcanised natural rubber was used in medical devices (such as surgical gloves) soon after the discovery of the vulcanisation process.16 Elastomeric materials have since been used in many medical products for their biocompatibility, design adaptability, mechanical compliance and economy of cost. Both natural and synthetic rubbers, e.g., polyurethanes (PU), poly hydroxyalkanoate (PHA), polysaccharides and elastin have been used in a range of implantables including cardiovascular devices, prosthetic devices, transdermal therapeutic systems and orthodontics,17 as listed in Table 2.
| Elastomers | Advantages or disadvantages | Tissue engineering applications | Reviews |
|---|---|---|---|
| Polyurethanes (PU) (physically cross-linked) | 1. Enormous diversity of chemical compositions and physical-mechanical properties | Wound dressings and scaffolds for tissue engineering could permit new developments | 17,53,54 |
| 2. Tissue-specific biocompatibility | |||
| 3. Tuneable biodegradability | |||
| 4. Mechanical flexibility | |||
| Polyhydroxyalkanoate (PHA) (physically cross-linked) | 1. Excellent biocompatibility | Scaffolds or template for tissue engineering including bone and skin | 55 |
| 2. A wide range of mechanical properties and biodegradability | |||
| 3. Expensive | |||
| Polyesters and copolyesters (physically cross-linked) | 1. Natural softness, good contact to surfaces | Suture wound care dressings | 17 |
| 2. Pleasant feeling, comparable to that of dry human skin | |||
| 3. No contamination to wounds, no plasticizers used | |||
| 4. Excellent resistance to a wide range of solvents and chemicals | |||
| 5. High tear strength | |||
| 6. Low-friction surfaces—no problems of adhering to wound areas, clothing, or bedding | |||
| 7. Extremely breathable | |||
| 8. Good processability | |||
| 9. Good sterilisability | |||
| 10. Low toxicity | |||
| Polyolefin and polydiene (chemically cross-linked) | 1. Excellent biostability | Hexsyn™: ethylene–propylene rubber: finger joint prostheses | 17,56 |
| 2. Functionality | |||
| 3. Biocompatibility | |||
| Poly(polyol sebacate) (PPS) (chemically cross-linked) | 1. Soft elasticity | Scaffolds or template for soft tissue engineering, including nerve, vessel and heart muscle | 4,18,23 |
| 2. Moderate biocompatibility | |||
| 3. Tuneable degradability | |||
| 4. Mild cytotoxicity | |||
| Biological polymers (elastic proteins) (hydrogen bonding or chemically cross-linked, such as elastins) | 1. Excellent biocompatibility | Templates or scaffolds for tissue engineering and regeneration of all types | 57 |
| 2. Concerns of disease transfer |
Naturally occurring elastomers
The natural extracellular matrices (ECMs) of soft tissues are composed of various collagens; thus, collagens have been widely used in soft tissue engineering.40,41 They provide a natural substrate for cellular attachment, proliferation and differentiation. Collagen, gelatine, fibrin and alginate have been extensively investigated in tissue engineering of almost all types, including bone,42 cartilage,43 skin,44 ligaments,45 vessels,46 heart muscles,47 the liver48 and nerves.49 Selected properties of them are listed in Table 3. Although natural polymers have advantages such as biodegradability and cytobiocompatibility as well as unique chemical, physical and mechanical properties (e.g. non-linear elasticity), they are subject to tedious purification techniques, processing variability and regulatory issues.5,50 In addition, they have unreliable mechanical properties and variable physical properties with different sources of protein matrices.18 These shortcomings of natural materials form the motivation for developing synthetic biodegradable polymers for use in tissue engineering.| Polymers | Young's modulus (MPa) | Tensile strength (MPa) | Elongations (%) | Refs |
|---|---|---|---|---|
| a PEUU = polyester urethane urea. b P3HB = poly (3-hydroxybutyrate). c P4HB = poly (4-hydroxybutyrate). | ||||
| Synthetic elastomers | ||||
| PUs (PEUUa) | 7–70 | 4–60 | 100–950 | 98 |
| PHAs (P3HBb-co-P4HBc) | 50–300 | 20–50 | 400–1000 | 5 |
| PCL-co-LA | 30 | 32 | 120 | 87 |
| PGS | 0.05–1.5 | 0.2–0.7 | 120–270 | 4,5,10,18 |
| Other PPS | 0.4–380 | 10–200 | 23 | |
| PGSA | 0.05–1.4 | 0.05–0.5 | 40–190 | 25 |
| Naturally occurring | ||||
| Elastin (bovine ligament) | 1.1 | 2 | 150 | 99,100 |
| Resilin (dragonfly tendon) | 2 | 4 | 190 | 99 |
| Knee articular cartilage | 2.1–11.8 | 52 | ||
| Smooth muscle (relaxed) | 0.006 | 300 | ||
| Smooth muscle (contracted) | 0.01 | 300 | ||
| Human bladder | 0.25 | 0.27 | 0.7 | |
| Heart muscle (at the end of diastole) | 18 | |||
| Myocardium of rat | 0.14 | 0.03–0.07 | ||
| Myocardium of human | 0.2–0.5 | 0.003–0.015 | ||
Synthetic elastomers
From a biological and clinical standpoint, synthetic elastomers should exhibit both biocompatibility and mechano-compatibility with natural ECM proteins. From a polymer perspective, these materials should be able to be synthesized in large batches and exhibit tuneable properties that could be realized by altering synthetic schemes and polymer processing conditions. To satisfy these criteria, the design of these materials should follow certain principles, including: (1) incorporation of ester bonds in polymer chains to promote degradation by hydrolysis and enzymatic activity; (2) tuneable elastomeric mechanical properties; and (3) use of non-toxic monomers that can be metabolized or excreted by the host.5There are two types of synthetic elastomers: thermoplastic elastomers (physically cross-linked) and cross-linked elastomers (chemically cross-linked).51 In thermoplastic elastomers, hard and rigid block segments alternate with block segments of a soft and flexible segments.51 At ambient temperatures, the soft, amorphous segments impart the rubbery, elastic behaviour to the material. At temperatures below Tg or melting temperature (Tm) of the hard segments, the hard chain segments from numerous adjacent chains aggregate together to form rigid amorphous or crystalline domains. These domains are ‘physical cross-links’ that act as anchor points that restrict soft chain segment motions; they function in much the same way as ‘chemical cross-links’ in cross-linked elastomers.51
The major advantage of thermoplastic elastomers over cross-linked elastomers is that upon heating above Tg or Tm of the hard phase, they soften and flow (i.e., the physical cross-links disappear) and can thus be processed easily. Unlike thermoplastic elastomers, cross-linked elastomers gain their elasticity from a chemical cross-linking process that occurs when the compound is subjected to heat or radiation, a process often called ‘curing’. Once cured, these compounds obtain the necessary physical properties. Reheating chemically cross-linked polymers will not cause them to flow as thermoplastics do consequently, conventional reforming is normally difficult or impossible with chemically cross-linked elastomers.51
Thermoplastic elastomers have good processability while cross-linked elastomers will not flow once cured.51 However, they often contain crystalline regions that cause heterogeneous degradation and a non-linear loss of mechanical strength with time. Moreover, the 3D geometry of a thermoplastic scaffold is commonly lost during the hydrolysis period. In this regard, cross-linked elastomers offer advantages for biomedical applications over thermoplastic materials,5,52 as listed in Table 4.
| Materials | Advantages | Disadvantages |
|---|---|---|
| Thermoplastic elastomers (e.g. PU, PHA) | 1. Good processability | 1. Heterogeneous degradation |
| 2. Tuneable mechanical properties | 2. Non-predictable loss of mechanical strength | |
| 3. Tuneable degradation rate through modification of the soft and hard segments. | 3. 3D geometry lost during the hydrolysis period | |
| Cross-linked elastomers (e.g. PPS) | 1. Retention of the 3D structure | 1. Poor processability |
| 2. Predictable degradation rates and a linear loss of mechanical strength with time | 2. Curing conditions limit the application of cross-linked elastomers as drug-delivery vehicles |
In contrast, cross-linked elastomeric polyesters undergo a combination of bulk and surface erosion degradation, which results in the maintenance of the 3D structure throughout the hydrolytic process.52 In addition, cross-linked elastomeric polyesters, which can breakdown to natural metabolic products by simple hydrolysis, have potential to be tailed in their degradation rates to match the healing kinetics of injured tissue. The rest of this article is thus devoted to synthetic rubbers, with a special attention being paid to chemically cross-linked elastomers.
3. Thermoplastic elastomers
PU (e.g. PEUU) and PHA (e.g. P3HB-co-P4HB and P4HB) represent two dominant thermoplastic rubbers. Their typical mechanical properties are provided in Table 3. An immediate and frequently utilised strategy for achieving biodegradation of PU elastomers is to incorporate a polyester macrodiol soft segment that hydrolyses in vitro and in vivo, such as poly(lactic acid) (PLA) or poly(glycolic acid) (PGA).58 In general, PUs and PHAs degrade more slowly than PGA/PLA/PLGA, with degradation rates being typically 2–3 years. From the degradation point of view, PUs and PHAs could be useful in tissue engineering for implants that require a longer retention time or a higher stability in the surrounding environment, but which are eventually absorbed. This might be useful for tissues with slower healing and remodelling times, or with an inability to maintain innate structural integrity (e.g., large wound). In addition to cardiovascular tissue engineering,59,60 PUs and PHAs have proven highly versatile, having been investigated for the repair of soft tissue (e.g., nerve,61–68 blood vessel,69 intestine,70) as well as harder load-bearing tissue (e.g., bone,71–76 cartilage,77 meniscus78–83 and ligament84,85).Polycaprolactone (PCL) has been copolymerized with PGA and PLA to produce thermoplastic rubber-like biomaterials.86–89 Most of these copolymers are synthesized by ring-opening polymerization in the presence of an initiator such as stannous (II) octoate [Sn(Oct)2]. Their mechanical properties are listed in Table 3. PLA, PGA, PCL and their copolymers remain popular for a variety of reasons, but primarily their biocompatibility and biodegradability.35 The degradation rates of the PCL-based block copolymers can vary over a wide range, depending on the ratio of co-monomers. In general, the copolymers degrade faster than the homopolymer alone. PGA-co-CL (PGACL) scaffolds lost 50% after 6-week incubation, whereas it took 6–12 months for PGA to degrade.90
PGACL and PLACL copolymers have both been investigated for potential applications in tissue engineering.12 In an in vivo work, cell-seeded scaffolds were implanted subcutaneously in nude mice. After three weeks, the PGACL scaffolds were found to provide adequate physical characteristics to induce new tissue formation.90 PLACL scaffolds have shown encouraging results as vascular grafts both in canine91,92 and human experimental subjects.87,92
As summarised in Table 4, the thermoplastic elastomers have advantages, being biocompatible and having a good processibility. One of major drawbacks of thermoplastic elastomers is their heterogeneous degradation properties.93,94 This heterogeneous degradation is presented as a localised loss of structural integrity of the materials with time (Fig. 1) such that the loss of mechanical strength is less predictable.95 This becomes more critical when thermoplastic materials are used to build a three-dimensional (3D) structure. The 3D geometry of a thermoplastic is lost during degradation of foam struts and the architecture of the network is difficult to maintain.96 Chemically cross-linked elastomers, on the other hand, provide more consistent degradation rates and predictable decline in mechanical properties due to their homogeneity (Fig. 1). Over the past decade, chemically cross-linked, biodegradable elastomers have gained increasingly attention in the field of tissue engineering, as discussed in Sections 4–6. Among them, poly(polyol sebacate) (PPS) is a family of soft elastomers showing great potential in organ engineering of soft types.4,7,97
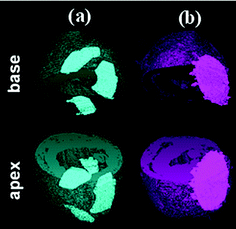 | ||
| Fig. 1 Elastomeric heart patches attached to the pericardium of rat hearts. Premature failure of a thermoplastic rubber patch (a), and maintenance of the physical integrity of a chemically cross-linked elastomer patch (b). | ||
4. Poly(polyol sebacate) (PPS)
Much attention has recently been paid to soft, biodegradable chemically cross-linked elastomers. PPS is a new family of cross-linked elastomers developed by Langer's group at MIT.4,7,97 These polymers are biocompatible and inexpensive,101 and they have already shown potential applications in nerve102 and vascular tissue engineering.103–105 A polyol is a sugar alcohol containing multiple hydroxyl groups (e.g., glycerol, mannitol, sorbitol and xylitol). Sebacic acid, a dicarboxylic acid with structure (HOOC)(CH2)8(COOH), is a naturally occurring chemical derived from castor oil which has been proven safe in vivo.106–108 Polyol and sebacic acid are both endogenous monomers found in human metabolism.Poly(glycerol sebacate) (PGS)
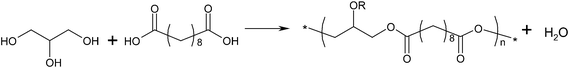 | ||
| Fig. 2 Reaction scheme for poly(glycerol sebacate), R=H or polymer chain. | ||
Thermal treatment is a common method of synthesising PGS due to the ease of processing, and it is typically conducted in two steps: pre-polymerisation and cross-linking. The prepolymer is formed by heating a mixture of glycerol and sebacic acid under a purge of inert gas through the reacting chamber to prevent the reactants' oxidation and at atmospheric pressure and to minimize evaporation of the volatile polyol monomers. The pre-polymerisation conditions vary from 110 to 150 °C for 1 to 12 h.
The level of linear versus cross-linking depends on the reaction kinetics of primary and secondary hydroxyl (–OH) groups of glycerol (Fig. 2 and 3). The primary –OH groups react much faster than the secondary ones at 100–150 °C,110 and form PGS polymer chains. The cross-linking process, which involves the secondary –OH groups (Fig. 3), becomes prominent only when there is a severe lack of primary –OH groups in the reactant system. The PGS pre-polymer can be dissolved in organic solvent (e.g., tetrahydrofuran (THF), 1,3-dioxolane and ethanol), which can subsequently be processed into various forms of tissue engineering substitutes, including 3D porous scaffolds,111 thin films or sheets104 and even fibres. The resultant polymer material is a transparent elastomer, which is a covalently cross-linked 3D network of random coils with hydroxyl groups attached to the backbone.
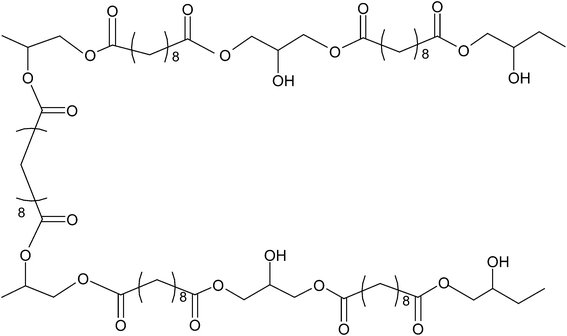 | ||
| Fig. 3 Cross-linking scheme between two PGS polymer chains. | ||
PGS has been synthesised over a range of temperatures and durations.4,7,10,101,104,111–115 It must be mentioned that the polymerisation kinetics of PGS reported from various studies have not proven to be consistent.10 In the work on surgical sealants,116 a non-cross-linked pre-polymer, which can either dissolve in THF or flow at a temperature around 40–50 °C, was produced after synthesis at 110 °C for three days, whereas a cross-linked PGS solid was formed under the same synthesis conditions in another work.10 Similar discrepancies can also be found in studies produced by other research groups. In the work of Wang et al.,4 for instance, a cross-linked PGS elastomer was produced after synthesis at 120 °C for three days, while in Yi and Lavan's work,117 an uncross-linked PGS pre-polymer was synthesised under the same conditions, and the pre-polymer was subsequently dissolved into THF for the production of PGS fibres.
The above discrepancy was potentially caused by a number of factors. On the one hand, the cross-linking kinetics of PGS are influenced by the synthesis conditions, especially temperature and the molar ratio of glycerol and sebacic acid. On the other hand, the synthesis conditions can vary significantly from one research laboratory/group to another. Firstly, the temperature uniformity of a vacuum oven is ±1 °C at best, typically 2.5 °C for most ovens. A difference of 5 °C can greatly change the cross-link kinetics of PGS, according to our experience. Secondly, the loss of volatile glycerol monomer is inevitable, and both the flow rate of the (nitrogen) gas purge and the capacity of the vacuum pump can greatly affect the loss of glycerol during synthesis. Consequently, different research groups report different cross-linking kinetics for the ‘same’ PGS polymers under the ‘same’ conditions. Hence, the reproducibility of PGS materials remains to be improved.
PGS has an advantage over other elastomers in terms of degradation behaviour. PGS predominantly degrades through surface erosion, which means the rate of degradation is predictable.121 Both mechanical strength and mass decrease almost linearly with time as the polymer degrades. During in vivo degradation, it maintains its original shape, with relatively low swelling. The water content of PGS implants rises and reaches 15% in 35 days, at which point the polymer degrades to >70 wt%.101 This manner of degradation is desired in many biomedical applications such as drug-delivery devices,122 scaffolding7,104 and soft tissue engineering.10,109,113,115,123
However, the rapid degradation of PGS materials tend cause cytotoxicity109 due to either excess non-reacted carboxylic acid groups or excessive amount of carboxylic acids produced by the aqueous hydrolysis of the PGS ester groups, which lowers the local extracellular pH to below physiological values (7.2–7.4). Another drawback is that the rapid degradation rate of PGS is believed to limit their application as a scaffold material in engineering tissues that have a healing rate of several months or years (e.g., the cardiac muscle). Hence, alternative chemical approaches are needed to decrease the enzymatic hydrolysis rate of the ester bonds in PGS polymers.114 Nonetheless, its fast degradation kinetics match the healing rate of bone (typically 4–6 weeks124), and it is thus desired as a replacement of collagen component in artificial bone matrix.
Poly(xylitol sebacate) (PXS) and other PPS elastomers
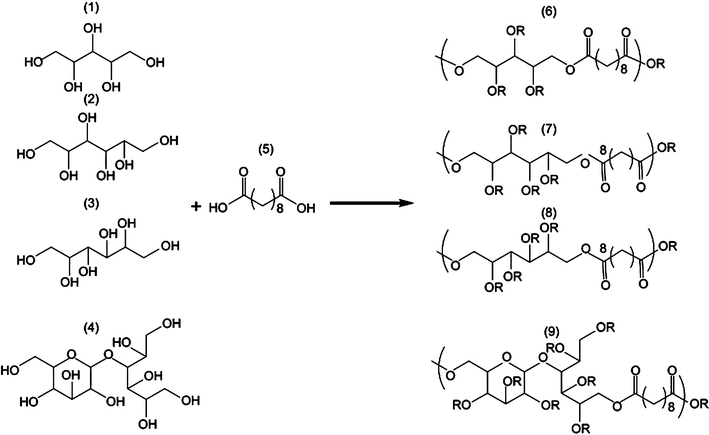 | ||
| Fig. 4 General synthetic scheme of polyol-based polymers. Xylitol (1), sorbitol (2), mannitol (3) and maltitol (4) were polymerised with sebacic acid (5) in different stoichiometries, yielding PXS (6), PSS (7), PMS(8) and PMtS (9).23 Reprinted with permission of Elsevier. | ||
| Synthesis of pre-polymer | Curing conditions | E (MPa) | UTS (MPa) | Maximal elongation (%) | |
|---|---|---|---|---|---|
a PSS = poly(sorbitol sebacate).
b PMS = poly(mannitol sebacate).
c PMtS = poly(maltitol sebacate).
d PES = poly(erythritol sebacate).
e Molar ratio of polyol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :sebacate. :sebacate.
|
|||||
| PGS 1:1e |
120 °C/24 h (inert gas), then 120 °C/5 h (40 mTorr) | 120 °C/48h (40 mTorr) | 0.282 ± 0.03 | >0.5 | 267 ± 59 |
| PXS 1:1 |
150 °C/2 h (inert gas), then 2–12 h (50 mTorr) | 120 °C/4 days (2 Pa) | 0.82 ± 0.15 | 0.61 ± 0.19 | 205 ± 56 |
| PXS 1:2 |
5.33 ± 0.40 | 1.43 ± 0.15 | 33 ± 5 | ||
| PSSa 1:1 |
120 °C/5 days (2 Pa) | 0.37 ± 0.08 | 0.57 ± 0.15 | 192 ± 60 | |
| PSSa 1:2 |
120 °C/4 days (2 Pa) | 2.67 ± 0.12 | 1.16 ± 0.33 | 66 ± 25 | |
| PMSb 1:1 |
120 °C/5 days (2 Pa) | 2.21 ± 0.21 | 0.79 ± 0.10 | 51 ± 9 | |
| PMS 1:2 |
12.82 ± 2.9 | 3.32 ± 0.76 | 45 ± 12 | ||
| PMtSc 1:4 |
378.0 ± 33 | 17.64 ± 1.3 | 11 ± 1 | ||
| PESd 1:1 |
145 °C/2 h (inert gas) then 7 h (50 mTorr) | 120 °C/3 days | 0.08 ± 0.01 | 0.14 ± 0.03 | 446 ± 41 |
| 140 °C/3 days | 1.92 ± 0.12 | 1.03 ± 0.19 | 85 ± 14 | ||
The biocompatibility of the PPS polymers is similar to that of PLGA.23,126 Similar to PGS, all PPS members show faster degradation kinetics in vivo than in vitro under physiological conditions, compared with PLGA. A most recent study has demonstrated that the enzyme-mediated degradation rate of PXS is significantly slower, approximately half of that of PGS.127
Photopolymerised poly(glycerol sebacate-co-acrylate) (PGSA)
One limitation to the thermal cross-linking procedure of the PGS system is the harsh processing conditions (long-term, high-temperature curing), which could limit its ability to polymerise directly in a tissue or incorporate cells or temperature-sensitive molecules. To overcome this shortcoming, photo-crosslinkable elastomers were developed by Nijst et al., and they demonstrated the ability to form an elastomeric network from poly(glycerol sebacate-co-acrylate) (PGSA) under mild conditions while preserving a wide range of physical properties.25 The advantages and disadvantages of both synthesis methods are presented in Table 6.| PPS synthesis processes | Advantages | Disadvantages |
|---|---|---|
| Thermal cross-linking | 1. Properties can be easily controlled by varying temperature and time | 1. Harsh processing conditions (long-term high-temperature curing, vacuum conditions) |
| 2. Cheap and easy to produce large batches of elastomers | ||
| 3. Various shapes can be cast from pre-polymers | ||
| Photo cross-linking | 1. Short reaction time | 1. UV light required for polymerisation |
| 2. Mild reaction conditions (room temperature) | ||
| 3. In situ curing | 2. Long-term UV light exposure may damage cell | |
| 4. Properties tuneable by varying degree of acrylation and combination with other acrylated molecules | 3. Non-polymerised PGSA may be toxic to cells |
Photo-crosslinkable PGSA was synthesised by reacting PGS precursors with acryloyl chloride (deprotonating free hydroxyl groups and appending acrylate groups) to form a PGSA pre-polymer (in Fig. 5). A PGSA pre-polymer was cast into its final form and then cross-linked using a photo-initiator and UV light by photo-induced free radical polymerisation at room temperature. By deprotonating free hydroxyl groups and appending acrylate groups, the PGS pre-polymer was modified with acrylate moieties that participate in chemical cross-linking between polymer chains. The materials were rapidly polymerised at room temperature after UV light irradiation.25
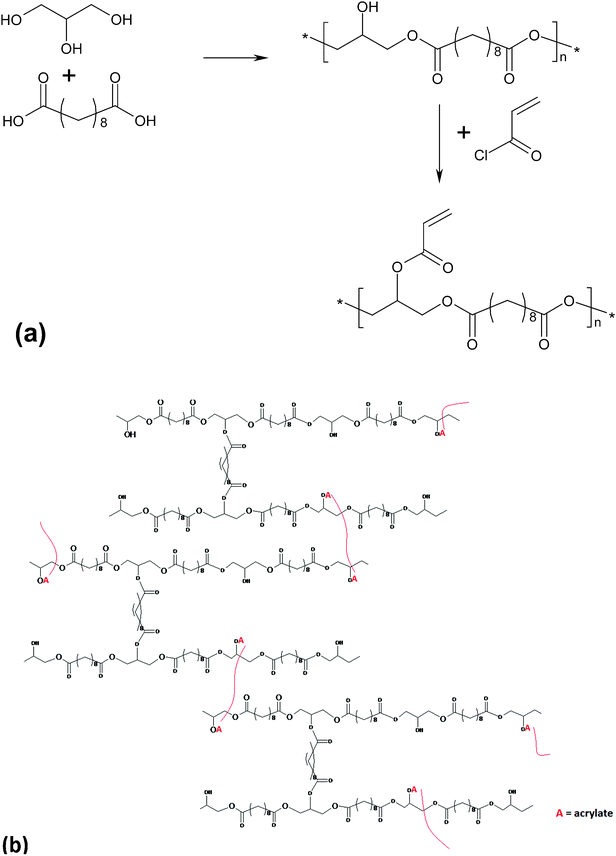 | ||
| Fig. 5 (a) Modification of poly(glycerol sebacate) pre-polymer with acrylate moieties for photo-initiated curing; (b) PGSA after polymerisation under UV light.25 Reprinted with permission of ACS publications. | ||
By varying the degree of acrylation of PGSA, these elastomers exhibited a Young's modulus, UTS and elongation at break ranging from 0.05 to 1.4MPa, 0.05 to 0.50 MPa, and 40 to 190%, respectively.25 The spectrum of PGSA properties could be expanded by the copolymerisation of PGSA with other acrylated molecules such as PEG-diacrylate. Increasing the concentration of PEG-diacrylate of this polymer system increases the Young's modulus and the UTS, while decreasing the maximum elongation, the swelling ratio and degradation rate.25In vitro assessments showed that photo-cured PGSA films supported human primary cell adherence and subsequent proliferation.25 However, a mild to moderate inflammatory response of implanted PGSA was observed in vivo.128
PGS-based elastomeric composites
One strategy to expand the properties of a class of materials is to develop a composite. Although PPS has a wide range of properties, the spectrum can be further increased by developing a PPS composite.118,119,129 A PPS-based bioactive glass/ceramic-filled composite was developed to simultaneously increase the PPS modulus and elongation and to minimise the drawbacks of PPS (e.g., the cytotoxicity caused by the acidic degradation products of very soft PGS materials mentioned above).119The composites of PGS and alkaline Bioglass® filler have demonstrated both improved cytocompatibility and enhanced mechanical properties.118,119 Firstly, the addition of micro-sized Bioglass® to PGS increased the elongation at break from 160 to 550%. Secondly, the moduli of PGS/Bioglass® drops abruptly in a physiological environment (culture medium), and the level of the drop can be tuned such that Bioglass® does not harden the composite in vivo but rather maintains the desired compliance of biomaterials required for soft tissue engineering.118,119
A potential electronically active PGS-based composite has been fabricated through incorporating multi-walled carbon nanotubes (MWCNTs).129 The motivation behind the work is that increasing the amount of MWCNTs may eventually lead to a percolating network of these conductive elements to form an electronically active PGS composite that may be utilised as a conducting biomaterial system in neural repair (e.g., nerve-bridging devices) or nerve-tissue engineering. Compared with pure poly(glycerol-sebacate-citrate)(PGSC), (PGSC)-based MWCNT-filled elastomeric composite exhibits increased mechanical properties with strength and modulus being 4.4 and 9.2 MPa, respectively. The composite materials demonstrated little cytotoxicity and slow degradation rates.129
Tissue engineering applications of PPS and PPS-based elastomers
The major reason why PPS is of major interest in tissue engineering and regeneration is that PPS have been reported to be non-toxic based on in vitro cell-based outcomes7 and in vivo findings using animal models.95,101 PGS is the most well studied PPS elastomer, with specific benefits shown in cardiovascular tissue engineering103,109 and small diameter nerve grafting.1135. Citric acid based elastomers
Synthesis
Citric acid is a natural preservative and has been widely used in the food industry. Diols or glycols are chemical compounds containing two hydroxyl (–OH) groups. The synthesis of poly(diol citrate) (PDC) is similar to PGS. Firstly, a prepolymer is produced via thermal polycondensation of three acidic group monomers from citric acid and an aliphatic diol, which are typically 1,6-hexanediol, 1,8-octanediol, 1,10-decanediol, or 1,12-dodecanediol. This is followed by post-polymerization at designed conditions (temperature/cure duration). Unlike other polycondensation reactions, PDC can be synthesized under relatively low temperature (as low as 37 °C) and even without vacuum.3,6Mechanical properties, degradation and compatibility of PDC
Four PDCs have been made separately with the four diols mentioned above, among which poly (1,8-octanediol) citrate (POC) is the most studied mainly because of its desirable mechanical properties: Young's modulus ranging from 0.92–16.4 MPa, UTS of 6.1 MPa and elongation at break of 117–265%.6 By altering the post-polymerization temperature, time, and monomer molar ratio, one can optimise both the mechanical properties and the degradation rate simultaneously.In vitro evaluation conducted with human aortic smooth muscle cells (HASMCs) and human aortic endothelial cells (HAECs) indicate that POC is as good as PLLA in terms of the growth and viability of the cells.4 Subcutaneous implantation in Sprague–Dawley rats has demonstrated that the thickness of the fibrous capsule induced by POC was similar to that induced by PGS, but significantly smaller than that of PLGA.4 Meanwhile, no chronic inflammatory response was observed. POC has been was reported to have a life time of 6 months when incubated in PBS at 37 °C in vitro, and to undergo both degradation and enzyme-catalysed hydrolysis in vivo, with implants remaining in situ for 2 months following subcutaneous implantation.6
Applications of POCs in tissue engineering
One of the potential applications of POC is as a tubular scaffold used in blood vessel engineering. Biphasic tubular scaffolds are constructed of two layers: a nonporous, skin-like interior with 1,12-dodecanediol) citrate (PDDC) and a porous exterior with POC. The developed PDC tubes are very well matched mechanically with native blood vessels,130 with an ability to undergo elastic recovery from deformation.6. Other biodegradable soft elastomers
Polyester amides
In vitro assessments have revealed that APS had excellent biocompatibility, showing healthy cell morphology and maintained metabolic activity of primary human foreskin fibroblasts when cultured on APS. In vivo trials have demonstrated that APS produces less foreign body responses than PLGA, with reduced fibrosis and fewer macrophages around APS implants compared to PLGA.131
Cross-linked poly(1,3-trimethylene carbonate)
Poly(1,3-trimethylene carbonate) (PTMC) was initially developed as a linear thermoplastic elastomer, displaying poor dimensional stability, tackiness and inadequate mechanical properties.133 To address these problems, cross-linked PTMC was developed.133–137 Cross-linked PTMC and their blends have been fabricated into flexible scaffolds with inter-connected pores and extensive micro-porosity by the fused deposition modelling followed by cross-linking with UV.1357. Fabrication of soft elastomeric scaffolds
In an organ, cells and their extracellular matrix (ECM) are usually organized into three-dimensional tissues. Therefore, in tissue engineering a highly porous 3D matrix (scaffold) is often necessary to accommodate cells and to guide their growth and tissue regeneration in three dimensions. Numerous techniques have been developed to process porous polymer scaffolds for use in tissue engineering. Polymeric scaffold processing methods could be classified into four groups, as listed in Table 7: (i) melted-polymer based, (ii) solvent based, (iii) gas-foaming, and (iv) rapid prototyping (RP). The choice of the correct technique is critical because the fabrication can significantly alter the properties of the implant and its degradation characteristics. A number of reviews are available in the literature.18,138–141| Fabrication technology | Required properties of materials | Available pore size (μm) | Porosity (%) | Porous structure |
|---|---|---|---|---|
| Melting-based | ||||
| Melt moulding | Thermoplastic | 50–500 | <80 | Spherical pores/low interconnectivity |
| Extrusion/particle leaching | Thermoplastic | <100 | <84 | Spherical pores/low interconnectivity |
| Solvent-based | ||||
| Solvent casting/particle leaching | Soluble | 30–300 | 20–50 | Irregular pores |
| Paraffin template | Soluble | 100–70 | >90 | Spherical pores |
| Emulsion freeze drying | Soluble | <200 | <97 | High volume of interconnected micropores |
| Thermally induced phase separation/freeze drying | Soluble | <200 | <97 | High volume of interconnected micropores |
| Electrospinning | Soluble | 20–100 | <95 | Fibrous |
| Membrane lamination | Soluble | 30–300 | <85 | Irregular pores |
| Gas-foaming | ||||
| Chemical reactant gas foaming | Amorphous | 100–700 | >90 | High volume of non-interconnected micropores |
| Physical induced gas foaming/particle leaching | Amorphous | Micropores<50 Macropores < 400 | <97 | Low volume of non-interconnected micropores combined with high volume of interconnected macropores |
| Rapid prototyping | ||||
| 3D printing | Soluble | 45–150 | <70 | 100% interconnected macropores |
| Fused deposition modelling | Thermoplastic | 160–700 | 48–77 | Regular geometrical honeycomb pores |
| Selective laser sintering | Polymer/ceramic and polymer composite | 0.006–200 | — | High volume of interconnectivity micropores |
As aforementioned, chemically cross-linked elastomers can be neither melted nor dissolved in any solvent. Hence, conventional reforming is normally difficult or impossible with chemically cross-linked elastomers.51 This limitation, however, can be addressed with a prepolymer of elastomers, which can be dissolved in organic solvent and subsequently processed into various forms of tissue engineering substitutes, including porous scaffolds,111 thin films or sheets104 and even fibres. This section is thus devoted to the scaffold fabrication of cross-linked elastomers, which has been advanced with the development of the PPS family, especially PGS.
Solvent casting and particle leaching
Solvent and particle leaching technology involves casting a dissolved prepolymer around packed leachable particles. After drying and cross-linking the prepolymer and leaching out the porogen, a porous elastomeric scaffold with interconnected pores forms. 3D PGS scaffolds with a random network of interconnected pores have been fabricated with this technology using a two-phase system of pores in which the large macroscopic pores (75–150 μm) are produced using sieved salt particles while the small microscopic pores (5–20 μm) are produced by vapour formation of glycerol.111 Specifically, the PGS pre-polymer is dissolved in THF and cast to the fused template of sodium chloride particles. Then, the PGS prepolymer is thermally cured at an elevated temperature (e.g. 150 °C) while being dispersed throughout the fused salt template. During the cross-linking treatment, volatile glycerol partially evaporates generating micropores. The dissolution of the salt and subsequent lyophilisation produced elastomer sponges with approximately 90% porosity, interconnected macropores and extensive micropores.Melt-based replica moulding and lamination technique
Controlling the cellular microenvironment within the scaffolds is critical for eliciting desirable biological responses such as proliferation, migration and maturation. However, the micro- and nano-structure of the scaffold is difficult to achieve via the traditional fabrication techniques such as solvent casting and particle leaching, gas foaming and 3D printing. Polymer scaffold replica moulding on micro-machined silicon substrate can achieve a feature resolution of less than 10 μm,142 the same length-scale of mammalian cells. Melt-based replica moulding fabrication technologies have been employed to achieve well-defined feature geometries in polycrystalline or amorphous thermoplastic biomaterials.Since high-temperature curing is required after melt-casting an PPS prepolymers, this processing requires the use of a sacrificial mould release layer consisting of a biologically benign material (e.g., sucrose) to prevent adhesion of these adhesive materials to the mould used during the final polymerisation.7Fig. 6 illustrates such a process of 3D microfluidic tissue engineering scaffolds. In this process, a negative silicon mould is fabricated using the standard photolithographic and plasma-etching techniques. Prior to replica moulding of PGS, a sacrificial sucrose release layer is spin-coated on the silicon master. For replica moulding of PGS, the melted PGS prepolymer is applied to the silicon substrate coated with sucrose. The PGS prepolymer sheet is then thermally cross-linked, forming the PGS sheet with the micro-pattern printed on it. The sheet is delaminated with the mould in water (Fig. 6). Finally, the PGS sheet is laminated by further curing (ester linkages from active hydroxyl and acid groups form between layers) to form a long-range ordered 3D microfluidic scaffold.7
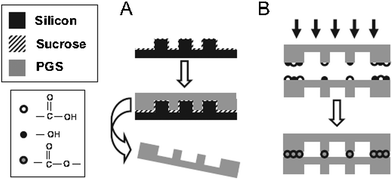 | ||
| Fig. 6 Overview of fabrication strategy for PGS microfluidic devices: A) replica moulding of PGS layers. PGS pre-polymer is cured into micro-patterned sheets and delaminated in water. B) Multiple layers are bound by physically adhering individual sheets and curing the films together.7 Reprinted with permission of John Wiley and Sons. | ||
Electrospinning
Electrospinning is a convenient method of producing arrays of loose nano-scale fibrous networks. Aligned textures produced by this technique are favoured in some tissue engineering applications to guide cells (e.g., in myocardial tissue with an aligned texture). However, the major technical hurdle in electrospinning cross-linked elastomers is that these polymers cannot dissolve into any solvents for spinning once cross-linked, and any fibres spun from uncross-linked pre-polymers would distort and flow in the subsequent cross-linking treatment.To address this challenge, a core–shell electrospinning technique has been applied. In this technique, the core is fed with the pre-polymer of elastomer, and the shell is fed with a thermoplastic polymer that could remain solid and maintain the fibrous shape at an elevated temperature during the subsequent curing process. The core–shell electrospinning technique has been applied for fabrication of PGS fibres. In this technique, the core is fed with the pre-polymer of PGS, and the shell is fed with PLLA that could remain solid and maintain the fibrous shape during the subsequent curing process of PGS.117 Subsequently, PLLA was removed from the fibres to yield a PGS scaffold.
Electrospinning can be more appropriately applied to photo-crosslinkable biodegradable elastomers, which do not require a thermal curing process. PGSA macromers were used to produce a nanofibrous scaffold using electrospinning, with gelatine as a carrier polymer to facilitate fibre formation and cell adhesion. Post-UV light exposure was used to facilitate PGSA cross-linking. The properties of the bulk fibrous scaffold were tested: mechanics (tensile modulus varied from 60 kPa to 1 MPa) and degradation (mass losses between 45 and 70% by week 12).143
Rapid-prototyping
The rapid-prototyping or solid free-form (SFF) technique is a computer-aided fabrication process. Using the rapid-prototyping techniques of fused deposition modelling (FDM), selective laser sintering (SNL), 3D printing (3D-P), stereo lithography or extrusion free forming, 3D objects are constructed layer by layer. These methods have an advantage over conventional fabrication techniques due to their ability to create geometries with accurately designed complex architectures.144For cartilage tissue engineering, a3D PGS scaffold was created145 with designed pore shapes, pore sizes, porosities and architectures through a sacrificial mould produced using SFF techniques (Fig. 7).145 In this technique, SFF methods are used to create inverse wax moulds. Subsequently, the wax moulds are used to form sacrificial hydroxyapatite moulds, which are then used to mould the PGS pre-polymer. After curing, the hydroxyapatite phase is dissolved using a rapid decalcifying agent to achieve the final PGS scaffold.145 The designed PGS 3D scaffold can be used in cartilage tissue engineering, and it has been demonstrated that the scaffold can support robust cartilage formation when seeded with porcine chondrocytes in vitro.145
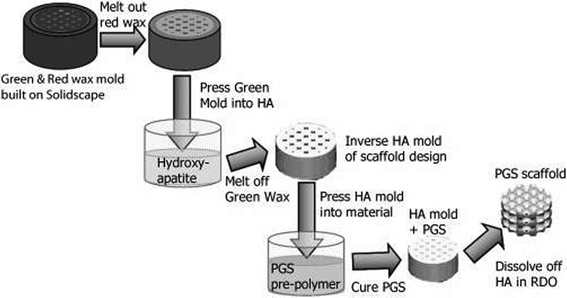 | ||
| Fig. 7 Fabrication of 3D designed PGS scaffolds involves creating wax moulds that are cast into hydroxyapatite to create an inverse mould and then cast into a PGS pre-polymer and cured, resulting in a PGS scaffold.145 Reprinted with permission of John Wiley and Sons. | ||
Recently, a micro-fabrication technique has also been used to produce accordion-like honeycomb microstructure 3D scaffolds for myocardial repair applications (Fig. 8).115 The 3D scaffold (highly porous and elastomeric with controllable stiffness and anisotropy) was fabricated by micro-ablating lateral array troughs in one PGS lamina, overlaying another PGS lamina, micro-ablating top-to-bottom pores through both lamina and then stabilizing the resultant scaffold by thermally cross-linking through autoclaving (121 °C for 30 min). The scaffold mechanically matches with the heart tissue in terms of stiffness and can guide the orientation of healthy cardiac cells. In addition, under electric field stimulation, heart muscle cells were encouraged to grow and contract with a directionally dependent electrical excitation threshold.
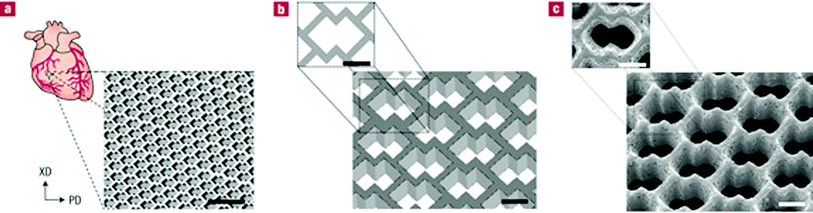 | ||
| Fig. 8 Accordion-like honeycomb scaffolds for myocardium. (a, b) Computer-designed image of the scaffolds. Scale bars: 1 mm (a) and 200 μm (b). (c) Scanning electron micrographs demonstrating the fidelity of excimer laser micro-ablation in rendering an accordion-like honeycomb design in PGS. Scale bar: 200 μm.115 Reprinted with permission of Nature Publishing Group. | ||
Stereo lithography is another popular method of fabrication by rapid-prototyping that uses light to polymerise, cross-link or harden a photosensitive material. A laser, which is guided by computer-assisted design software in the desired pattern, selectively polymerises material from a bath of photo-polymeric solution in a layer-by-layer process. The final product is washed to remove unreacted polymer and yield a 3D structure with specific micro architectures.140 This method has yet been applied to soft biodegradable elastomers. There is a great deal of potential in applying stereo lithography to photo-cross-linkable PGSA elastomers with rapid photo-activation and robust mechanical properties, as mentioned above.
Summary of scaffolding techniques of cross-linked elastomers
A number of fabrication techniques have been developed to produce PPS scaffolds for tissue engineering. In choosing a certain fabrication technique to produce scaffolds for different applications, it is necessary to weigh up the advantages and disadvantages of the techniques. Table 8 provides a comparison of the PPS fabrication techniques discussed in this review.| Techniques | Advantages | Disadvantages |
|---|---|---|
| Melt-based replicate moulding and lamination | Achievable high-feature resolution < 10 μm | Laminated 3D scaffold may lack require mechanical strength for load-bearing tissue |
| Solvent casting and particle-leaching | Control over porosity, pore size and high porosity | Poor quality control uses organic chemicals and residual solvents |
| Simple, low cost | ||
| Electrospinning | Fabricate nanofibrous porous scaffolds | Aligned texture may not always be desirable |
| Simple, low cost | Post-processing curing required | |
| Aligned structure | Low productivity | |
| Rapid-prototyping | Controlled quality, pore size and porosity | Expensive |
| Advanced in fabricating scaffolds with precise and complex structures |
8. Summary and future perspectives
Biodegradable, soft elastomers play an important role in the field of soft tissue engineering, and the studies of these elastomers for many applications have bloomed in the past two decades. In this article we have focused on the synthesis, materials properties of soft, degradable elastomers, and their scaffold fabrication techniques. Although these biomaterials are generally biocompatible and compliant, a number of issues associated with their degradation and synthesis remain to be addressed. First, very soft degradable elastomers can generate acidic degradation products, causing cytotoxicity, which hamper their application as a cell delivery vehicle. Second, the in vivo degradation rates of these materials are generally too fast to be applicable to tissue engineering of soft types. Last, the synthesis techniques of these materials (e.g. PPS) need to be improved to meet the highly demanded quality control in tissue engineering. Hence, future work is needed prior to applying these soft degradable elastomers to clinical applications, especially in the following two directions: (i) development of new synthesis techniques of degradable elastomers to achieve high reproducibility and expand fabrication capabilities; (ii) reduction of their degradation rates to meet the clinical requirement of specific tissue by developing new hybrid elastomeric biomaterials.References
- R. Langer and J. Vacanti, Science, 1993, 260, 920–926 CAS.
- L. E. Niklason, J. Gao, W. M. Abbott, K. K. Hirschi, S. Houser, R. Marini and R. Langer, Science, 1999, 284, 489–493 CrossRef CAS.
- J. Yang, A. R. Webb, S. J. Pickerill, G. Hageman and G. A. Ameer, Biomaterials, 2006, 27, 1889–1898 CrossRef CAS.
- Y. Wang, G. A. Ameer, B. J. Sheppard and R. Langer, Nat. Biotechnol., 2002, 20, 602–606 CrossRef CAS.
- C. J. Bettinger, Macromol. Biosci., 2011, 467–482 CrossRef CAS.
- J. Yang, A. Webb and G. Ameer, Adv. Mater., 2004, 16, 511–516 CrossRef CAS.
- C. Bettinger, E. Weinberg, K. Kulig, J. Vacanti, Y. Wang, J. Borenstein and R. Langer, Adv. Mater., 2006, 18, 165–169 CrossRef CAS.
- L.-Q. Zhang and R. Shi, in Current Topics in Elastomers Research, ed. A. K. Bhowmick, CRC Press, Boca Raton, 2008, ch. 8, pp. 222-247 Search PubMed.
- J. J. Stankus, J. Guan and W. R. Wagner, Encyclopedia of biomaterials and biomedical engineering, 2004, pp. 484–494 Search PubMed.
- Q. Z. Chen, A. Bismarck, U. Hansen, S. Junaid, M. Q. Tran, S. E. Harding, N. N. Ali and A. R. Boccaccini, Biomaterials, 2008, 29, 47–57 CrossRef CAS.
- A. N. Gent in Science and Technology of Rubber, ed. J. E. Mark, B. Erman and F. R. Eirich, Academic Press, Burlington, 3rd edn, 2005, pp. 1-27 Search PubMed.
- A. R. Webb, J. Yang and G. A. Ameer, Expert Opin. Biol. Ther., 2004, 4, 801–812 CrossRef CAS.
- R. J. Zdrahala and I. J. Zdrahala, Journal of Biomaterials Applications, 1999, 14, 67–90 CAS.
- M. Martina and D. W. Hutmacher, Polym. Int., 2007, 56, 145–157 CrossRef CAS.
- H. M. Nugent and E. R. Edelman, Circ. Res., 2003, 92, 1068–1078 CrossRef CAS.
- http://www.hopkinsmedicine.org/news/media/releases/rubber_gloves_born___and_now_banished___at_johns_hopkins, accessed on 22 March 2011.
- R. Yoda, J. Biomater. Sci., Polym. Ed., 1998, 9, 561–626 CrossRef CAS.
- Q. Z. Chen, S. E. Harding, N. N. Ali, A. R. Lyon and A. R. Boccaccini, Mater. Sci. Eng., R, 2008, 59, 1–37 CrossRef.
- G.-Q. Chen and Q. Wu, Biomaterials, 2005, 26, 6565–6578 CrossRef CAS.
- D. P. Martin and S. F. Williams, Biochem. Eng. J., 2003, 16, 97–105 CrossRef CAS.
- R. Shi, D. F. Chen, Q. Y. Liu, Y. Wu, X. C. Xu, L. Q. Zhang and W. Tian, Int. J. Mol. Sci., 2009, 10, 4223–4256 CrossRef CAS.
- A. Lendlein and R. Langer, Science, 2002, 296, 1673–1676 CrossRef.
- J. P. Bruggeman, B. J. de Bruin, C. J. Bettinger and R. Langer, Biomaterials, 2008, 29, 4726–4735 CrossRef CAS.
- J. P. Bruggeman, C. J. Bettinger, C. L. Nijst, D. S. Kohane and R. Langer, Adv. Mater., 2008, 20, 1922–1927 CrossRef CAS.
- C. L. E. Nijst, J. P. Bruggeman, J. M. Karp, L. Ferreira, A. Zumbuehl, C. J. Bettinger and R. Langer, Biomacromolecules, 2007, 8, 3067–3073 CrossRef CAS.
- J. Yang, Y. Zhang, S. Gautam, L. Liu, J. Dey, W. Chen, R. P. Mason, C. A. Serrano, K. A. Schug and L. Tang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10086–10091 CrossRef CAS.
- L. J. Lei, T. Ding, R. Shi, Q. Y. Liu, L. Q. Zhang, D. F. Chen and W. Tian, Polym. Degrad. Stab., 2007, 92, 389–396 CrossRef CAS.
- R. T. Tran, P. Thevenot, D. Gyawali, J.-C. Chiao, L. Tang and J. Yang, Soft Matter, 2010, 6, 2449–2461 RSC.
- D. Gyawali, P. Nair, Y. Zhang, R. T. Tran, C. Zhang, M. Samchukov, M. Makarov, H. K. W. Kim and J. A. Yang, Biomaterials, 2010, 31, 9092–9105 CrossRef CAS.
- H. M. Younes, E. Bravo-Grimaldo and B. G. Amsden, Biomaterials, 2004, 25, 5261–5269 CrossRef CAS.
- B. G. Amsden, G. Misra, F. Gu and H. M. Younes, Biomacromolecules, 2004, 5, 2479–2486 CrossRef CAS.
- C. J. Bettinger, J. P. Bruggeman, J. T. Borenstein and R. Langer, J. Biomed. Mater. Res., Part A, 2009, 91A, 1077–1088 CrossRef CAS.
- Y. Dausse, L. Grossin, G. Miralles, S. Pelletier, D. Mainard, P. Hubert, D. Baptiste, P. Gillet, E. Dellacherie, P. Netter and E. Payan, Osteoarthritis Cartilage, 2003, 11, 16–28 CrossRef CAS.
- A. D. Augst, H. J. Kong and D. J. Mooney, Macromol. Biosci., 2006, 6, 623–633 CrossRef CAS.
- B. L. Seal, T. C. Otero and A. Panitch, Mater. Sci. Eng., R, 2001, 34, 147–230 CrossRef.
- C. N. Grover, R. E. Cameron and S. M. Best, J. Mech. Behav. Biomed. Mater., 2012, 10, 62–74 CrossRef.
- T. B. William, N. Karthik and L. C. Elliot, in Encyclopedia of Biomaterials and Biomedical Engineering, ed. G. E. Wnek and G. L. Bowlin, Informa Healthcare, New York, 2008, ch. 85, pp. 891–896 Search PubMed.
- D. L. Nettles, A. Chilkoti and L. A. Setton, Adv. Drug Delivery Rev., 2010, 62, 1479–1485 CrossRef CAS.
- R. Dandu, A. Von Cresce, R. Briber, P. Dowell, J. Cappello and H. Ghandehari, Polymer, 2009, 50, 366–374 CrossRef CAS.
- M. L. Siriwardane and B. J. Pfister, Ieee 36th Annual Northeast Bioengineering Conference, 2010 Search PubMed.
- C. Du, F. Z. Cui, X. D. Zhu and K. de Groot, J. Biomed. Mater. Res., 1999, 44, 407–415 CrossRef CAS.
- M. Sittinger, D. W. Hutmacher and M. V. Risbud, Curr. Opin. Biotechnol., 2004, 15, 411–418 CrossRef CAS.
- J. Raghunath, J. Rollo, K. M. Sales, P. E. Butler and A. M. Seifalian, Biotechnol. Appl. Biochem., 2007, 46, 73–84 CrossRef CAS.
- B. Balakrishnan, M. Mohanty, P. R. Umashankar and A. Jayakrishnan, Biomaterials, 2005, 26, 6335–6342 CrossRef CAS.
- N. Hunt and L. Grover, Biotechnol. Lett., 2010, 32, 733–742 CrossRef CAS.
- J. Yu, Y. Gu, K. T. Du, S. Mihardja, R. E. Sievers and R. J. Lee, Biomaterials, 2009, 30, 751–756 CrossRef CAS.
- W.-H. Zimmermann, I. Melnychenko and T. Eschenhagen, Biomaterials, 2004, 25, 1639–1647 CrossRef CAS.
- Y. Miura, T. Akimoto and K. Yagi, Ann. N. Y. Acad. Sci., 1988, 542, 521–532 CrossRef CAS.
- T. Hashimoto, Y. Suzuki, M. Kitada, K. Kataoka, S. Wu, K. Suzuki, K. Endo, Y. Nishimura and C. Ide, Exp. Brain Res., 2002, 146, 356–368 CrossRef CAS.
- C. A. Vacanti, L. J. Bonassar, and J. P. Vacanti, inPrinciples of tissue engineering, ed. R. P. Lanza., R. Langer. and J. P. Vacanti, Academic Press, California, 2nd edn, 2000, pp. 671–682 Search PubMed.
- W. Callister, Materials science and engineering: an introduction, John Wiley and Sons, New York, 7th edn, 2007 Search PubMed.
- D. G. Barrett and M. N. Yousaf, Molecules, 2009, 14, 4022–4050 CrossRef CAS.
- M. R. Toub, Elastomerics, 1987, 119, 20 CAS.
- J. Curtis and A. Colas, in Biomaterials science: an introduction to materials in medicine, ed. B. D. Ratner, A. S. Hoffman, F. J. Schoen and J. E. Lemons, Academic Press, San Diego, 2nd edn, 2005, pp. 697–707 Search PubMed.
- T. Freier, Adv. Polym. Sci., 2006, 203, 1–61 CrossRef CAS.
- H. B. Lee, H. Quach, D. B. Berry and W. J. Stith, ACS Symp. Ser., 1984, 256, 99–109 CrossRef CAS.
- P. R. Shewry, A. S. Tatham and A. J. Bailey, Elastomeric Proteins: Structures, Biomechanical Properties and Biological Roles, Cambridge University Press, Cambridge, 2003 Search PubMed.
- P. Bruin, G. J. Veenstra, A. J. Nijenhuis and A. J. Pennings, Makromol. Chem. Rapid Commun., 1988, 9, 589–594 CrossRef CAS.
- O. Duvernoy, T. Malm, J. Ramstrom and S. Bowald, Thorac. Cardiovasc. Surg., 1995, 43, 271–274 CrossRef CAS.
- A. Kalangos and B. Faidutti, J. Thorac. Cardiovasc. Surg., 1996, 112, 1401–1402 CrossRef CAS.
- V. Chiono, S. Sartori, A. Rechichi, C. Tonda-Turo, G. Vozzi, F. Vozzi, M. D'Acunto, C. Salvadori, F. Dini, G. Barsotti, F. Carlucci, S. Burchielli, S. Nicolino, C. Audisio, I. Perroteau, P. Giusti and G. Ciardelli, Macromol. Biosci., 2011, 11, 245–256 CrossRef CAS.
- X. H. Wang, T. K. Cui, Y. N. Yan and R. J. Zhang, J. Bioact. Compat. Polym., 2009, 24, 109–127 CrossRef CAS.
- L. A. Pfister, M. Papaloizos, H. P. Merkle and B. Gander, J. Peripher. Nerv. Syst., 2007, 12, 65–82 CrossRef CAS.
- G. Soldani, G. Varelli, A. Minnocci and P. Dario, Biomaterials, 1998, 19, 1919–1924 CrossRef CAS.
- M. Borkenhagen, R. C. Stoll, P. Neuenschwander, U. W. Suter and P. Aebischer, Biomaterials, 1998, 19, 2155–2165 CrossRef CAS.
- A. Hazari, M. Wiberg, G. Johansson-Ruden, C. Green and G. Terenghi, Br. J. Plast. Surg., 1999, 52, 653–657 CrossRef CAS.
- A. Hazari, G. Johansson-Ruden, K. Junemo-Bostrom, C. Ljungberg, G. Terenghi, C. Green and M. Wiberg, J. Hand Surg., Br. Eur. Vol., 1999, 24, 291–295 CrossRef CAS.
- C. Ljungberg, G. Johansson-Ruden, K. J. Bostrom, L. Novikov and M. Wiberg, Microsurgery, 1999, 19, 259–264 CrossRef CAS.
- L. Soletti, A. Nieponice, Y. Hong, S. H. Ye, J. J. Stankus, W. R. Wagner and D. A. Vorp, J. Biomed. Mater. Res., Part A, 2011, 96A, 436–448 CrossRef CAS.
- G. Winkeltau, K. H. Treutner, P. Bertram, M. M. Lerch and V. Schumpelick, Langenbecks Arch. Chir., 1989, 374, 32–38 CrossRef CAS.
- C. Schlickewei, S. Verrier, S. Lippross, S. Pearce, M. Alini and S. Gogolewski, Macromol. Symp., 2007, 253, 162–171 CrossRef CAS.
- C. M. Hill, Y. H. An, Q. K. Kang, L. A. Hartsock, S. Gogolewski and K. Gorna, Macromol. Symp., 2007, 253, 94–97 CrossRef CAS.
- S. Gogolewski and K. Gorna, J. Biomed. Mater. Res., Part A, 2007, 80A, 94–101 CrossRef CAS.
- S. Gogolewski, K. Gorna and A. S. Turner, J. Biomed. Mater. Res., Part A, 2006, 77A, 802–810 CrossRef CAS.
- N. Galego, C. Rozsa, R. Sanchez, J. Fung, A. Vazquez and J. S. Tomas, Polym. Test., 2000, 19, 485–492 CrossRef CAS.
- N. R. Boeree, J. Dove, J. J. Cooper, J. Knowles and G. W. Hastings, Biomaterials, 1993, 14, 793–796 CrossRef CAS.
- D. Eglin, S. Grad, S. Gogolewski and M. Alini, J. Biomed. Mater. Res., Part A, 2010, 92A, 393–408 CrossRef CAS.
- J. H. deGroot, F. M. Zijlstra, H. W. Kuipers, A. J. Pennings, J. Klompmaker, R. P. H. Veth and H. W. B. Jansen, Biomaterials, 1997, 18, 613–622 CrossRef CAS.
- J. Klompmaker, R. P. H. Veth, H. W. B. Jansen, H. K. L. Nielsen, J. H. deGroot, A. J. Pennings and R. Kuijer, Biomaterials, 1996, 17, 1685–1691 CrossRef CAS.
- J. Klompmaker, R. P. H. Veth, H. W. B. Jansen, H. K. L. Nielsen, J. H. deGroot and A. J. Pennings, Biomaterials, 1996, 17, 1169–1175 CrossRef CAS.
- J. H. deGroot, R. deVrijer, A. J. Pennings, J. Klompmaker, R. P. H. Veth and H. W. B. Jansen, Biomaterials, 1996, 17, 163–173 CrossRef CAS.
- J. Klompmaker, H. W. B. Jansen, R. P. H. Veth, H. K. L. Nielsen, J. H. Degroot, A. J. Pennings and R. Kuijer, J. Orthop. Res., 1992, 10, 359–370 CrossRef CAS.
- J. Klompmaker, H. W. B. Jansen, R. P. H. Veth, J. H. Degroot, A. J. Nijenhuis and A. J. Pennings, Biomaterials, 1991, 12, 810–816 CrossRef CAS.
- E. Liljensten, K. Gisselfalt, B. Edberg, H. Bertilsson, P. Flodin, A. Nilsson, A. Lindahl and L. Peterson, J. Mater. Sci.: Mater. Med., 2002, 13, 351–359 CrossRef CAS.
- K. Gisselfalt, B. Edberg and P. Flodin, Biomacromolecules, 2002, 3, 951 CrossRef CAS.
- O. Jeon, S. H. Lee, S. H. Kim, Y. M. Lee and Y. H. Kim, Macromolecules, 2003, 36, 5585–5592 CrossRef CAS.
- D. Cohn and A. F. Salomon, Biomaterials, 2005, 26, 2297–2305 CrossRef CAS.
- B. Asplund, J. Sperens, T. Mathisen and J. Hilborn, J. Biomater. Sci., Polym. Ed., 2006, 17, 615–630 CrossRef CAS.
- L. S. Wang, Z. P. Zhang, H. C. Chen, S. L. Zhang and C. D. Xiong, J. Polym. Res., 2010, 17, 77–82 CrossRef CAS.
- S.-H. Lee, B.-S. Kim, S. H. Kim, S. W. Choi, S. I. Jeong, I. K. Kwon, S. W. Kang, J. Nikolovski, D. J. Mooney, Y.-K. Han and Y. H. Kim, J. Biomed. Mater. Res., 2003, 66A, 29–37 CrossRef CAS.
- G. Matsumura, S. Miyagawa-Tomita, T. Shin'oka, Y. Ikada and H. Kurosawa, Circulation, 2003, 108, 1729–1734 CrossRef.
- G. Matsumura, N. Hibino, Y. Ikada, H. Kurosawa and T. Shin'oka, Biomaterials, 2003, 24, 2303–2308 CrossRef CAS.
- G. G. Pitt, M. M. Gratzl, G. L. Kimmel, J. Surles and A. Sohindler, Biomaterials, 1981, 2, 215–220 CrossRef.
- R. F. Storey and T. P. Hickey, Polymer, 1994, 35, 830–838 CrossRef CAS.
- D. J. Stuckey, H. Ishii, Q. Z. Chen, A. R. Boccaccini, U. Hansen, C. A. Carr, J. A. Roether, H. Jawad, D. J. Tyler, N. N. Ali, K. Clarke and S. E. Harding, Tissue Eng. A, 2010, 16, 3395–3402 CrossRef CAS.
- B. Amsden, Soft Matter, 2007, 3, 1335–1348 RSC.
- C. J. Bettinger, J. T. Borenstein and R. S. Langer, Nanoscale Materials Science in Biology and Medicine, 2005, 845, 25–30 CAS.
- ed. K. Gorna and S. Gogolewski, Novel biodegradable polyurethane for biomedical applications, ASTM International, West Conshohochen, PA, USA, 2000 Search PubMed.
- J. Gosline, M. Lillie, E. Carrington, P. Guerette, C. Ortlepp and K. Savage, Philos. Trans. R. Soc. London, Ser. B, 2002, 357, 121–132 CrossRef CAS.
- J. C. Isenburg, D. T. Simionescu and N. R. Vyavahare, Biomaterials, 2004, 25, 3293–3302 CrossRef CAS.
- Y. D. Wang, Y. M. Kim and R. Langer, J. Biomed. Mater. Res., 2003, 66A, 192–197 CrossRef CAS.
- C. A. Sundback, J. Y. Shyu, Y. D. Wang, W. C. Faquin, R. S. Langer, J. P. Vacanti and T. A. Hadlock, Biomaterials, 2005, 26, 5454–5464 CrossRef CAS.
- D. Motlagh, J. Yang, K. Y. Lui, A. R. Webb and G. A. Ameer, Biomaterials, 2006, 27, 4315–4324 CrossRef CAS.
- C. J. Bettinger, B. Orrick, A. Misra, R. Langer and J. T. Borenstein, Biomaterials, 2006, 27, 2558–2565 CrossRef CAS.
- J. M. Kemppainen and S. J. Hollister, J Biomed Mater Res A, 2010, 94, 9–18 CrossRef.
- G. Liu, B. Hinch and A. D. Beavis, J. Biol. Chem., 1996, 271, 25338–25344 CrossRef CAS.
- A. V. Grego and G. Mingrone, Clin. Nutr., 1995, 14, 143–148 CrossRef CAS.
- J. Tamada and R. Langer, J. Biomater. Sci., Polym. Ed., 1992, 3, 315–353 CrossRef CAS.
- Q. Z. Chen, H. Ishii, G. A. Thouas, A. R. Lyon, J. S. Wright, J. J. Blaker, W. Chrzanowski, A. R. Boccaccini, N. N. Ali, J. C. Knowles and S. E. Harding, Biomaterials, 2010, 31, 3885–3893 CrossRef CAS.
- ed. W. Stockmayer,Advancing fronts in chemistry, Reinhold, New York, 1945 Search PubMed.
- J. Gao, P. M. Crapo and Y. D. Wang, Tissue Eng., 2006, 12, 917–925 CrossRef CAS.
- F. Christina, M. R. Kaazempur-Mofrad, B. Jeffrey, J. P. Vacanti, R. Langer and Y. Wang, Tissue Eng., 2005, 11, 302–309 CrossRef.
- C. A. Sundback, J. Y. Shyu, Y. Wang, W. C. Faquin, R. S. Langer, J. P. Vacanti and T. A. Hadlock, Biomaterials, 2005, 26, 5454–5464 CrossRef CAS.
- I. Pomerantseva, N. Krebs, A. Hart, C. M. Neville, A. Y. Huang and C. A. Sundback, J. Biomed. Mater. Res., Part A, 2009, 91A, 1038–1047 CrossRef CAS.
- G. C. Engelmayr, M. Cheng, C. J. Bettinger, J. T. Borenstein, R. Langer and L. E. Freed, Nat. Mater., 2008, 7, 1003–1010 CrossRef CAS.
- Q. Z. Chen, S. L. Liang and G. A. Thouas, Soft Matter, 2011, 7, 6484–6492 RSC.
- F. Yi and D. A. Lavan, Macromol. Biosci., 2008, 8, 803–806 CrossRef CAS.
- Q. Z. Chen, L. Y. Jin, W. D. Cook, D. Mohn, E. L. Lagerqvist, D. A. Elliott, J. M. Haynes, N. Boyd, W. J. Stark, C. W. Pouton, E. G. Stanley and A. G. Elefanty, Soft Matter, 2010, 6, 4715–4726 RSC.
- S. L. Liang, W. D. Cook, G. A. Thouas and Q. Z. Chen, Biomaterials, 2010, 31, 8516–8529 CrossRef CAS.
- S.-L. Liang, X.-Y. Yang, X.-Y. Fang, W. D. Cook, G. A. Thouas and Q. Z. Chen, Biomaterials, 2011, 32, 8486–8496 CrossRef CAS.
- A. Göpferich, Biomaterials, 1996, 17, 103–114 CrossRef.
- Z. J. Sun, C. Chen, M. Z. Sun, C.-H. Ai, X. L. Lu, Y. F. Zheng, B. F. Yang and D. L. Dong, Biomaterials, 2009, 30, 5209–5214 CrossRef CAS.
- J. Gao, A. E. Ensley, R. M. Nerem and Y. Wang, J. Biomed. Mater. Res., Part A, 2007, 83A, 1070–1075 CrossRef CAS.
- S. Kakar, and T. A. Einhorn, in Skeletal trauma: basic science, management, and reconstruction, ed. B. D. Browner, A. M. Levine, J. B. Jupiter, P. G. Trafton and C. Krettek, W B Saunders Co, Elsevier, Philadelphia, USA, 4th edn 2008, vol. 1, ch. 2, pp. 33–50 Search PubMed.
- I. Jaafar, M. Ammar, S. Jedlicka, R. Pearson and J. Coulter, J. Mater. Sci., 2010, 45, 2525–2529 CrossRef CAS.
- D. G. Barrett, W. Luo and M. N. Yousaf, Polym. Chem., 2010, 1, 296–302 RSC.
- Q. Z. Chen, X. Y. Yang, Y. Li and G. A. Thouas, RSC Adv., 2012, 2, 4125–4134 RSC.
- J. L. Ifkovits, R. F. Padera and J. A. Burdick, Biomedical Materials, 2008, 3 Search PubMed.
- Q. Liu, J. Wu, T. Tan, L. Zhang, D. Chen and W. Tian, Polym. Degrad. Stab., 2009, 94, 1427–1435 CrossRef CAS.
- R. Roeder, J. Wolfe, N. Lianakis, T. Hinson, L. A. Geddes and J. Obermiller, J. Biomed. Mater. Res., 1999, 47, 65–70 CrossRef CAS.
- C. J. Bettinger, J. P. Bruggeman, J. T. Borenstein and R. S. Langer, Biomaterials, 2008, 29, 2315–2325 CrossRef CAS.
- K. D. Daniel, G. Y. Kim, C. C. Vassiliou, F. Jalali-Yazdi, R. Langer and M. J. Cima, Lab Chip, 2007, 7, 1288–1293 RSC.
- A. P. Pêgo, D. W. Grijpma and J. Feijen, Polymer, 2003, 44, 6495–6504 CrossRef.
- E. Bat, J. A. Plantinga, M. C. Harmsen, M. J. A. van Luyn, Z. Zhang, D. W. Grijpma and J. Feijen, Biomacromolecules, 2008, 9, 3208–3215 CrossRef CAS.
- E. Bat, B. H. M. Kothman, G. A. Higuera, C. A. van Blitterswijk, J. Feijen and D. W. Grijpma, Biomaterials, 2010, 31, 8696–8705 CrossRef CAS.
- R. Chapanian, M. Y. Tse, S. C. Pang and B. G. Amsden, Biomaterials, 2009, 30, 295–306 CrossRef CAS.
- D. W. Grijpma, Q. Hou and J. Feijen, Biomaterials, 2005, 26, 2795–2802 CrossRef CAS.
- D. W. Hutmacher, Biomaterials, 2000, 21, 2529–2543 CrossRef CAS.
- M. S. Widmer and A. G. Mikos,in Frontiers in tissue engineering, ed. W. P. Charles, Jr, G. M. Antonios, V. M. Larry and R. S. Langer, Pergamon, Oxford, 1998, pp. 107–120 Search PubMed.
- M. B. Murphy and A. G. Mikos,in Principles of tissue engineering, ed. L. Robert, L. Robert and V. Joseph, Academic Press, Burlington, 3rd edn, 2007, pp. 309–321 Search PubMed.
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413–3431 CrossRef CAS.
- J. T. Borenstein, H. Terai, K. R. King, E. J. Weinberg, M. R. Kaazempur-Mofrad and J. P. Vacanti, Biomed. Microdevices, 2002, 4, 167–175 CrossRef CAS.
- J. L. Ifkovits, J. J. Devlin, G. Eng, T. P. Martens, G. Vunjak-Novakovic and J. A. Burdick, ACS Appl. Mater. Interfaces, 2009, 1, 1878–1886 CAS.
- K. F. Leong, C. M. Cheah and C. K. Chua, Biomaterials, 2003, 24, 2363–2378 CrossRef CAS.
- J. M. Kemppainen and S. J. Hollister, J. Biomed. Mater. Res., Part A, 2010, 94A, 9–18 CrossRef CAS.
- I. Zein, D. W. Hutmacher, K. C. Tan and S. H. Teoh, Biomaterials, 2002, 23, 1169–1185 CrossRef CAS.
- Q. Z. Chen, Bubble Sci., Eng. Technol., 2011, 3, 34–47 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |