Reversible encapsulation of a nitrate guest via hydrogen-bonded self-assembled capsule formation by a flexible tripodal receptor in polar solvent through dynamic self-assembly†
Ashutosh S.
Singh
and
Shih-Sheng
Sun
*
Institute of Chemistry, Academia Sinica, 115 Nankang, Taipei, Taiwan, Republic of China. E-mail: sssun@chem.sinica.edu.tw; Fax: +011-886-2-27831237; Tel: +011-886-2-27898596
First published on 3rd August 2012
Abstract
Reversible formation of a self-assembled capsule by dimerization of two units of discrete and highly flexible tripodal receptors through multiple hydrogen bonds in polar solvents is reported. The structural scaffold responsible for the capsule formation was prepared by in situ nitration reaction of the N-terminal aromatic ring of the tripodal receptor. The studies show that the nitro group at the ortho position of the N-terminal aromatic ring is mandatory for hydrogen bonded capsule formation and the mode of hydrogen bonding depends upon the electronic nature of the N-terminal aromatic ring. The present approach has been utilized for reversible encapsulation of a weakly coordinating and hydrophilic nitrate anion through a dynamic self-assembly process.
Introduction
Encapsulation of guest molecules from surrounding media into a confined space created by assembly of complementary building block(s) has shown numerous applications in various facets of chemistry.1–3 The topology of the building blocks and the nature of their binding sites for the assembly process determine the final structures of self-assembled supramolecular species and their subsequent practical utilities. Molecular building blocks with cyclic arrays, such as calixarenes, cucurbitals, cyclodextrins, porphyrins, etc., possess a well-defined structural conformation (cyclic receptors), but a limited degree of freedom and are thus feasible for assembly processes in solution to entrap suitable guests by selective recognition. In contrast, design of acyclic receptors with highly flexible conformation and a lack of pre-organized cavity for guest encapsulation is very interesting, yet a very challenging task. The ability to encapsulate a guest molecule by an artificial receptor through non-covalent interactions would render these supramolecular assemblies suitable for applications such as storage, transport and delivery processes. Nevertheless, the nature of the binding sites plays a crucial role to accomplish these phenomena and the hydrogen bond provides the simplest tool to recover the valuable host depending upon the surrounding environment.Hydrogen bonding is one of the most common intermolecular interactions for constructing self-assembly supramolecular hosts4 and subsequent guest recognition.5 Hydrogen-bonded assemblies are usually stable in less polar organic solvents, but prone to dissociate in polar or protic solvents. With few exceptions,6,7 the formation of self-assembly capsules by purely hydrogen bonding interactions and subsequent anionic guest recognition is mainly explored in non-polar solvents4 and dissociates in polar solvents.8
The large hydration energy and weak basicity of nitrate anions results in low affinity of nitrate anions for hydrogen bonding interactions. Nitrate anion recognition has been mostly studied in less-polar solvents to favor the hydrogen bonding interactions between nitrate anion synthetic hosts and, very commonly, poor binding affinity has been reported in polar solvents. Recognition of an anionic substrate in the biological process occurs in a specific hydrophobic recognition environment with complimentary induced-fit conformation and, thus, free energy lost on dehydration of the anion upon binding is compensated by the interaction of the anion with available binding sites.9 The lack of such a unique binding nature in a typical artificial system renders recognition of weakly coordinating anions, like nitrate anions, a highly challenging task. Moreover, for practical applications with fidelity and feasibility, stimuli-responsive control over the hydrogen bond system is mandatory, especially for transport and delivery processes by artificial receptors for suitable guest molecules.
In general, an acyclic receptor without a specific complementary structure favours interaction with basic anions via hydrogen bonding interactions or, in some extreme cases, a neat proton transfer process.10 Herein, we report the encapsulation of a weakly coordinating nitrate anion by a series of highly flexible, acyclic tripodal receptors through amide N–H hydrogen bonds (Fig. 2). The amide N–H bond is well explored in the literature to form hydrogen bonding interactions with various anions in both natural11 and artificial systems.12 Encapsulation of a nitrate anion can be reversibly switched between the nitrate-bound discrete nitrate complex and the self-assembled capsule, where the reversibility is modulated by a polarity change of the solvent media. The interconversion between two or more discrete and structurally well defined assemblies is known to be regulated by external stimuli, such as solvents13a,c,d,g, concentration13a,b,e,f,h and temperature.13f,h,i Due to the labile nature of the metal–ligand coordination bond and the possible existence of solvated ion pairs,13g interconversion between two metal coordination species is known through dynamic self-assembly by varying solvent polarity.13a,b,d,e Apart from π–π interactions, however, several other non-covalent interactions are possible in purely hydrogen-bonded organic supramolecular assemblies, which makes it difficult to achieve the switching between two assembled species in competitive polar solvents. Interestingly, the self-assembled capsule reported here is stabilized by seaming of hydrogen bonds on its curvature in polar solvents, such as acetone-d6 and CD3NO2. The mode of binding in self-assembled capsule formation depends upon the electronic nature of the N-terminal aromatic ring of tripodal receptors. The nitrate anion encapsulation and self-assembled capsule formation in polar solvents have been characterized in detail through 1H NMR titrations, high resolution ESI mass spectra and the crystal structure of the self-assembled dimer (52).
Results and discussion
Design principle and synthesis of tripodal receptors
The design principle of these receptors was motivated from our ongoing research interest in tripodal receptors for anion recognition.14 In our previous report, we have shown that halide functionality is dependent on capsule formation and reversible binding of perchlorate anion, both in the solid state and in solution. Nevertheless, no selectivity was achieved because the driving force for perchlorate anion encapsulation was only electrostatic interactions. Later, we have shown that incorporating amide functionality at each branch of the tripodal framework resulted in receptors that reversibly bind nitrate anions via self-assembled capsule formation by decreasing the polarity of the solvent media (Fig. 1).15 A nitro group at the ortho position with respect to the amide functionality (on the N-terminal aromatic ring) on each tripodal branch was mandatory for the formation of a self-assembled dimeric capsule with a pseudo trigonal bipyramidal topology. To study in depth and generalize the present results, a series of tripodal receptors (1–7, Fig. 2) were synthesized and fully characterized. An electron donating group at the para-position with respect to the amide functionality was designed for in situ electrophilic aromatic substitution at the N-terminal aromatic ring upon treatment with nitric acid and subsequent solvent polarity-modulated capsule formation (vide infra). No nitro-substitution occurs with either unsubstituted derivative (receptor 7), the electron-donating group at the ortho and meta positions (receptors 2 and 3, respectively), or the electron-withdrawing group at the para-position (receptor 4).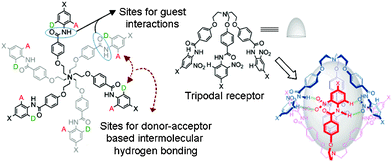 | ||
| Fig. 1 The design principle for a hydrogen bonded self-assembled capsule. | ||
Tripodal receptors 1–7 were synthesized in three steps starting from tris-(chloroethyl)amine hydrochloride14a (Scheme 1) in 70–95% overall yields.15 Receptor 1′ was synthesized by dropwise addition of 20% HNO3 to a suspension of receptor 1 in acetonitrile at room temperature (25 °C) and stirring the solution mixture for 10–15 min when an orange-colored precipitate appears. The precipitate was filtered out and washed with 20% K2CO3 solution and extracted with ethyl acetate. After removing the solvent under reduced pressure, 1′ was obtained as a shiny yellow solid. Unlike receptor 1′,15 receptor 6′ was synthesized by dropwise addition of 20% HNO3 in acetonitrile to a suspension of receptor 6 in acetonitrile and the resulting mixture solution was heated at 80 °C for 30–45 min. After cooling, the precipitate was filtered and washed with 20% K2CO3 and extracted with ethyl acetate. After removing ethyl acetate at reduced pressure, receptor 6′ was obtained as shiny yellow needles in 95% yield. Receptor 5 has been used as a model receptor for capsule formation and also for comparative studies with respect to receptors 1′ and 6′.15
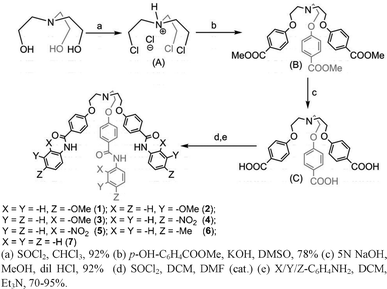 | ||
| Scheme 1 Synthetic procedures for receptors 1–7. | ||
Synthesis of nitrate-encapsulated complexes and crystal structure analysis
The nitrate-encapsulated complexes 2a–5a and 7a were prepared by slow addition of dilute nitric acid in MeOH to a suspension of corresponding receptors 2–5 and 7 in CHCl3 and isolated with 65–87% yields. The purities and identities for all of the complexes were confirmed by NMR spectroscopy and high-resolution electrospray mass spectrometry (HRESMS). Unlike the straightforward preparation of complexes 2a–5a and 7a, the formation of nitrate-encapsulated complexes from receptors 1 and 6 were found to be solvent-dependent (Fig. 3). The preparation of nitrate-encapsulated complexes 1a and 1b were reported earlier.15 As similar observation was noted when nitric acid in aqueous methanol was added to a suspension of receptor 6 in chloroform and the solution color changed from off-white to creamy yellow after 1 h with solid precipitation. The precipitate was filtered off and dried in the open air. The 1H NMR spectrum measured in a DMSO-d6 solution (see supplementary information†) showed two well-separated amide N–H peaks at 10.6 ppm (the major peak) and 10.0 ppm (a very tiny peak), which implies the formation of two different species. The experiment was repeated in pure acetonitrile at room temperature (25 °C) for 24 h and in this case also two well-separated peaks appeared for the amide N–H peak, with the peak integrated area of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The ratio was in contrast to what was observed for receptor 1, which showed a 1:2 ratio between the peaks at 10.6 and 10.0 ppm.15
:1 ratio. The ratio was in contrast to what was observed for receptor 1, which showed a 1:2 ratio between the peaks at 10.6 and 10.0 ppm.15
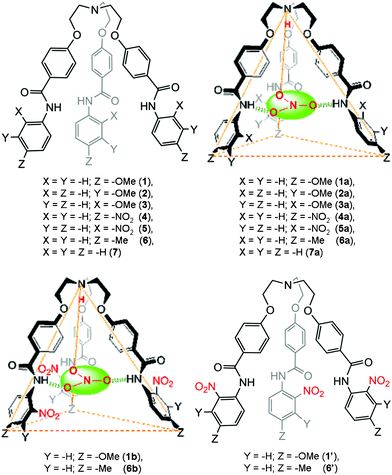 | ||
| Fig. 2 A schematic representation of receptors designed for studies (top left and bottom right) and a topographical representation of the nitrate-encapsulated complexes (top right and bottom left). | ||
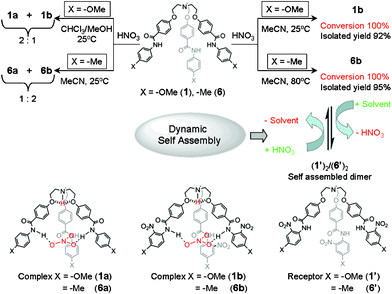 | ||
| Fig. 3 A schematic representation of the solvent-dependent formation of the nitrate-encapsulated complex and reversible binding of the nitrate anion through interconversion between a nitrate-encapsulated complex and a self-assembled capsule. | ||
The analytically pure nitrate encapsulated complex 6a was obtained as a milky white precipitate by addition of nitric acid in MeOH to a suspension of receptor 6 in MeOH, whereas the deep orange-colored 6b was obtained as the precipitate by addition of nitric acid in acetonitrile to a suspension of receptor 6 in acetonitrile followed by heating at 80 °C for 30–45 min. In contrast to the facile synthesis of 1b at room temperature, the nitrate-encapsulated complex 6b was prepared from an acetonitrile solution of receptor 6 by heating at 80 °C for 30 min because of the poor electron donating nature of the methyl substituent that resulted in a less activated aromatic C–H bond for nitration. 1H NMR spectra of 6a and 6b recorded in DMSO-d6 showed amide N–H peaks at 10.1 and 10.6 ppm for 6a and 6b, respectively. Their identities were also confirmed by HRESI mass spectra. HRESI mass spectrometry has been successfully utilized for the detection of non-covalently bonded supramolecular species in solution.16 Negative mode HRESMS of acetonitrile solution showed the expected peak cluster at m/z 838.3453, corresponding to the nitrate-encapsulated species [6a − H]− (calcd. m/z 838.3452) and another peak cluster at m/z 973.3002, which corresponds to the nitrate-encapsulated nitro-substituted species [6b − H]− (calcd. m/z 973.3004).
The single crystals of 6a suitable for X-ray diffraction were grown from a DMSO-d6 solution at room temperature over a period of four weeks. Fig. 4 shows the crystal structure of complex 6a. The crystal structure of 6a is C3 symmetric and isostructural to 1a, as reported previously.15 The centrally bridged N-atom is protonated, as evidenced in the 1H NMR spectrum of nitrate complex 6a, where the aliphatic protons close to the centrally bridged N-atom show a 0.80 ppm downfield shift in comparison to the free receptor. The O-atom of each aliphatic branch of the receptor is hydrogen-bonded to the proton attached to the centrally bridged N-atom (dN–H⋯O 2.213 Å, θN–H⋯O 114.5°). The end of each branch lies at the apex of the isosceles showing a perfect trigonal pyramidal shape of a discrete nitrate anion encapsulated complex. One ortho hydrogen atom of the C-terminal aromatic ring at each branches shows C–H⋯π interaction with the C-terminal aromatic ring of the neighboring branch (dC–H⋯π 3.253 Å and θC–H⋯π 178.8°) in a tilted edge-to-face fashion.17 The amide N–H bonds of each branch protrude inside towards the center in plane. A nitrate anion is bound inside the cavity by strong H-bonds between the O-atoms of the nitrate anion and three amide N–Hs (dN–H⋯O 2.234 Å, θN–H⋯O 159.8°, Fig. 4). In the discrete nitrate complex, the distance between the bridged N-atom and the center of the encapsulated nitrate anion is 7.90 Å (θN–H⋯N 180°).
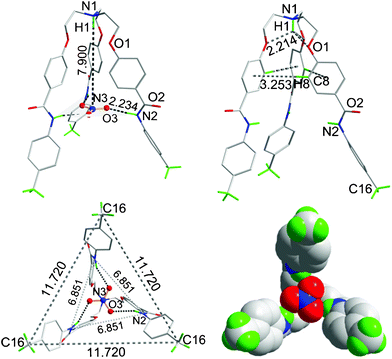 | ||
| Fig. 4 The crystal structure of 6a showing a discrete nitrate anion encapsulated complex (top, left); formation of the cone-shape conformation through N–H⋯O hydrogen bonding and C–H⋯π interactions (top, right), respectively; the end of each branch lies at the apex of an equilateral triangle (bottom, left); and a space-filled diagram of the nitrate anion-encapsulated complex (bottom, right). All H-atoms have been omitted for clarity except for the methyl C–Hs and those (shown in green color) interacting with the nitrate anion. All bond distances are given in Å. | ||
Nitrate anion binding in solution
Binding of nitrate anions in solution by these tripodal receptors was evaluated through 1H NMR titrations by addition of nitrate anions as a tetrabutylammonium salt to an acetone-d6 solution of the perchlorate complexes 1c–7c.18 The perchlorate complex was selected for solution binding studies because of its monobasic nature and good solubility in common deuterated solvents. The weak basicity, large ionic radii and lipophilic nature of the perchlorate anion also ensures minimal interference for nitrate binding in polar solvents. Table 1 summarizes the binding constants of nitrate anions with protonated receptors 1c, 2c, 4c, 6c and 7c of the corresponding receptors 1, 2, 4, 6 and 7, respectively, in acetone-d6 solutions. As anticipated, the obtained binding constant data suggest that the binding affinities for nitrate anions generally depend upon the acidic nature of the amide N–H protons in these receptors. In the case of receptors 2c and 4c, the methoxy groups and the electron withdrawing nitro groups at the meta- and para- positions, respectively, relative to the amide functionality render amide N–Hs more polarized and as a result receptors 2c and 4c exhibit higher binding affinities than the others. The protonated receptors bearing a nitro group at the ortho position of the N-terminal aromatic ring (receptors 1c′, 5c, and 6c′) showed no peak changes, even after the addition of 20 equivalents of nitrate anions (Fig. S86–S93). It implies that the amide N–Hs of these receptors are not available for nitrate binding because of engaging in either intramolecular hydrogen bonds with the ortho substituent of N-terminal aromatic rings or intermolecular hydrogen bonds with another molecule. In particular, significant downfield shifts in signals of the ortho protons of N-terminal aromatic ring for receptors 1′ and 6′, as well as the signal of meta protons of the N-terminal aromatic ring for receptor 5 that are very similar to the spectra obtained in CDCl3 suggest the existence of self-assembled dimers for these three receptors, even in polar solvents like acetone-d6. This fact was finally confirmed through HRESMS and single crystal structure analysis, as discussed below.| Complexes | 1c | 2c | 4c | 6c | 7c |
|---|---|---|---|---|---|
|
a Titration experiments were performed in acetone-d6 solutions at 298 K. Binding constants were obtained by nonlinear fitting of the chemical shifts of amide N–H peak versus incoming nitrate anion concentration based on a 1:1 binding mode on the basis of the crystal structures. The concentrations of perchlorate complexes used were 1.48 mM.
|
|||||
| K a | 370 ± 20 | 1550 ± 70 | 1080 ± 120 | 340 ± 10 | 400 ± 20 |
Dynamic self-assembly of molecular capsule
1H NMR titration in a DMSO-d6 solution of a mixture of nitrate-encapsulated complexes 6a and 6b upon addition of CDCl3 showed a similar pattern of peak shift to that of 1a and 1b as observed previously.15 The amide N–H peak of complex 6b was initially upfield shifted until the ratio of DMSO-d6/CDCl3 (v/v) is at 1:1 followed by a downfield shift with a further increasing amount of CDCl3 (see Fig. S80 in the supplementary information†). The peak of proton h in complex 6a gradually downfield shifted upon addition of CDCl3, showing a typical pattern of self-assembled capsule formation, whereas the amide N–H peak of complex 6a is constantly upfield shifted with increasing concentration of CDCl3. The upfield shift of the amide N–H peak is attributed to the polarity change after addition of CDCl3 to a DMSO-d6 solution.15 The 1H NMR titration spectra of pure complex 6b in a DMSO-d6 solution with addition of CDCl3 is depicted in Fig. S82†. The 1H NMR spectrum shown in red color of Fig. S82† after partial evaporation of CDCl3 from a DMSO-d6/CDCl3 mixture solution almost merges with the parent spectrum that demonstrates the reversible switching between two discrete forms, the nitrate-encapsulated complex 6b and the self-assembled capsule 6′2.
1H NMR titration spectra of neutral receptor 6′ (Fig. 5a) in a DMSO-d6 solution with addition of CDCl3 also showed a similar pattern of peak shift to that of complex 6b, which suggests that protonation of the centrally bridged N-atom is not mandatory for self-assembled capsule formation. The 1H NMR spectrum shown in red color of Fig. 5a after partial evaporation of CDCl3 from a mixture solution almost merged with the parent spectrum of receptor 6′, which indicates the capsule formation from neutral receptor 6′ through dynamic self-assembly regulated by the solvent polarity. The formation of a self-assembled capsule with increasing concentration of CDCl3 can be rationalized as with increasing concentration of less polar solvent, such as CDCl3, the hydrogen-bonding interactions became more favorable and resulted in more stable hydrogen-bonded assemblies in solution. However, similar observations were noted upon titration of a DMSO-d6 solution of receptor 6′, even with in the presence of a polar solvent, such as acetone-d6 (Fig. 5b) or CD3NO2 (Fig. S81†). This observation is unique in comparison to previously reported examples, where hydrogen-bonded capsules are stable in polar solvents either because of hindered bulky groups at the end of each branch of the receptor6 or because of the participation of the polar solvent molecules in stabilizing the resulting capsule.7 Upon partial evaporation of acetone-d6 from a mixture solution, the resulting 1H NMR spectrum shown in red color of Fig. 5b nearly merged with the original one recorded in DMSO-d6, which illustrates the reversibility of the capsule formation from a neutral receptor in a polar solvent. A similar observation was noted when the corresponding nitrate complex 6b in a DMSO-d6 solution was titrated either with CDCl3, acetone-d6 or CD3NO2 (see Fig. S82–S84 in the supplementary information†).
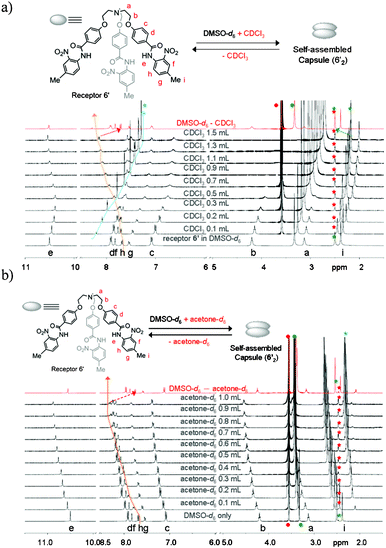 | ||
| Fig. 5 1H NMR (400 MHz, 20 °C) titration spectra of receptor 6′ (10.3 mM) in a DMSO-d6 solution upon the addition of varying amounts of CDCl3 (a) and acetone-d6 (b), respectively. After partial evaporation of CDCl3 (top) and acetone-d6 (bottom), the resulting spectra (red color in a and b) almost merged with the original one recorded in DMSO-d6 only. Star marks in green and red color represent the peaks for DMSO-d6 from the solvent and from the internal reference (from TMS in DMSO-d6), respectively. Star marks in blue color represent the solvent peaks (CDCl3, top), (acetone-d6, bottom) added to DMSO-d6 solution of receptor 6′. | ||
On the other hand, a different binding mode was observed for the formation of homodimer (52) in contrast to 1′2 or 6′2 (Fig. 7). The 1H NMR titration experiments of either receptor 5 or its corresponding nitrate-encapsulated complex 5a in a DMSO-d6 solution showed a downfield shift of the meta C–H protons of N-terminal aromatic rings (proton f of receptor 5, see Fig. 6) with an increasing amount of CDCl3. Similar observations were noted in a 1H NMR titration experiment of a DMSO-d6 solution of receptor 5 (Fig. S75 and S76†) or its corresponding nitrate complex (Fig. S77–S79†) with polar solvents, such as acetone-d6 or CD3NO2. The pattern of peak shifting of the amide N–H protons was similar to the cases observed in receptors 1′ and 6′. The different binding modes along the seam of the capsule can be rationalized based on the electronic property of the N-terminal aromatic rings of receptors 1′, 5, and 6′. In receptor 5, a nitro group at the ortho position of the N-terminal aromatic ring makes the meta position (ortho position with respect to the nitro group) electron deficient and thus protons f in receptor 5 participate in the intermolecular hydrogen bonds. However, an electron donating group at the para position of receptors 1′ and 6′ renders the ortho position with respect to the amide groups of the N-terminal aromatic ring the most electron deficient and thus protons h of receptors 1′ and 6′ are most susceptible to the intermolecular hydrogen bonding interactions to form the self-assembled homodimeric capsules (1′2 and 6′2).
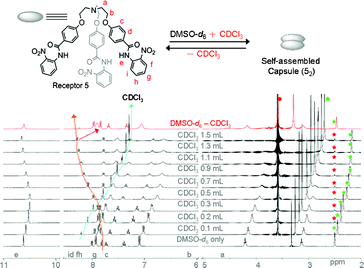 | ||
| Fig. 6 1H NMR (400 MHz, 20 °C) titration spectra of receptor 5 (10.3 mM) in a DMSO-d6 solution upon addition of varying amounts of CDCl3. After partial evaporation of CDCl3, the resulting spectra (in red color) merged with the original one recorded in DMSO-d6. Star marks in green and red colors in all spectra represent peaks for DMSO-d6 solvent and from the internal reference in TMS, respectively. Circle marks in green and red colors represent the residual water peaks from deuterated solvents. | ||
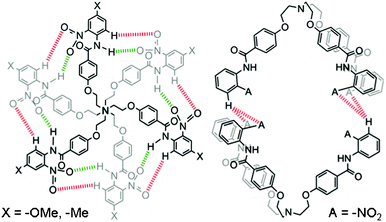 | ||
| Fig. 7 Different binding modes of hydrogen bonds in self-assembled capsules 1′2, 6′2 (left), and 52 (right). Receptors 1′ and 6′ form self-assembled capsules by seaming through cyclic array of twelve hydrogen bonds; six between aromatic C-Hs and O-atoms of the nitro groups, as shown in red color and the other six between amide N–Hs and O-atoms of the nitro groups, as shown in green color. Receptor 5 forms a self-assembled dimer by four hydrogen bonds with two hydrogen bonds between the O-atoms of two nitro groups of one molecule and meta H-atoms of the N-terminal aromatic ring of another molecule. | ||
Thus, receptors 1′ and 6′ with an electron donating functional group at the para-position of the N-terminal aromatic ring form self-assembled capsules by seaming of the periphery through a cyclic array of a total of twelve hydrogen bonds; six between aromatic C–Hs and O-atoms of the nitro groups and the other six between amide N–Hs and O-atoms of the ortho-substituted nitro groups, as illustrated by red and green colors, respectively in Fig 7. On the other hand, according to crystal structure (Fig. 8) obtained from an acetone-d6 solution of receptor 5 at room temperature over a period of two weeks, receptor 5 forms a dimer by a total of four hydrogen bonds with two hydrogen bonds between the aromatic C–H of one molecule and O-atoms of the two nitro groups of another molecule. Fig. 8 shows the crystal structure of the hydrogen-bonded homodimer 52. In the absence of a suitable guest, the crystal packing effect dominates in the crystal structure and two branches of receptor 5 are stacked to each other through π⋯π interactions. The intramolecular hydrogen bonds between the amide N–Hs and O-atoms of the ortho-substituted nitro groups lie in the range of 1.90–1.97 Å (θN–H⋯O 132.1–136.9°). The intermolecular hydrogen bonds between the protons f of N-terminal aromatic rings lie in the range of 2.88–3.01 Å (θN–H⋯O 127.1–136.9°).
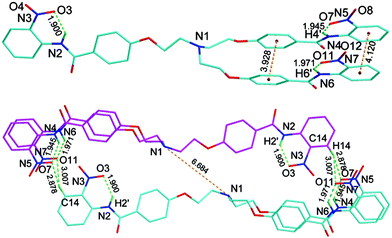 | ||
| Fig. 8 The crystal structure of homodimer 52 showing intramolecular hydrogen bonds between amide N–Hs and O-atoms of the nitro group (top) and hydrogen-bonded homodimer 52 (bottom). All hydrogen atoms are omitted for clarity except for amide N–Hs. | ||
The formation of the self-assembled capsule in a polar solvent was confirmed through high resolution ESI-mass spectra. Positive mode HRESIMS of a mixture solution of DMSO/acetone (1:1, v/v) showed the expected peak clusters at m/z 1942.5692 (calcd. m/z 1942.5879), 1761.5193 (calcd. m/z 1761.5211) and 1846.6067 (calcd. m/z 1846.6184), corresponding to [M + Na]+ peaks for 1′2, 52 and 6′2, respectively. Positive mode HRESIMS acquired in a mixture solution of DMSO/nitromethane (1:1, v/v) also showed the expected peak clusters at m/z 1958.5551 (calcd. m/z 1958.5618), 1777.4955 (calcd. m/z 1777.4950), and 1862.5892 (calcd. m/z 1862.5923) corresponding to [M + K]+ peaks for 1′2, 52 and 6′2, respectively.
Formation of a self-assembled capsule in polar solvents, such as acetone-d6 or CD3NO2, through seaming of hydrogen bonds is quite surprising since hydrogen bond formation is expected to be unfavored in a polar solvent due to the solvent competitive environment. Recently, Schmuke et al. have shown that self-assembled supramolecular species can be stabilized through charged hydrogen bonding interactions much better than their corresponding neutral receptors.19 The present work shows unique examples of formation of purely hydrogen-bonded self-assembled capsules in polar solvents.
Nitrate anion binding in polar solvents is also a reversible process. Upon treatment of a DMSO-d6 solution of nitrate complex 1b with 5 equivalents of KOH and sonicating the solution for 2–3 min, the amide N–H peak disappeared (Fig. S95†) and the solution color changed from yellow to a deep orange color. The disappearance of the amide peak and the color change may be attributed to the deprotonation of amide protons upon the addition of base. Upon the addition of trifluoroacetic acid to the same solution, the solution color again retained its yellow color. HRESI mass spectra of this sample confirmed that its identity was the same as the original sample, the nitrate complex 1b.
Thus, although the binding mode of homodimer 52 is different from capsules 1′2 and 6′2, the reversibility of the capsules can be simply tuned by changing the polarity of the solvent media. Self-assembled capsule formation in a polar solvent is a rarely observed phenomenon with significant practical utilities.1–3 Since these three receptors 1′, 5 and 6′ exist in a capsule form, even in polar solvents such as acetone-d6, addition of 20 equivalents of tetrabutylammonium nitrate did not show a notable shift of the amide N–H peak during the titration of their corresponding perchlorate complexes (see Fig. S87 and S91†). Furthermore, the self-assembled 52 is extremely stable in CDCl3 solution, even upon dilution up to 1 × 10−8 M without a sign of dissociation or formation of a new species (see Fig. S85 in the supplementary information†). This observation is in sharp contrast to a literature example, where some ill-defined aggregates started to appear in dilute solution below 2 mM.20
Conclusions
In conclusion, solvent polarity-dependent reversible binding of highly hydrated and weakly coordinating nitrate anions and solvent-polarity-controlled self-assembled capsule formation by highly flexible tripodal receptors through dynamic self-assembly is reported. The self-assembled capsules reported here show some distinct features. The dynamic self-assembly process can be easily controlled by solvent-induced transformation between discrete tripodal receptors and their hydrogen-bonded homodimeric capsules. The homodimeric capsule 52 is remarkably stable up to 1 × 10−8 M, without dissociation or forming any new species (aggregates). The reversible encapsulation-releasing of nitrate anions can be achieved through dynamic self-assembly/disassembly via conversion between a nitrate complex and its self-assembled homodimer by simply modulating the surrounding medium polarity from a polar DMSO to a less polar CHCl3 or acetone. Thus, the current concept for capsule formation and dissociation is expected to be applicable in potential delivery systems. It can also find applications in environmental remediation of nitrate anions, which are notoriously difficult to bind. Current research efforts are being conducted along these directions.Experimental section
Materials and methods
Triethanolamine, thionyl chloride, 4-hydroxybenzoic acid and all substituted amines were commercially available and used as received. All solvents were purified by standard procedures. All NMR spectra were recorded with an AV 400 spectrometer. Single crystal data were collected with a Brücker Kappa CCD diffractometer. CCDC-777053 and CCDC-863976 contain the supplementary crystallographic data for nitrate complexes 6a and 52, respectively†. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.1H NMR titration of all samples were measured on a Brüker AV400 at 400 MHz and deuterated solvent used from a freshly opened bottle. Titration experiments for the calculation of binding constants were performed in acetone-d6 solutions and binding constants were calculated from the shifting of the amide N–H peak. Samples were prepared just prior to titration experiments and the nitrate anion was used in the form of tetrabutylammonium salt. Titration experiments for capsule formation were performed in DMSO-d6 solutions with an increasing amount of deuterated solvents, such as CDCl3, acetone-d6 and CD3NO2, with TMS in DMSO-d6 as the internal reference.
General procedures for the synthesis of receptors 1–7
Receptors 1–7 were synthesized from previously reported15 tris(2-chloroethyl)amine hydrochloride (A) by simple SN2 substitution with 4-hydroxy methylbenzoate in DMSO followed by basic hydrolysis to yield the corresponding triacid (C). Subsequent condensation of the triacid with thionyl chloride in dichloromethane yielded the corresponding acid chloride. Subsequently, the acid chloride solution in dichloromethane was slowly added to a mixture of the corresponding amino derivative and Et3N in dichloromethane and refluxed for 8–12 h. The solvent was removed under reduced pressure. Ice-cold water was added to the residue and stirred at room temperature for 2–3 h to precipitate the target compounds (1, 4, 6 and 7). The crude products were purified by flash chromatography with CHCl3/MeOH mixture solution as the eluent to yield the desired receptors. Crude products of receptors 2, 3 and 5 were extracted with CHCl3. Solvent was evaporated under reduced pressure after drying over anhydrous MgSO4 and a short pad column was flashed with 1% MeOH in CHCl3 to get the pure product. The desired receptors 1–7 were isolated in 70–95% yields.1: creamy white solid (yield 95%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.12 (t, 6H, J = 5.6 Hz), 3.73 (s, 9H), 4.18 (t, 6H, J = 5.6 Hz), 6.90 (d, 6H, J = 8.8 Hz), 7.03 (d, 6H, J = 8.8 Hz), 7.65 (d, 6H, J = 8.8 Hz), 7.93 (d, 6H, J = 8.8 Hz), 9.96 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.5, 55.2, 66.8, 113.7, 114.1, 122.0, 127.0, 129.4, 132.4, 155.4, 161.0, 164.5. HRESIMS m/z 825.3494 (calcd m/z 825.3500 for [M + H]+).
1a: pale yellow solid (yield 92%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.74 (s, 9H), 3.92 (s, 6H), 4.57 (s, 6H), 6.92 (d, 6H, J = 8.4 Hz), 7.10 (d, 6H, J = 8.4 Hz), 7.67 (d, 6H, J = 8.4 Hz), 8.00 (d, 6H, J = 8.4 Hz), 10.03 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.4, 55.2, 62.6, 113.7, 114.3, 122.1, 127.9, 129.5, 132.3, 155.5, 160.0, 164.4. HRESIMS m/z 886.3306 (calcd m/z 886.3299 for [M − H]−).
1b: orange color solid (yield 92%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.86 (s, 9H), 3.92 (s, 6H), 4.58 (s, 6H), 7.15 (d, 6H, J = 8.8 Hz), 7.35 (d, 3H, J = 9.2 Hz), 7.52 (s, 3H), 7.64 (d, 3H, J = 9.2 Hz), 7.98 (d, 6H, J = 8.8 Hz) 10.44 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.3, 56.1, 62.7, 109.2, 114.6, 120.2, 124.5, 126.6, 128.0, 129.7, 143.9, 156.4, 160.5, 164.4. HRESIMS m/z 1021.2855 (calcd m/z 1021.2852 for [M − H]−).
1′: deep yellow solid (yield 87%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.13 (t, 6H, J = 5.2 Hz), 3.85 (s, 9H), 4.20 (t, 6H, J = 5.2 Hz),7.08 (d, 6H, J = 8.8 Hz), 7.33 (d, 3H, J = 9 Hz), 7.51 (s, 3H), 7.65 (d, 3H, J = 8.8 Hz), 7.92 (d, 6H, J = 8.8 Hz), 10.39 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.4, 56.0, 66.9, 109.1, 114.3, 120.2, 124.6, 125.7, 127.7, 129.6, 143.6, 156.1, 161.5, 164.6. HRESIMS m/z 960.3060 (calcd m/z 960.3052 for [M + H]+).
1′2: 1H NMR (CDCl3, 400 MHz, 20 °C) δ 3.18 (t, 6H, J = 5.2 Hz), 3.83 (s, 9H), 4.16 (t, 6H, J = 5.2 Hz), 6.93 (d, 6H, J = 8.8 Hz), 7.21 (d, 3H, J = 9.2 Hz), 7.64 (s, 3H), 7.86 (d, 6H, J = 8.8 Hz), 8.80 (d, 3H, J = 9.2 Hz), 10.97 (s, 3H). 13C NMR (CDCl3, 100 MHz, 20 °C) δ 54.6, 56.1, 67.5, 108.7, 114.9, 123.7, 123.9, 126.7, 129.5, 129.6, 137.0, 154.9, 162.3, 165.1. HRESIMS m/z 1942.5861 (calcd m/z 1942.5879 for [M + Na]+).
2: ochre brown powder (yield 71%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 2.71 (t, 6H, J = 5.2 Hz), 3.33 (s, 9H), 3.78 (t, 6H, J = 5.2 Hz), 6.25 (d, 3H, J = 8.0 Hz), 6.64 (d, 6H, J = 8.8 Hz), 6.81 (t, 3H, J = 8.0 Hz), 6.94 (d, 3H, J = 8.0 Hz), 7.05 (s, 3H), 7.53 (d, 6H, J = 8.8 Hz), 9.62 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.5, 55.0, 66.9, 106.0, 109.0, 112.6, 114.1, 126.9, 129.3, 129.6, 140.5, 159.4, 161.2, 164.9. HRESIMS m/z 847.3323 (calcd m/z 847.3319 for [M + Na]+).
2a: dark brown solid (yield 65%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.12 (t, 6H, J = 5.6 Hz), 3.74 (s, 9H), 4.19 (t, 6H, J = 5.6 Hz), 6.66 (d, 3H, J = 8.0 Hz), 7.04 (d, 6H, J = 8.8 Hz), 7.22 (t, 3H, J = 8.0 Hz), 7.35 (d, 3H, J = 8.0 Hz), 7.46 (s, 3H), 7.94 (d, 6H, J = 8.8 Hz), 10.04 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.5, 55.0, 66.9, 106.0, 109.0, 112.6, 114.1, 126.9, 129.3, 129.6, 140.5, 159.4, 161.2, 164.9. HRESIMS m/z 886.3308 (calcd m/z 886.3299 for [M − H]−).
3: maroon color viscous liquid (yield 73%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.11 (t, 6H, J = 5.6 Hz), 3.82 (s, 9H), 4.17 (t, 6H, J = 5.6 Hz), 6.93–6.96 (m, 3H), 7.03 (d, 6H, J = 8.8 Hz), 7.06 (s, 3H), 7.12–7.16 (m, 3H), 7.82 (d, 3H, J = 7.6 Hz), 7.93 (d, 6H, J = 8.8 Hz), 9.22 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.6, 55.8, 66.9, 111.3, 114.3, 120.3, 123.8, 125.4, 126.7, 127.1, 129.4, 151.2, 161.3, 164.5. HRESIMS m/z 825.3497 (calcd m/z 825.3500 for [M + H]+).
3a: yellow color viscous liquid (yield 65%).1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.83 (s, 9H), 3.95 (s, 6H), 4.59 (s, 6H), 6.97 (t, 3H, J = 7.6 Hz), 7.07 (d, 6H, J = 8.0 Hz), 7.12 (d, 6H, J = 8.8 Hz), 7.17 (d, 3H, J = 8.8 Hz), 7.84 (d, 3H, J = 8.0 Hz), 8.02 (d, 6H, J = 8.8 Hz), 9.32 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.5, 55.8, 62.7, 111.4, 114.6, 120.3, 124.0, 125.6, 127.1, 127.6, 129.5, 151.3, 160.2, 164.5. HRESIMS m/z 886.3290 (calcd m/z 886.3299 for [M−H]−).
4: yellow solid (yield 85%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.88 (s, 6H), 4.64 (s, 6H), 7.13 (d, 6H, J = 8.4 Hz), 8.04–8.09 (m, 9H), 8.21 (d, 6H, J = 9.2 Hz), 10.75 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 52.6, 62.7, 114.4, 119.8, 124.6, 126.8, 130.1, 142.2, 145.7, 160.5, 165.3. HRESIMS m/z 870.2730 (calcd m/z 870.2735 for [M + H]+).
4a: yellow solid (yield 87%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.93 (s, 6H), 4.58 (s, 6H), 7.13 (d, 6H, J = 8.8 Hz), 8.01–8.05 (m, 9H), 8.22 (d, 6H, J = 9.2 Hz), 10.65 (s, 3 H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.4, 62.7, 114.4, 119.8, 124.7, 126.9, 130.0, 142.3, 145.6, 160.5, 165.4. HRESIMS m/z 931.2539 (calcd m/z 931.2535 for [M − H]−).
5: brown solid (yield 70%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.13 (t, 6H, J = 5.2 Hz), 4.21 (t, 6H, J = 5.2 Hz), 7.10 (d, 6H, J = 8.8 Hz), 7.38 (t, 3H, J = 7.6 Hz), 7.73 (t, 3H, J = 7.6 Hz), 7.84 (d, 3H, J = 8.0 Hz), 7.94 (d, 6H, J = 8.8 Hz), 8.00 (d, 3H, J = 8.0 Hz), 10.63 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.4, 66.9, 114.4, 124.9, 125.1, 125.5, 129.7, 132.0, 134.0, 142.3, 161.7, 164.6. HRESIMS m/z 870.2739 (calcd m/z 870.2735 for [M + H]+).
52: 1H NMR (CDCl3, 400 MHz, 20 °C) δ 3.28 (s, 6H), 4.24 (s, 6H), 6.96 (d, 6H, J = 8.8 Hz), 7.14 (t, 3H, J = 8.0 Hz), 7.63 (t, 3H, J = 8.0 Hz), 7.89 (d, 6H, J = 8.8 Hz), 8.21 (d, 3H, J = 8.4 Hz), 8.91 (d, 3H, J = 8.4 Hz), 11.23 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 54.5, 67.0, 114.9, 122.2, 123.2, 126.0, 126.7, 129.6, 135.8, 136.3, 136.4, 162.2, 165.2. HRESIMS m/z 1739.5383 (calcd m/z 1739.5392 for [M + H]+).
5a: yellow powder (yield 85%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.93 (s, 6H), 4.59 (s, 6H), 7.16 (d, 6H, J = 8.8 Hz), 7.40 (t, 3H, J = 8.0 Hz), 7.74 (t, 3H, J = 8.0 Hz), 7.81 (d, 3H, J = 7.6 Hz), 7.98–8.02 (m, 9H), 10.69 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.4, 62.7, 114.7, 125.1, 125.5, 125.8, 126.5, 129.9, 131.9, 134.2, 142.7, 160.7, 164.7. HRESIMS m/z 931.2542 (calcd m/z 931.2535 for [M − H]−).
6: white solid (yield 90%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 2.27 (s, 9H), 3.12 (t, 6H, J = 5.2 Hz), 4.18 (t, 6H, J = 5.2 Hz), 7.04 (d, 6H, J = 8.8 Hz), 7.13 (d, 6H, J = 8.4 Hz), 7.65 (d, 6H, J = 8.4 Hz), 7.95 (d, 6H, J = 8.8 Hz), 10.01 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 20.5, 53.5, 66.8, 114.0, 120.4, 127.0, 128.9, 129.5, 132.3, 136.8, 161.1, 164.6. HRESIMS m/z 799.3467 (calcd m/z 799.3472 for [M + Na]+).
6a: white solid (yield 96%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 2.27 (s, 9H), 3.92 (s, 6H), 4.57 (s, 6H), 7.11 (d, 6H, J = 8.8 Hz), 7.14 (d, 6H, J = 8.4 Hz), 7.65 (d, 6H, J = 8.4 Hz), 8.00 (d, 6H, J = 8.8 Hz), 10.06 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 20.5, 53.4, 62.6, 114.3, 120.5, 127.9, 129.0, 129.6, 132.5, 136.7, 160.0, 164.5. HRESIMS m/z 838.3453 (calcd m/z 838.3452 for [M − H]−).
6b: yellow solid (yield 91%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 2.35 (s, 9H), 3.91 (s, 6H), 4.57 (s, 6H), 7.12 (d, 6H, J = 8.8 Hz), 7.52 (d, 3H, J = 7.6 Hz), 7.65 (d, 3H, J = 8.4 Hz), 7.80 (s, 3H), 7.95 (d, 6H, J = 8.4 Hz), 10.57 (s, 3 H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 20.2, 53.5, 62.8, 114.8, 125.0, 126.0, 126.7, 129.5, 129.9, 134.8, 135.6, 142.7, 160.7, 164.8. HRESIMS m/z 973.3002 (calcd m/z 973.3004 for [M − H]−).
6′: orange solid (yield 93%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 2.38 (s, 9H), 3.91 (s, 6H), 4.58 (s, 6H), 7.15 (d, 6H, J = 6.8 Hz), 7.56 (s, 3H), 7.68 (d, 3 H, J = 7.2 Hz), 7.83 (s, 3H), 7.98 (d, 6H, J = 6.8 Hz), 10.56 (s, 3 H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 20.0, 53.3, 62.7, 114.6, 124.8, 125.8, 126.5, 129.3, 129.7, 134.6, 135.4, 142.5, 160.5, 164.5. HRFABMS m/z 912.3196 (calcd m/z 912.3204 for [M + H]+).
6′2: 1H NMR (CDCl3, 400 MHz, 20 °C) δ 2.37 (s, 9H), 3.20 (s, 6H), 4.17 (s, 6H), 6.95 (s, 6H), 7.46 (s, 3H), 7.89 (s, 6 H), 8.01 (s, 3H), 8.80 (s, 3H), 11.13 (s, 3H). 13C NMR (CDCl3, 100 MHz, 20 °C) δ 20.8, 54.6, 67.5, 114.9, 122.2, 125.8, 126.7, 129.6, 133.5, 136.4, 137.3, 162.3, 165.2. HRESIMS m/z 1846.6067 (calcd m/z 1846.6184 for [M + Na]+).
7: creamy white solid (yield 89%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.13 (t, 6H, J = 5.6 Hz), 4.19 (s, 6H, J = 5.6 Hz), 7.07 (m, 9H), 7.33 (3, 6H, J = 7.6 Hz), 7.76 (d, 6H, J = 8.0 Hz), 7.95 (d, 6H, J = 8.0 Hz), 10.07 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.6, 66.9, 114.2, 120.5, 123.5, 127.0, 128.6, 129.7, 139.3, 161.2, 165.0. HRESIMS m/z 735.3174 (calcd m/z 735.3183 for [M + H]+).
7a: creamy yellow solid (yield 85%). 1H NMR (DMSO-d6, 400 MHz, 20 °C) δ 3.90 (s, 6H), 4.56 (s, 6H), 7.07–7.13 (m, 9H), 7.34 (t, 6H, J = 8.0 Hz), 7.77 (d, 6H, J = 8.0 Hz), 8.01 (d, 6H, J = 8.4 Hz), 10.12 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, 20 °C) δ 53.4, 62.6, 114.3, 120.5, 123.6, 127.8, 128.6, 129.7, 139.2, 160.1, 164.7. HRESIMS m/z 796.2977 (calcd m/z 796.2983 for [M − H]−).
Crystallographic data for 6a (C45H49N5O9): Mr = 839.92, trigonal, space group P-3, a = b = 15.5323 (6) Å; c = 11.4631(16) Å; α = β = 90.00; γ = 120.00°; V = 2394.99 (18) Å3; Z = 2; ρcalcd = 1.165 Mg/m3; λ = 0.71073 Å; μ = 0.081 mm−1; T = 200(2) K; 15166 reflections collected, 2791 unique (Rint = 0.0626) R1 = 0.1807; wR2 = 0.4985; goodness-of-fit on F2 1.079. Half a CHCl3 molecule has been squeezed. Crystallographic data for 52 (C45H39N7O12): Mr = 869.83, monoclinic, space group P21/c, a = 23.327 (14) Å; b = 11.333 (6) Å; c = 15.245 (9) Å; α = 90; β = 99.7; γ = 90°; V = 3973 (4) Å3; Z = 4; ρcalcd = 1.454 Mg m−3; λ = 0.71073 Å; μ = 0.108 mm−1; T = 200(2) K; 6537 reflections collected, 4927 unique (Rint = 0.0554) R1 = 0.1700; wR2 = 0.2315; goodness-of-fit on F2 0.4923.
Acknowledgements
We are grateful to the National Council of Taiwan (Grant No 100-2113-M-001-024-MY3) and Academia Sinica for support of this research. A. S. S. thanks the postdoctoral fellowship sponsored by the National Science Council of Taiwan (Grant No 100-2811-M-001-090). Mass spectrometry analyses were performed by Mass Spectrometry facility of the Institute of Chemistry, Academia Sinica.References
- (a) J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS; (b) J. Kang and J. Rebek Jr., Nature, 1997, 385, 50–52 CrossRef CAS; (c) S. Rieth, K. Hermann, B.-Y. Wang and J. D. Badjić, Chem. Soc. Rev., 2011, 40, 1609–1622 RSC; (d) T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC; (e) S. Liu and B. C. Gibb, Chem. Commun., 2008, 3709–3716 RSC; (f) N. Kameta, A. Tanaka, H. Akiyama, H. Minamikawa, M. Masuda and T. Shimizu, Chem. Eur. J., 2011, 17, 5251–5255 CrossRef CAS.
- (a) M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS; (b) L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2005, 127, 3674–3675 CrossRef CAS; (c) S. Liu, H. Gan, A. T. Hermann, S. W. Rick and B. C. Gibb, Nature Chem., 2010, 2, 847–852 CrossRef CAS; (d) M. D. Pluth, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2007, 46, 8587–8589 CrossRef CAS.
- (a) Self-assembled hosts for stabilization of reactive intermediates see : T. N. Parac, D. L. Caulder and K. N. Raymond, J. Am. Chem. Soc., 1998, 120, 8003–8004 CrossRef; (b) M. Yoshizawa, T. Kuskawa, M. Fujita and K. Yamaguchi, J. Am. Chem. Soc., 2000, 122, 6311–6312 CrossRef CAS; (c) D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2006, 45, 745–748 CrossRef CAS; (d) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS.
- (a) L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469–472 CrossRef CAS; (b) T. Heinz, D. M. Rudkevich and J. Rebek Jr., Nature, 1998, 394, 764–766 CrossRef CAS; (c) L. J. Prins, J. Huskens, F. de Jong, P. Timmerman and D. N. Reinhoudt, Nature, 1999, 398, 498–504 CrossRef CAS; (d) J. Rebek Jr., Acc. Chem. Res, 1999, 32, 278–286 CrossRef and references therein; (e) M. O. Vysotsky, I. Thondrof and V. Böhmer, Angew. Chem., Int. Ed., 2000, 39, 1264–1267 CrossRef CAS; (f) L. J. Prins, J. Huskens, F. de Jong, P. Timmerman and D. N. Reinhoudt, Nature, 2000, 408, 181–184 CrossRef CAS; (g) A. M. Rincón, P. Prados and J. de Mendoza, J. Am. Chem. Soc., 2001, 123, 3493–3498 CrossRef; (h) J. L. Atwood and A. Szumna, J. Am. Chem. Soc., 2002, 124, 10646–10647 CrossRef CAS; (i) J. L. Atwood, L. J. Barbour and A. Jerga, Proc. Natl. Acad. Sci. USA, 2002, 99, 4837–4841 CrossRef CAS; (j) G. W. V. Cave, J. Antesberger, L. J. Barbour, R. M. McKinlay and J. L. Atwood, Angew. Chem., Int. Ed., 2004, 43, 5263–5266 CrossRef CAS; (k) L. Avram and Y. Cohen, J. Am. Chem. Soc., 2004, 126, 11556–11563 CrossRef CAS; (l) J. Rebek Jr., Angew. Chem., Int. Ed., 2005, 44, 2068–2078 CrossRef; (m) O. Kasyan, V. Kalchenko, M. Bolte and V. Böhmer, Chem. Commun., 2006, 1932–1934 RSC; (n) A. Piermattei, M. Giesbers, A. T. M. Marcelis, E. Mendes, S. J. Picken, M. Crego-Valama and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2006, 45, 7543–7546 CrossRef CAS; (o) L. Avram and Y. Cohen, Org. Lett., 2006, 8, 219–222 CrossRef CAS; (p) S. Shimizu, T. Kiuchi and N. Pan, Angew. Chem., Int. Ed., 2007, 46, 6442–6445 CrossRef CAS; (q) A. Shivanyuk, J. Am. Chem. Soc., 2007, 129, 14199–14199 CrossRef; (r) B.-Y. Wang, X. Bao, Z. Yan, V. Maslak, C. M. Hadad and J. D. Badjić, J. Am. Chem. Soc., 2008, 130, 15127–15133 CrossRef CAS; (s) Y. S. Park and K. Paek, Org. Lett., 2008, 10, 4867–4870 CrossRef CAS; (t) D. Ajami and J. Rebek Jr., J. Org. Chem., 2009, 74, 6584–6591 CrossRef CAS; (u) P. Ballester and G. Gil-Ramirez, Proc. Natl. Acad. Sci. USA, 2009, 106, 10455–10459 CrossRef CAS; (v) M. Alajarin, R.-A. Orenes, J. W. Steed and A. Pastor, Chem. Commun., 2010, 46, 1394–1403 RSC; (w) A. Asadi, D. Ajami and J. Rebek Jr., J. Am. Chem. Soc., 2011, 133, 10682–10684 CrossRef CAS.
- P. Ballester, Chem. Soc. Rev., 2010, 39, 3810–3830 RSC and references therein..
- M. O. Vysotsky, I. Thondorf and V. Böhmer, Chem. Commun., 2001, 1890–1891 RSC.
- (a) A. Shivanyuk and J. Rebek Jr., Chem. Commun., 2001, 2424–2425 RSC; (b) A. Shivanyuk and J. Rebek Jr., Chem. Commun., 2001, 2374–2375 RSC; (c) J. L. Atwood, L. J. Barbour and A. Jerga, Chem. Commun., 2001, 2376–2377 RSC.
- (a) S. K. Körner, F. C. Tucci, D. M. Rudkevich, T. Heinz and J. Rebek, Chem. Eur. J., 2000, 6, 187–195 CrossRef; (b) A. Shivanyuk and J. Rebek Jr., Chem. Commun., 2002, 2326–2327 RSC; (c) S. Kubik, C. Reyheller and S. Stuewe, J. Inclusion Phenom. Macrocyclic Chem., 2005, 52, 137–187 CrossRef CAS; (d) G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS.
- S. Mangani and M. Ferraroni in Supramolecular Chemistry for Anions (Eds.; A. Bianchi, K. Bowman-James and E. Garcia-España), Wiley-VCH, New York, 1997, p. 63 Search PubMed.
- (a) S. Nishizawa, P. Bühlmann, M. Iwao and Y. Umezawa, Tetrahedron Lett., 1995, 36, 6483–6486 CrossRef CAS; (b) P. Bühlmann, S. Nishizawa, K. P. Xiao and Y. Umezawa, Tetrahedron Lett., 1997, 53, 1647–1654 Search PubMed; (c) T. W. Bell, N. M. Hext and A. B. Khasanov, Pure Appl. Chem., 1998, 70, 2371–2377 CrossRef CAS; (d) S. Sasaki, M. Mizuno, K. Naemura and Y. Tobe, J. Org. Chem., 2000, 65, 275–283 CrossRef CAS; (e) C.-Y. Wu, M.-S. Chen, C.-A. Lin, S.-C. Lin and S.-S. Sun, Chem. Eur. J., 2006, 12, 2263–2269 CrossRef CAS; (f) C.-Y. Chen, T.-P. Lin, C.-K. Chen, S.-C. Lin, M.-C. Tseng, Y.-S. Wen and S.-S. Sun, J. Org. Chem., 2008, 73, 900–911 CrossRef CAS; (g) Z. Lin, H.-C. Chen, S.-S. Sun, C.-P. Hsu and T. J. Chow, Tetrahedron, 2009, 65, 5216–5221 CrossRef CAS.
- T. Martin, U. Obst and J. Rebek Jr., Science, 1998, 281, 1842–1845 CrossRef CAS.
- (a) F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609–1646 CrossRef CAS; (b) P. D. Beer and P. A. Gale, Angew. Chem. Int. Ed., 2001, 40, 486–516 CrossRef CAS; (c) C. R. Bondy and S. J. Loeb, Coord. Chem. Rev., 2003, 240, 77–99 CrossRef CAS; (d) S. O. Kang, J. M. Llinares, D. Powell, D. VanderVelde and K. Bowman-James, J. Am. Chem. Soc., 2003, 125, 10152–10153 CrossRef CAS and references therein; (e) K. Choi and A. D. Hamilton, J. Am. Chem. Soc., 2003, 125, 10241–10249 CrossRef CAS; (f) S. O. Kang, R. A. Begum and K. Bowman-James, Angew. Chem. Int. Ed., 2006, 45, 7882–7894 CrossRef CAS; (g) C.-L. Chen, Y.-H. Chen, C.-Y. Chen and S.-S. Sun, Org. Lett., 2006, 8, 5053–5056 CrossRef CAS; (h) C.-L. Chen, T.-P. Lin, Y.-S. Chen and S.-S. Sun, Eur. J. Org. Chem., 2007, 3999–4010 CrossRef CAS; (i) S. O. Kang, V. W. Day and K. Bowman-James, J. Org. Chem., 2010, 75, 277–283 CrossRef CAS; (j) V. Amendola, G. Bergamaschi, A. Buttafava, L. Fabbrizzi and E. Monzani, J. Am. Chem. Soc., 2010, 132, 147–156 CrossRef CAS.
- (a) C. Provent, E. Rivara-Minten, S. Hewage, G. Brunner and A. F. Williams, Chem. Eur. J., 1999, 5, 3487–3494 CrossRef CAS; (b) S. Hiraoka, T. Yi, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2002, 124, 14510–14511 CrossRef CAS; (c) L. Pirondini, A. G. Stendardo, S. Geremia, M. Campagnolo, P. Samori, J. P. Rabe, R. Fokkens and E. Dalcanale, Angew. Chem. Int. Ed., 2003, 42, 1384–1387 CrossRef CAS; (d) K. Suzuki, M. Kawano and M. Fujita, Angew. Chem. Int. Ed., 2007, 46, 2819–2822 CrossRef CAS; (e) K. Harano, S. Hiraoka and M. Shionoya, J. Am. Chem. Soc., 2007, 129, 5300–5301 CrossRef CAS; (f) T. Haino, T. Fujii, A. Watanabe and U. Takayanagi, Proc. Natl. Acad. Sci. USA, 2009, 106, 10477–10481 CrossRef CAS; (g) J. Betancourt, M. Martin-Hidalgo, V. Gubala and J. M. Rivera, J. Am. Chem. Soc., 2009, 131, 3186–3188 CrossRef CAS; (h) F. Weng, J. Zhang, X. Ding, S. Dong, M. Liu, B. Zheng, S. Li, L. Wu, Y. Yu, H. W. Gibson and F. Huang, Angew. Chem. Int. Ed., 2010, 49, 1090–1094 CrossRef; (i) L. D. Shirtcliff, H. Xu and F. Diederich, Eur. J. Org. Chem., 2010, 846–855 CrossRef CAS; (j) S. Hiraoka, T. Nakamura, M. Shiro and M. Shionoya, J. Am Chem. Soc., 2010, 132, 13223–13225 CrossRef CAS; (k) J. Tian, S. J. Dalgarno and J. L. Atwood, J. Am. Chem. Soc., 2011, 133, 1399–1404 CrossRef CAS.
- (a) A. S. Singh, B.-Y. Chen, Y.-S. Wen, C. Tsai and S.-S. Sun, Org. Lett., 2009, 11, 1867–1870 CrossRef CAS; (b) C.-Y. Hung, A. S. Singh, C.-W. Chen, Y.-S. Wen and S.-S. Sun, Chem. Commun., 2009, 1511–1513 RSC; (c) A. S. Singh and S.-S. Sun, J. Org. Chem., 2012, 77, 1880–1890 CrossRef CAS.
- A. S. Singh and S.-S. Sun, Chem. Commun., 2011, 47, 8563–8565 RSC.
- (a) K. C. Russell, E. Leize, A. V. Dorsselaer and J.-M. Lehn, Angew. Chem. Int. Ed. Engl., 1995, 34, 209–213 CrossRef CAS; (b) B. Olenyuk, J. A. Whiteford, A. Fechtenkotter and P. J. Stang, Nature, 1999, 398, 796–799 CrossRef CAS.
- J. M. Mahoney, K. A. Stucker, H. Jiang, I. Carmichael, N. R. Brinkmann, A. M. Beatty, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 2922–2928 CrossRef CAS.
- (a) All reported receptors are soluble in polar solvent such as DMSO-d6. However, binding of nitrate anion with artificial receptor in polar solvent such as DMSO-d6 is either very mild or no binding at all. See references: A. P. Bisson, V. M. Lynch, M. K. C. Monahan and E. V. Anslyn, Angew. Chem., Int. Ed., 1997, 36, 2340–2342 CrossRef CAS; (b) S. Mason, T. Clifford, L. Seib, K. Kuczera and K. Bowman-James, J. Am. Chem. Soc., 1998, 120, 8899–8900 CrossRef CAS; (c) K. Choi and A. D. Hamilton, J. Am. Chem. Soc., 2001, 123, 2456–2457 CrossRef CAS; (d) C. R. Bondy and S. J. Loeb, Coord. Chem. Rev., 2003, 240, 77–99 CrossRef CAS; (e) S. O. Kang, R. A. Begum and K. Bowman-James, Angew. Chem. Int. Ed., 2006, 45, 7882–7894 CrossRef CAS; (f) M. Arunachalam and P. Ghosh, Inorg. Chem., 2010, 49, 943–951 CrossRef CAS.
- (a) C. Schmuck and W. Wienand, J. Am. Chem. Soc., 2003, 125, 452–459 CrossRef CAS; (b) C. Rether, E. Verheggen and C. Schmuck, Chem. Commun., 2011, 47, 9078–9079 RSC.
- O. Mogck, M. Pons, V. Böhmer and W. Vogt, J. Am. Chem. Soc., 1997, 119, 5706–5712 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: experimental characterization data, titration spectra and crystallographic details. CCDC 777053 and 863976 for nitrate complex 6a and self-assembled dimer 52 respectively. See DOI: 10.1039/c2ra20759a/ |
| This journal is © The Royal Society of Chemistry 2012 |