Oxidation of diclofenac catalyzed by manganese porphyrins: synthesis of novel diclofenac derivatives†
Cláudia M. B.
Neves
a,
Mário M. Q.
Simões
*a,
M.Rosário M.
Domíngues
a,
Isabel C. M. S.
Santos
a,
M.Graça P. M. S.
Neves
*a,
Filipe A. Almeida
Paz
b,
Artur M. S.
Silva
a and
José A. S.
Cavaleiro
a
aQOPNA, Chemistry Department, University of Aveiro, Aveiro 3810-193, Portugal. E-mail: msimoes@ua.pt (M.M.Q. Simões); Fax: +351 234 370 084; Tel: +351 234 370 713
bCICECO, Chemistry Department, University of Aveiro, Aveiro 3810-193, Portugal
First published on 13th July 2012
Abstract
The oxidation of drugs using metalloporphyrins has been the subject of several studies in recent years. Diclofenac, one of the most frequently used anti-inflammatory drugs, is metabolized in humans by cytochrome P450 (CYP) enzymes to hydroxy-derivatives and to some metabolites resulting from oxidative decarboxylation. In this paper, the in vitro formation of several new diclofenac derivatives, initially resulting from oxidative decarboxylation, similar to what happens in vivo, is revealed. Chloro [5,10,15,20-tetrakis(2,6-dichlorophenyl)porphyrinato]manganese(III), [Mn(TDCPP)Cl], and chloro [5,10,15,20-tetrakis(pentafluorophenyl)porphyrinato]manganese(III), [Mn(TPFPP)Cl], are tested in the presence of hydrogen peroxide at room temperature. The new products obtained are fully characterized, three of them being characterized in the solid state using X-ray diffraction studies.
Introduction
Diclofenac, 2-[(2,6-dichlorophenyl)amino]benzeneacetic acid (1), one of the most frequently used anti-inflammatory drugs, is metabolized in humans by cytochrome P450 (CYP) enzymes to hydroxyl-derivatives, among which 4′-hydroxydiclofenac, the major metabolite, and 5-hydroxydiclofenac were isolated and characterized.1–3 Some metabolites resulting from oxidative decarboxylation of diclofenac mediated by CYP enzymes have also been reported.4Pharmaceuticals, as potential environmental contaminants, have received increasing scientific and public attention in the last years.5–7 In particular, diclofenac (1) and its metabolites are recurrently detected in many environmental matrices and much effort has been made to remove these compounds from the environment, usually by oxidative processes.8–10 Synthetic models are helpful, not only for understanding the behaviour of pharmaceuticals in the environment, but also in the prediction of drugs' mechanism of action. Thus, biomimetic models may allow the production of metabolites or metabolite candidates, and even the isolation and identification of unstable in vivo intermediates.11 Metalloporphyrins (MPs) are recognized as excellent catalysts able to mimic the oxidation reactions catalyzed by CYP enzymes, including epoxidation or hydroxylation.11–13 Some authors have reported the hydroxylation of diclofenac catalyzed by MPs with ammonium acetate as co-catalyst and hydrogen peroxide or t-butyl hydroperoxide as oxidants.14–16 Moreover, in the presence of oxidants such as periodates, iodosylbenzene, N-oxides or hydrogen peroxide, metalloporphyrins have shown to be good catalysts for the oxidative decarboxylation of carboxylic acids and some anti-inflammatory drugs having a carboxyl moiety.17–23
Our group was able to build up a line of research in the field of biomimetic oxidation of organic compounds using H2O2, a cheap and waste-avoiding oxidant, and Mn(III) or Fe(III) porphyrins as catalysts.23–34 In this paper, our recent studies on the oxidation of diclofenac (1) using hydrogen peroxide as oxidant and manganese(III) porphyrins as catalysts are presented. Chloro [5,10,15,20-tetrakis(2,6-dichlorophenyl)porphyrinato]manganese(III), [Mn(TDCPP)Cl] (CAT-1), chloro [5,10,15,20-tetrakis(pentafluorophenyl)porphyrinato]manganese(III), [Mn(TPFPP)Cl] (CAT-2) (Fig. 1) and three different co-catalysts were used, namely ammonium acetate, sodium acetate/acetic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and imidazole. Several products have been observed, mainly resulting from an initial oxidative decarboxylation process (Fig. 2).
:1) and imidazole. Several products have been observed, mainly resulting from an initial oxidative decarboxylation process (Fig. 2).
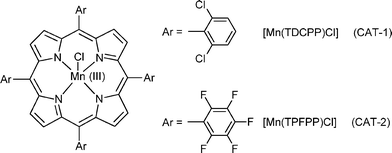 | ||
| Fig. 1 Structures of the catalysts used in the present study. | ||
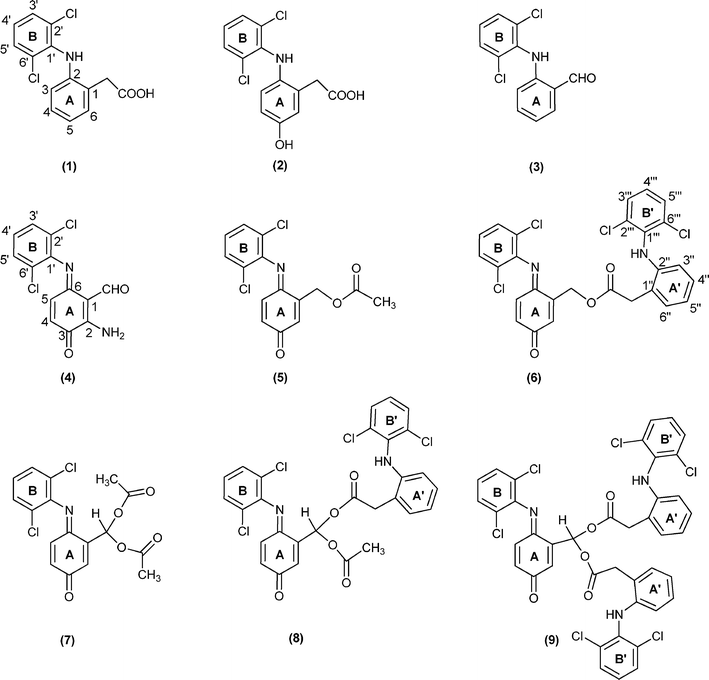 | ||
| Fig. 2 Structures of diclofenac and diclofenac derivatives. | ||
Results and discussion
The oxidation reactions of diclofenac (1) using the selected manganese(III) porphyrins as catalysts (Fig. 1) were performed with progressive additions of diluted H2O2, under normal atmosphere, in a mixture of acetonitrile/water at 30 °C. The reactions were monitored both by TLC and by reverse-phase HPLC and the conversion of 1 was determined by HPLC using p-chloroacetophenone as internal standard. The addition of H2O2 was stopped when no further conversion of 1 was observed after two successive HPLC analyses. Compounds 3–9 (Fig. 2) were isolated by TLC and fully characterized by mass spectrometry, 1D and 2D NMR studies. The structures of compounds 4, 6 and 7 were unambiguously elucidated by X-ray crystallography (see details below). The structural details of compound 4 have recently been described.35The quantification of the products obtained under the different catalytic conditions studied (Table 1) was based on the analysis of the NMR spectra of the final reaction mixtures; the ratio between products 3–9 was determined using selected diagnostic signals for each derivative (vide infra). These analyses were performed after passing the reaction mixtures through a column to remove the paramagnetic manganese catalyst.
| Catalyst | Co-catalyst | H2O2 (equiv.) | Time (h) | Conv. (%)b | Ratio (%)c | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 5 | 6 | 7 | 8 | 9 | |||||
|
a Reaction conditions: 0.1 mmol of diclofenac, 1.33 × 10−3 mmol of catalyst (a solution in CH3CN) and 0.2 mmol of ammonium acetate, or 0.2 mmol imidazole, or sodium acetate/acetic acid mixture (0.1 mmol + 0.1 mmol) in CH3CN:H2O (10:1), for a total volume of 2.0 mL, under normal atmosphere at 30 °C; the oxidant used was 30% H2O2 (w/w) diluted (1:5) in CH3CN and 0.05 mmol were added to the reaction mixtures every 15 min.
b Determined by reverse phase HPLC.
c Determined by 1H NMR.
|
|||||||||||
| CAT-1 | Ammonium acetate | 16 | 8 | 78 | 6 | 23 | 11 | 25 | 7 | 18 | 10 |
| Sodium acetate/acetic acid (1:1) |
16 | 8 | 59 | 48 | 0 | 4 | 10 | 8 | 18 | 12 | |
| Imidazole | 16 | 8 | 64 | 13 | 0 | 0 | 44 | 0 | 0 | 42 | |
| CAT-2 | Ammonium acetate | 6 | 3 | 22 | 12 | 12 | 3 | 17 | 3 | 18 | 35 |
| Without | Ammonium acetate | 8 | 4 | 0 | — | — | — | — | — | — | — |
Under the present conditions, besides the aromatic ring (A) hydroxylation and subsequent quinone-imine formation, the main products result from the oxidative decarboxylation of diclofenac; this then leads to the formation of several ester (5 and 6) and geminal diester (7–9) derivatives.
The analysis of Table 1 shows that the best conversions (59–78%) of 1 were achieved when [Mn(TDCPP)Cl] was used as catalyst. The formation and the relative proportions of the products seem to be related with the co-catalyst used. Compound 4 was only observed in the presence of ammonium acetate. The presence of the amino group in the structure of this compound let us to conclude that its formation results from the reaction with ammonium acetate. Therefore, in order to avoid this side product, further reactions using a sodium acetate/acetic acid mixture (1:1) as co-catalyst were performed.36 Under these conditions the absence of compound 4 was accompanied by an increase in the amount of compound 3 (48% vs. 6%). However, products 5, 7 and 8 resulting from esterification with the acetic acid present in the reaction medium were still obtained. In order to avoid the plethora of products due to the esterification pathway with acetic acid, imidazole was used as the co-catalyst. Under these conditions the formation of aldehyde 3 was accompanied by the formation of only the monoester 6 and of the geminal diester 9, both resulting from the esterification with diclofenac.
The oxidation of diclofenac in the presence of [Mn(TPFPP)Cl] and ammonium acetate showed to be much less efficient. The conversion of diclofenac (22%) stopped after 3 h of reaction. This result can be related to the well known degradation of metalloporphyrin catalysts, namely when H2O2 is used as oxidant.37,38 The absence of the typical Soret band in the UV-vis spectrum of the reaction mixture after 3 h of reaction seems to corroborate the degradation of the catalyst as the chief reason for the lower efficiency of this catalyst when compared with [Mn(TDCPP)Cl]. Blank experiments were performed, without catalyst, and no diclofenac conversion was registered using HPLC.
Mechanistic considerations
Scheme 1 illustrates two putative mechanistic proposals for the formation of the obtained diclofenac derivatives 2–9. Mansuy and collaborators have isolated the quinone–imine intermediate 10 in the oxidation of diclofenac with metalloporphyrins and obtained 2 by the reduction of 10 with NaBH4.16 In the present work, compound 2 was observed using HPLC at the beginning of the oxidation reactions, almost disappearing during the oxidation process. However, we were able to isolate a small amount of compound 2 using HPLC and to obtain its mass spectrum, which is in accordance with the literature data.10 This let us to suggest that diclofenac derivatives may result from a pathway involving the formation of intermediate 2 that can be oxidized to the quinone–imine intermediate 10, which after decarboxylation affords intermediate 11 (Scheme 1 - pathway a). The oxidation of 11 originates alcohol 12, the putative precursor of monoesters 5 and 6, through esterification with acetic acid or another molecule of diclofenac, respectively. Further oxidation of 12 can afford aldehyde 13 or the geminal diol 14, potential precursor of the symmetric geminal diesters (also known as acylals) 7 and 9 and of the asymmetric geminal diester 8 obtained by esterification with the acids present, namely diclofenac and acetic acid. One possibility is the formation of geminal diol 14 during the oxidation process,39,40 being rapidly quenched by the acidic components present in the reaction medium. Geminal diesters are usually prepared from aldehydes and acid anhydrides using strong protic acids, a different approach being presented here.41,42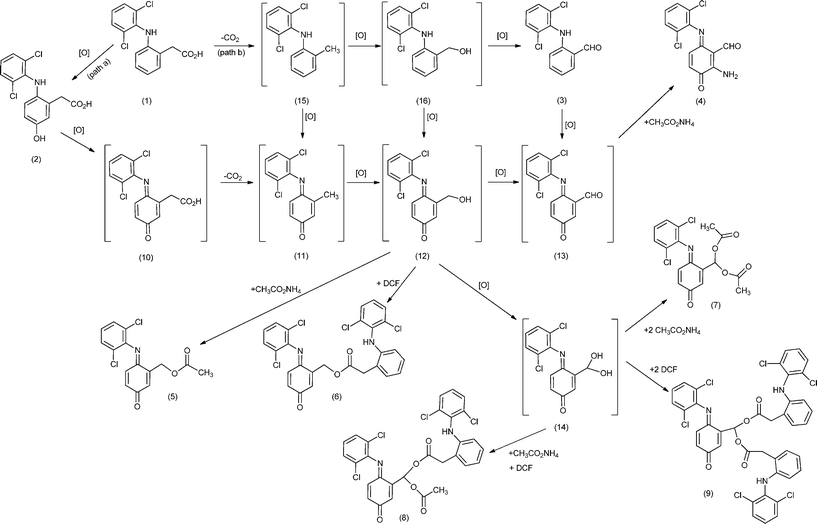 | ||
| Scheme 1 Putative mechanism for the formation of diclofenac (DCF) derivatives. The compounds between brackets are potential intermediates. | ||
The second mechanistic proposal is based on the formation and isolation of aldehyde 3 (Scheme 1 - pathway b), supporting the possibility of a decarboxylation process to occur before the hydroxylation of ring A, affording intermediate 15. The oxidation of 15 into 11 and/or 16 can justify, after further oxidation, the formation of 12, the precursor of the monoesters. Similarly, both the oxidation of 16 and/or 12 can justify the formation of geminal diol 14, the precursor of the geminal diesters. The formation of compound 4 can be justified through a Michael addition reaction between 13 and NH3 from ammonium acetate present in the reaction medium. So, compound 3 should result from the decarboxylation of 1 followed by oxidation, as already described in the literature for diclofenac, in vivo,4 and for other carboxylic acids, using a supported manganese(III) porphyrin.23
Structural elucidation
The structures of the new products 4–9 were established based on a careful analysis of their 1H and 13C NMR spectra and their molecular formulae were confirmed by HRMS. 2D NMR studies (COSY, HMBC and HSQC) were also carried out in order to unequivocally identify the proton and carbon resonances that support the proposed structures. In addition, the structures of compounds 4, 6 and 7 were unambiguously established from single crystal X-ray crystallography (see discussion below).The MS data obtained for 2 is in agreement with the literature.10 In fact, an electrospray (ESI) mass spectrum (MS) of 2 showed the [M+H]+ ion at m/z 312 and the corresponding sodium adduct [M+Na]+ ion at m/z 334. The formation of both ions confirmed the presence of a molecular species with 311 of molecular weight (compound 2). The MS/MS spectrum of the [M+H]+ showed the product ions at m/z 294 (due to loss of H2O), at m/z 266 (due to loss of HCOOH, confirming the presence of a carboxylic group) and at m/z 231 and 230, due to loss of Cl˙ and HCl, the latter ones confirming the presence of chlorine atoms in the molecule. In order to confirm the proposed structure based on the fragmentation pathways, and since the sample was very pure, pseudo MS3 spectra of ions at m/z 294 and 266 were acquired. These ions were formed in the source by increasing the cone voltage to 40 V, producing in-source fragmentation. This procedure gives similar information of MS3 spectra obtained in an ion trap mass spectrometer.43 The pseudo MS3 spectrum of the ion at m/z 294 showed the product ion at m/z 266 while the pseudo MS3 spectrum of the ion m/z 266 showed the ion at m/z 231.
The mass spectrum of 3 was obtained by GC-MS and is in accordance to the literature.44 Moreover, the 1H NMR spectrum of 3 shows, besides the singlets at δ 9.99 and 9.79 ppm identified as the CHO and NH protons, respectively, a triplet at δ 7.19 ppm and a doublet at δ 7.43 ppm corresponding, respectively, to the proton resonances of 4′-H and 3′,5′-H of the chlorinated B ring. The signals due to the A ring of 3 appear as a broad doublet at δ 6.35 ppm (3-H), a double triplet at δ 6.90 ppm (5-H), a double doublet of doublets at δ 7.35 ppm (4-H), and a double doublet at δ 7.62 ppm (6-H). This pattern is in agreement with that observed for diclofenac in the aromatic region and confirms that both aryl units A and B of 3 are intact.
The 1H NMR spectrum of aldehyde 4 presents a singlet at δ 10.69 ppm corresponding to the resonance of the CHO proton, and two doublets at δ 6.59 and 6.70 ppm corresponding to 4-H and 5-H, respectively. The resonances of the NH2 protons appear as two broad singlets at δ 6.49 and 9.70 ppm due to the different intramolecular N−H⋯O hydrogen bonds. The signals of ring B of 4 maintain the characteristic pattern already described for aldehyde 3.
The 1H NMR spectra of the more complex compounds 5–9 are consistent with the presence of the quinone-imine ring A showing resonances attributed to the protons 2-H, 4-H and 5-H between δ 6.4 and 6.7 ppm. The characteristic pattern of the B ring is maintained in all these derivatives confirming that the oxidation did not occur in this unit. In addition, the multiplicity of 2-H at ring A, due to the long-range coupling with the CH2 protons in derivatives 5 and 6, or with the CH proton in derivatives 7–9 directly linked to ring A can be used as a fingerprint tool to confirm the structure of these derivatives as an ester (5 and 6) or a diester (7–9).
In particular, the 1H NMR spectrum of compound 5 shows the resonances of methylene protons as a doublet (J = 1.9 Hz) at δ 5.33 ppm and the resonance of protons 2-H as a double triplet (J = 1.9 and J = 2.1 Hz), since it is also coupled with 4-H, at δ 6.70 ppm. The resonances of protons 4-H and 5-H appear as a double doublet and as a doublet at δ 6.47 and 6.65 ppm. The proton resonance of the methyl group of 5 appears as a singlet at δ 2.20 ppm. The 1H NMR spectrum of compound 6 shows the proton resonances of rings A (2-H, 4-H and 5-H) and B (3′-H, 4′-H and 5′-H) and of the methylene protons with a similar pattern to that described for compound 5. In the aliphatic region, the 1H NMR spectrum of 6 presents a singlet at δ 3.95 ppm assigned to the methylene protons of the extra diclofenac unit (CH2 linked to ring A'). Based on the 2D NMR studies, COSY, HSQC and HMBC spectra (Fig. 3 presents the main HMBC correlations), it was possible to identify all the proton and carbon resonances of 6, namely the signals of the two different 2,6-dichlorophenyl units (rings B and B').
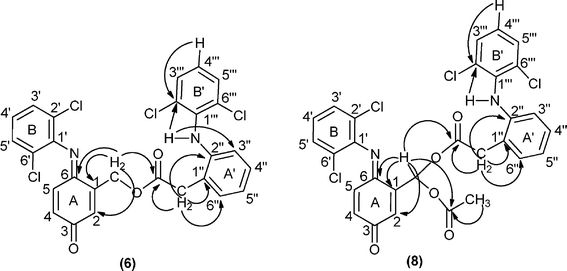 | ||
| Fig. 3 Main connectivities in the HMBC spectra of compounds 6 and 8. | ||
Considering the 1H NMR spectra of diesters 7–9, the proton resonance of the geminal linkage was identified as the doublet occurring at δ 8.07 ppm for 7, at δ 8.16 ppm for 8 and at δ 8.22 ppm for 9 due to the long-range coupling with 2-H of ring A. The resonance of proton 2-H of the quinone-imine ring A appears as a double doublet at δ 6.86 ppm for 7, at δ 6.79 ppm for 8 and at δ 6.68 ppm for 9, due to the long-range coupling with the methynic proton of the geminal linkage and with 4-H from ring A.
For the symmetric diester 7, the signal in the aliphatic region at δ 2.17 ppm integrating for six protons was attributed to the methyl protons of the acetate group. Considering the same region from the spectrum of the asymmetric diester 8, in addition to the singlet at δ 2.16 ppm also attributed to the acetate group, an AB spin system at δ 3.91 and 3.89 ppm were observed, which were assigned to the diastereotopic methylene protons of the extra diclofenac unit. A similar feature was observed for the methylenic protons of the two extra diclofenac units of diester 9. The resonances of these four aliphatic protons also appear as an AB spin system at δ 3.88 and 3.94 ppm.
Although the 1H and 13C NMR spectra of diesters 8 and 9 present a complex pattern in the aromatic region due to the extra diclofenac units, the assignment of the signals was possible based on correlations observed in the COSY and HSQC spectra, but the HMBC correlations were the most important ones to establish the connectivities between the moieties of these molecules, as exemplified for diester 8 in Fig. 3.
The most important features in the 13C NMR spectra of the new compounds are the carbon resonances assigned to: i) the imino (δ 158.2–159.3 ppm) and keto-carbonyl groups (δ 182.3–186.8 ppm) of the quinone-imine moiety for compounds 4–9; ii) the ester groups (δ 167.9–171.6 ppm) for compounds 5–9; iii) the methyne carbon of the geminal linkage (δ 85.0–88.6 ppm) for compounds 7–9; iv) the methylenic carbon directly linked to ring A (δ 60.8 and 61.6 ppm) for the monoesters 5 and 6; v) the aldehyde group (δ 193.5 ppm) for compound 4.
The signals used for the quantification of compounds 3–9 by 1H NMR were those of the aldehyde protons at δ 9.99 ppm for 3 and at δ 10.69 ppm for 4, those of the methylene protons at δ 5.33 ppm for 5 and at δ 5.40 ppm for 6, those of the methyne protons at δ 8.07 ppm for 7, at δ 8.16 ppm for 8 and at δ 8.22 ppm for 9.
Single-crystal X-ray diffraction studies
Compounds 2-amino-6-(2,6-dichlorophenylimino)-3-oxocyclohexa-1,4-dienecarbaldehyde (4), [6-(2,6-dichlorophenylimino)-3-oxocyclohexa-1,4-dien-1-yl]methyl 2-[2-(2,6-dichlorophenylamino)phenyl]acetate (6) and [6-(2,6-dichlorophenylimino)-3-oxocyclohexa-1,4-dien-1-yl]methylene diacetate (7) were successfully recrystallized as small single-crystals and their crystal structures unequivocally elucidated using X-ray diffraction. The structural details of compound 4 have recently been described and we refer the reader to this previous publication for a detailed structural description of its crystal features.35Compound 6 crystallizes in the centrosymmetric P21/n monoclinic space group with the asymmetric unit being composed of a whole molecular unit as depicted in Fig. 4. Despite the ability of the molecule to permit rotational movements around the central ester group, the combination of a strong intramolecular N−H⋯O hydrogen bond (between the amine group and the adjacent carbonyl moiety) [internuclear distance of 2.833(5) Å], and the presence of two peripheral bulky 2,6-dichlorobenzene substituents, leads to a robust molecular unit which close packs in the solid state driven by the need to effectively fill the available space (Fig. 5). Indeed, the existence of two chloride atoms in each peripheral section of the molecule induces, on the one hand, a rotation of the 2,6-dichlorobenzene substituents with respect to the neighbouring six-membered rings so order to minimize steric repulsion between such adjacent bulky moieties: the C16→C21 ring subtends an angle of ca. 64° with the C22→C27 ring, while the analogous angle between the C1→C6 and C7→C12 rings is of ca. 75°. On the other hand, due to the same steric hindrance effects, rotation is thus severely limited, hence leading to the aforementioned structural robustness of the molecular unit. We note the remarkable absence of π–π contacts in the crystal structure, most likely also due to the significantly twisted nature of the molecular unit. The close packing of individual molecules of 6 also leads to the concomitant formation of a relatively small number of weak C−H⋯O hydrogen bonding interactions. Worth of note is the C9−H9⋯O1 interaction which is rather strong [dD⋯A of 3.357(6) Å] and directional [∠(DHA) greater than 160°], ultimately establishing an effective bridge between adjacent molecular units of 6 (thus forming pseudo-dimers) via the creation of a R22(8) graph set motif (not shown).45 Additional C−H⋯O supramolecular contacts in conjunction with a weak Cl⋯π contact interconnect adjacent pseudo-dimers (Table 2) yielding the crystal structure of 6.
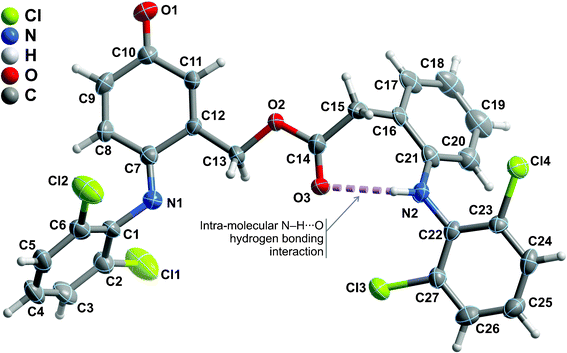 | ||
| Fig. 4 Schematic representation of the asymmetric unit of compound 6 which is composed by a whole molecular unit. Non-hydrogen atoms are represented as thermal ellipsoids drawn at the 50% probability level and hydrogen atoms as small spheres with arbitrary radius. The atomic labelling of all non-hydrogen atoms is provided. Intramolecular hydrogen bonding interactions are represented as dashed violet lines. For geometrical details on the represented supramolecular interactions see Table 2. | ||
![Ball-and-stick representation of the crystal packing of compound 6 viewed in perspective along the [010] direction of the unit cell. Supramolecular interactions (both intra- and intermolecular) have been omitted for clarity. For geometrical details on these supramolecular interactions see Table 2.](/image/article/2012/RA/c2ra20801f/c2ra20801f-f5.gif) | ||
| Fig. 5 Ball-and-stick representation of the crystal packing of compound 6 viewed in perspective along the [010] direction of the unit cell. Supramolecular interactions (both intra- and intermolecular) have been omitted for clarity. For geometrical details on these supramolecular interactions see Table 2. | ||
| D–H⋯A | d(D⋯A) | ∠(DHA) |
|---|---|---|
| a Symmetry transformations used to generate equivalent atoms: (i) 1 − x, 2 − y, −z; (ii) 1 − x, 1 − y, −z; (iii) x, −1 + y, z. b Centres of gravity (Cg) (see Fig. 4): Cg1 = C7, C8, C9, C10, C11 and C12. | ||
| N2−H2⋯O3 | 2.833(5) | 127 |
| C9−H9⋯O1i | 3.357(6) | 167 |
| C13−H13A⋯O1ii | 3.360(6) | 135 |
| C15−H15B⋯O1iii | 3.397(6) | 154 |
| Cl⋯π Contacts | d(Cl⋯π) | ∠(CClπ) |
|---|---|---|
| C27−Cl3⋯Cg1iii | 3.962(3) | 77.04(14) |
Compound 7 crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with the asymmetric unit being instead composed of two whole crystallographically independent molecular units as shown in Fig. 6a. As depicted in the overlay representation (Fig. 6b), the distinction between the two moieties concerns solely a rotation about the C−C bond of the tertiary carbon while the remaining portion of the molecules remains approximately unaltered: the C−H bond is, thus, in opposite sites of the plane defined by the nitrogen atom and the oxocyclohexa-1,4-dien-1-yl ring six-membered ring, leading to +syn-clinal and -syn-clinal conformations which correspond to torsion angles of ca. 58 and −55°, respectively. These two crystallographically independent moieties are at the genesis of two independent supramolecular one-dimensional chains which close pack in the ab plane of the unit cell in a typical brick-wall-like fashion to yield the structure of 7 (Fig. 7). Each chain is assembled by a combination of strong onset π–π contacts between symmetry-related 2,6-dichlorobenzene substituents [inter-centroid distances ranging from 3.464(4) to 3.620(4) – Fig. 7b] and weak C−H⋯O supramolecular contacts involving, mainly, the terminal –CH3 groups and adjacent carbonyl moieties (not shown). Despite the low directionality of these latter interactions [all ∠(DHA) interaction angles are smaller than 139° - Table 3] they induce nevertheless a close proximity between donor and acceptor [dD⋯A ranging from 3.217(10) to 3.413(8) Å], hence being of decisive importance in the supramolecular assembly of the crystal structure of 7. Connections between adjacent chains are mainly of the Cl⋯π type and, remarkably, involve only one chloride atom: while the moiety on the right of Fig. 6a interacts via Cl2 with the neighbouring C24→C29 ring, the moiety on the left is instead interacting with the C7→C12 ring via the chloride atom Cl3 (see Table 3 for additional geometric details). A note worth mentioning concerns the fact that these Cl⋯π interactions only occur with the oxocyclohexa-1,4-dien-1-yl ring, in particular with the localised double bonds belonging to the residue.
space group with the asymmetric unit being instead composed of two whole crystallographically independent molecular units as shown in Fig. 6a. As depicted in the overlay representation (Fig. 6b), the distinction between the two moieties concerns solely a rotation about the C−C bond of the tertiary carbon while the remaining portion of the molecules remains approximately unaltered: the C−H bond is, thus, in opposite sites of the plane defined by the nitrogen atom and the oxocyclohexa-1,4-dien-1-yl ring six-membered ring, leading to +syn-clinal and -syn-clinal conformations which correspond to torsion angles of ca. 58 and −55°, respectively. These two crystallographically independent moieties are at the genesis of two independent supramolecular one-dimensional chains which close pack in the ab plane of the unit cell in a typical brick-wall-like fashion to yield the structure of 7 (Fig. 7). Each chain is assembled by a combination of strong onset π–π contacts between symmetry-related 2,6-dichlorobenzene substituents [inter-centroid distances ranging from 3.464(4) to 3.620(4) – Fig. 7b] and weak C−H⋯O supramolecular contacts involving, mainly, the terminal –CH3 groups and adjacent carbonyl moieties (not shown). Despite the low directionality of these latter interactions [all ∠(DHA) interaction angles are smaller than 139° - Table 3] they induce nevertheless a close proximity between donor and acceptor [dD⋯A ranging from 3.217(10) to 3.413(8) Å], hence being of decisive importance in the supramolecular assembly of the crystal structure of 7. Connections between adjacent chains are mainly of the Cl⋯π type and, remarkably, involve only one chloride atom: while the moiety on the right of Fig. 6a interacts via Cl2 with the neighbouring C24→C29 ring, the moiety on the left is instead interacting with the C7→C12 ring via the chloride atom Cl3 (see Table 3 for additional geometric details). A note worth mentioning concerns the fact that these Cl⋯π interactions only occur with the oxocyclohexa-1,4-dien-1-yl ring, in particular with the localised double bonds belonging to the residue.
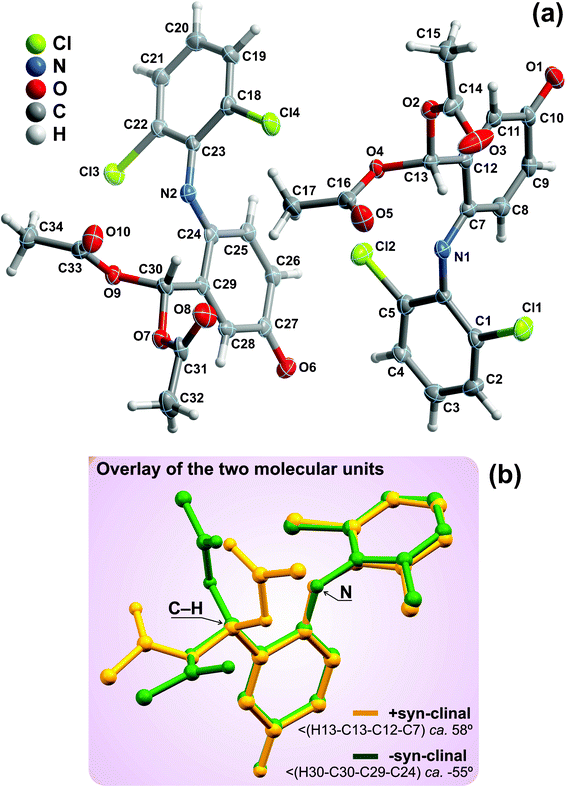 | ||
| Fig. 6 (a) Schematic representation of the two whole molecular units composing the asymmetric unit of compound 7. Non-hydrogen atoms are represented as thermal ellipsoids drawn at the 30% probability level and hydrogen atoms as small spheres with arbitrary radius. The atomic labelling of all non-hydrogen atoms is provided. (b) Structural overlap of the two crystallographically independent molecular units emphasising the two distinct conformations which arise from the rotation along the tertiary carbon atom. Structural drawing created using the software package Mercury.57 | ||
![(a) Ball-and-stick representation of the crystal packing of compound 7 viewed in perspective along the [001] direction of the unit cell, emphasising the close packing in a brick-wall-like fashion of supramolecular one-dimensional chains parallel to the c-axis of the unit cell. Supramolecular interactions have been omitted for clarity. (b) Detail view of one chain showing the close π–π contacts between aromatic rings belonging to adjacent molecular units. For geometric details on the represented (and also omitted) supramolecular interactions see Table 3.](/image/article/2012/RA/c2ra20801f/c2ra20801f-f7.gif) | ||
| Fig. 7 (a) Ball-and-stick representation of the crystal packing of compound 7 viewed in perspective along the [001] direction of the unit cell, emphasising the close packing in a brick-wall-like fashion of supramolecular one-dimensional chains parallel to the c-axis of the unit cell. Supramolecular interactions have been omitted for clarity. (b) Detail view of one chain showing the close π–π contacts between aromatic rings belonging to adjacent molecular units. For geometric details on the represented (and also omitted) supramolecular interactions see Table 3. | ||
| D–H⋯A | d(D⋯A) | ∠(DHA) |
|---|---|---|
| a Symmetry transformations used to generate equivalent atoms: (i) 1 − x, −y, 1 − z; (ii) 1 − x, 1 − y, 1 − z; (iii) x, 1 + y, z; (iv) −1 + x, 1 + y, z; (v) −x, 2 − y, −z; (vi) 1 − x, 1 − y, −z; (vii) −x, 2 − y, 1 − z. b Centres of gravity (Cg) (see Fig. 6):Cg1 = C24, C25, C26, C27, C28 and C29;Cg2 = C7, C8, C9, C10, C11 and C12;Cg3 = C1, C2, C3, C4, C5 and C6;Cg4 = C18, C19, C20, C21, C22 and C23. | ||
| C15−H15C⋯O3i | 3.413(8) | 152 |
| C17−H17C⋯O1ii | 3.343(7) | 139 |
| C25−H25⋯O5iii | 3.296(8) | 132 |
| C34−H34A⋯O3iv | 3.217(10) | 125 |
| C34−H34B⋯O6v | 3.309(9) | 132 |
| Cl⋯π Contacts | d(Cl⋯π) | ∠(CClπ) |
|---|---|---|
| C5−Cl2⋯Cg1 | 3.586(3) | 121.4(2) |
| C22−Cl3⋯ Cg2iv | 3.591(3) | 128.1(2) |
| π–π Contacts | Inter-centroid distance | |
|---|---|---|
| Cg 3⋯Cg3vi | 3.464(4) | |
| Cg 4⋯Cg4vii | 3.620(4) | |
Experimental
General oxidation procedureThe oxidation reactions were carried out using 0.1 mmol of diclofenac sodium salt, 1.33 × 10−3 mmol of catalyst, the co-catalyst (0.2 mmol of ammonium acetate, 0.2 mmol of imidazole or 0.1 mmol of sodium acetate + 0.1 mmol of acetic acid), and p-chloroacetophenone (I.S.; 0.23 mmol) in CH3CN:H2O (10:1) in a total volume of 2.0 mL under normal atmosphere at 30 °C and protected from light. The oxidant used was aqueous hydrogen peroxide 30% (w/w) diluted (1:5) in CH3CN, and 0.05 mmol were added to the reaction mixture every 15 min. Reactions were monitored by TLC using dichloromethane as eluent and by HPLC, which was carried out using a Merck model L-6200A instrument equipped with a L-4250 UV-Vis Merck detector and a LiChrospher 100 RP-18.5 μm reversed-phase column. The mobile phase consisted initially of 45% solvent A [acetic acid in water (0.05M)] and 55% solvent B [acetonitrile] for 20 min, followed by a 10 min linear gradient going from 55% solvent B to 90% solvent B. This 10:90 (A:B) ratio was kept for more 30 min, followed by a 10 min re-equilibration to the initial conditions. The HPLC flow rate was 0.65 mL min−1 and the detector was set to λ = 276 nm. At the end of the reactions, the mixtures were analyzed by UV-Vis (Uvikon 922) to evaluate the catalyst degradation, extracted with CH2Cl2 and the organic compounds were separated by preparative TLC using dichloromethane as eluent, and analyzed by 1H and 13C NMR (Bruker Avance 300 spectrometer at 300.13 and 75.47 MHz, respectively), using CDCl3 as solvent and TMS as the internal reference. Chemical shifts (δ) are given in ppm and coupling constants (J) are given in Hz. High resolution mass spectral analyses (HRMS-ESI+) were performed with a microTOF (focus) mass spectrometer. Ions were generated with an ApolloII (ESI) source. Ionization was achieved by electrospray, with the use of a voltage of 4500 V applied to the needle, and a counter voltage between 100 and 150 V applied to the capillary. Melting points were determined with a Büchi melting point apparatus and are uncorrected.
The conversion of 1 was determined by HPLC using an internal standard, and the ratio between the products 3–9 was based on the 1H NMR spectra of the final reaction mixtures, after column chromatography using dichloromethane with 5% methanol as eluent. The GC-MS analysis of 3 was performed on a Finnigan Trace GC-MS (Thermo Quest CE instruments) using helium as the carrier gas (35 cm s−1). The column used was a silica capillary DB-5 type (30 m × 0.25 mm id, 0.25 μm film thickness). The chromatographic conditions were as follows: split ratio: 1/40; initial temperature: 120 °C (2 min); temperature rate: 10 °C min−1; final temperature: 260 °C (10 min); injector temperature: 270 °C; MS interface temperature: 270 °C.
A small amount of compound 2 was collected by HPLC, dried under vacuum in a Speed-vac apparatus to remove the acetonitrile and then analyzed by ESI-MS, after being taken in methanol. Electrospray mass spectrometry (ESI-MS) and tandem mass spectrometry (ESI-MS/MS) spectra in positive ion mode were acquired in a Q-TOF 2 instrument (Micromass, Manchester, UK). For the acquisition of mass spectra the needle voltage was set at 3000 V with the ion source at 80 °C and cone voltage at 15 V. Nitrogen was used as nebulizer gas and argon as collision gas. Tandem mass spectra (MS/MS) of molecular ions were performed by collision induced decomposition using argon as the collision gas and by changing collision energy between 15–20 eV. Pseudo MS3 spectra was obtained increasing the cone voltage to 40 V and MS/MS spectra were obtained for the pseudo product ions obtained in the source using the same experimental condition reported for the MS2.43,46 Data acquisition was carried out with a Micromass Masslynx 4.0 data system.
All reagents were purchased from commercial sources unless otherwise noted.
Characterization
2-[2-(2,6-dichlorophenylamino)-5-hydroxyphenyl]acetic acid or 5-hydroxydiclofenac (2): ESI-MS m/z: 334.0 [M+Na]+, 312.0 [M+H]+.10,472-(2,6-Dichlorophenylamino)benzaldehyde (3): δH (300 MHz; CDCl3) 6.35 (1H, br d, J = 8.4 Hz, 3-H), 6.90 (1H, dt, J = 7.4 and 0.9 Hz, 5-H), 7.19 (1H, t, J = 8.1 Hz, 4′-H), 7.35 (1H, ddd, J = 8.4, 7.4 and 1.5 Hz, 4-H), 7.43 (2H, d, J = 8.1 Hz, 3′,5′-H), 7.62 (1H, dd, J = 7.4 and 1.5 Hz, 6-H), 9.79 (1H, br s, NH), 9.99 (1H, s, CHO) ppm. MS (EI) m/z: 267 (38%), 266 (9), 265 (M+. , 60), 232 (18), 231 (10), 230 (56), 229 (9), 204 (11), 203 (11), 202 (33), 201 (20), 195 (13), 168 (14), 167 (100), 166 (29), 165 (8), 164 (11), 161 (8), 139 (9), 119 (15).44
2-Amino-6-(2,6-dichlorophenylimino)-3-oxocyclohexa-1,4-dienecarbaldehyde (4): m.p. 135–136 °C; δH (300 MHz; CDCl3) 6.49 (1H, br s, NH2), 6.59 (1H, d, J = 10.3 Hz, 4-H), 6.70 (1H, d, J = 10.3 Hz, 5-H), 7.03 (1H, dd, J = 8.3 and 7.7 Hz, 4′-H), 7.37 (2H, d, J = 8.0 Hz, 3′,5′-H), 9.70 (1H, br s, NH2), 10.69 (1H, s, CHO) ppm. δC (75 MHz; CDCl3) 105.8 (C-1), 125.1 (C-4′, C-2′,6′), 128.2 (C-3′,5′), 130.5 (C-5), 130.9 (C-4), 144.4 (C-1′), 146.4 (C-2), 158.2 (C-6), 182.3 (C-3), 193.5 (CHO) ppm. HRMS-ESI-TOF mz calcd for C13H9Cl2N2O2 [M+H]+ 295.0036, found 295.0035.
[6-(2,6-Dichlorophenylimino)-3-oxocyclohexa-1,4-dien-1-yl]methyl acetate (5): m.p. 137–138 °C; δH (300 MHz; CDCl3) 2.20 (3H, s, CH3), 5.33 (2H, d, J = 1.9 Hz, CH2), 6.47 (1H, dd, J = 10.1 and 2.1 Hz, 4-H), 6.65 (1H, d, J = 10.1 Hz, 5-H), 6.70 (1H, dt, J = 2.1 and 1.9 Hz, 2-H), 7.08 (1H, dd, J = 8.4 and 7.8 Hz, 4′-H), 7.38 (2H, d, J = 8.1 Hz, 3′,5′-H) ppm. δC (75 MHz; CDCl3) 20.8 (CH3), 60.8 (CH2), 124.1 (C-2′,6′), 126.0 (C-4′), 128.3 (C-3′,5′), 128.8 (C-5), 129.3 (C-2), 133.1 (C-4), 143.7 (C-1′), 145.6 (C-1), 159.3 (C-6), 170.2 (CO2CH3), 186.8 (C-3) ppm. HRMS-ESI-TOF mz calcd for C15H12Cl2NO3 [M+H]+ 324.0189, found 324.0187.
[6-(2,6-Dichlorophenylimino)-3-oxocyclohexa-1,4-dien-1-yl]methyl 2-[2-(2,6-dichlorophenylamino)phenyl]acetate (6): m.p. 135–136 °C; δH (300 MHz; CDCl3) 3.95 (2H, s, A'-CH2COOCH2-A), 5.40 (2H, d, J = 1.7 Hz, A'-CH2COOCH2-A), 6.46 (1H, dd, J = 10.1 and 2.2 Hz, 4-H), 6.56 (1H, br d, J = 7.7 Hz, 3′′-H), 6.63 (1H, d, J = 10.1 Hz, 5-H), 6.67 (1H, dt, J = 2.2 and 1.7 Hz, 2-H), 6.80 (1H, br s, NH), 6.98 (1H, dd, J = 8.3 and 7.8 Hz, 4′′′-H), 6.99 (1H, dt, J = 7.7 and 1.0 Hz, 5′′-H), 7.05 (1H, dd, J = 8.2 and 8.1 Hz, 4′-H), 7.15 (1H, dt, J = 7.7 and 1.5 Hz, 4′′-H), 7.28 (1H, dd, J = 7.7 and 1.5 Hz, 6′′-H), 7.33 (2H, d, J = 8.1 Hz, 3′′′,5′′′-H), 7.36 (2H, d, J = 8.1 Hz, 3′,5′-H) ppm. δC (75 MHz; CDCl3) 38.4 (A'-CH2COOCH2-A), 61.6 (A'-CH2COOCH2-A), 118.4 (C-3′′), 122.2 (C-5′′), 124.0 and 124.1 (C-2′,6′ and C-4′′′), 126.0 (C-4′), 128.2 (C-4′′), 128.3 (C-3′,5′), 128.7 (C-5), 128.8 (C-3′′′,5′′′), 129.5 (C-2′′′,6′′′), 129.7 (C-2), 130.9 (C-6′′), 133.2 (C-4), 137.7 (C-1′′′), 142.7 (C-2′′), 143.7 (C-1′), 145.2 (C-1), 159.3 (C-6), 171.6 (A'-CH2CO2CH2-A), 186.7 (C-3) ppm. HRMS-ESI-TOF mz calcd for C27H18Cl4N2NaO3 [M+Na]+ 580.9964, found 580.9961.
[6-(2,6-Dichlorophenylimino)-3-oxocyclohexa-1,4-dien-1-yl]methylene diacetate (7): m.p. 158–160 °C; δH (300 MHz; CDCl3) 2.17 (6H, s, CH3), 6.52 (1H, dd, J = 10.2 and 2.2 Hz, 4-H), 6.68 (1H, d, J = 10.2 Hz, 5-H), 6.86 (1H, dd, J = 2.2 and 1.0 Hz, 2-H), 7.07 (1H, dd, J = 8.4 and 7.7 Hz, 4′-H), 7.37 (2H, d, J = 8.1 Hz, 3′,5′-H), 8.07 [1H, d, J = 1.0 Hz, CH(OAc)2] ppm. δC (75 MHz; CDCl3) 20.7 (CH3), 84.6 (C-H), 123.9 (C-2′,6′), 126.1 (C-4′), 128.3 (C-3′,5′), 128.9 (C-5), 130.6 (C-2), 133.2 (C-4), 143.7 (C-1′), 144.1 (C-1), 158.4 (C-6), 168.0 (OCOCH3), 186.7 (C-3) ppm. HRMS-ESI-TOF mz calcd for C17H13Cl2NNaO5 [M+Na]+ 404.0063, found 404.0061.
{Acetoxy[6-(2,6-dichlorophenylimino)-3-oxocyclohexa-1,4-dien-1-yl]}methyl 2-[2-(2,6-dichlorophenylamino)phenyl]acetate (8): m.p. 78–80 °C; δH (300 MHz; CDCl3) 2.16 (3H, s, CH3), 3.89 and 3.96 (2H, AB spin system, J = 15.5 Hz, CH2), 6.40 (1H, br s, NH), 6.50 (1H, dd, J = 10.2 and 2.2 Hz, 4-H), 6.54 (1H, br d, J = 7.7 Hz, 3′′-H), 6.65 (1H, d, J = 10.2 Hz, 5-H), 6.79 (1H, dd, J = 2.2 and 1.0 Hz, 2-H), 6.95 (1H, t, J = 8.0 Hz, 4′′′-H), 6.95–7.00 (1H, m, 5′′-H), 6.98 (1H, t, J = 8.1 Hz, 4′-H), 7.14 (1H, dt, J = 7.7 and 1.6 Hz, 4′′-H), 7.23 (1H, dd, J= 7.7 and 1.6 Hz, 6′′-H), 7.25–7.28 (1H, m, 3′-H or 5′-H), 7.28 (2H, d, J = 8.0 Hz, 3′′′,5′′′-H), 7.30 (1H, dd, J = 8.1 and 1.2 Hz, 3′-H or 5′-H), 8.16 (1H, d, J = 1.0 Hz, CHO2) ppm. δC (75 MHz; CDCl3) 20.7 (CH3), 38.1 (CH2), 85.0 (CHO2), 118.9 (C-3′′), 122.5 (C-5′′), 123.7 (C-1′′), 123.87 (C-4′′′), 123.93 (C-2′,6′), 126.0 (C-4′), 128.2 (C-3′,5′), 128.3 (C-4′′), 128.7 (C-3′′′,C-5′′′), 128.9 (C-5), 129.3 (C-2′′′,6′′′), 130.7 (C-2), 133.2 (C-4), 137.9 (C-1′′′), 142.7 (C-2′′), 143.5 (C-1), 143.8 (C-1′), 158.4 (C-6), 167.9 (CO2-CH3), 169.4 (CO2CH2), 186.5 (C-3) ppm. HRMS-ESI-TOF mz calcd for C29H20Cl4N2NaO5 [M+Na]+ 639.0018, found 639.0016.
[6-(2,6-Dichlorophenylimino)-3-oxocyclohexa-1,4-dien-1-yl]methylene bis{2-[2-(2,6-dichlorophenylamino)phenyl]acetate} (9): m.p. 73–75 °C; δH (300 MHz; CDCl3) 3.88 and 3.94 (4H, AB spin system, J = 15.4 Hz, CH2), 6.38 (2H, br s, NH), 6.47 (1H, dd, J = 10.2 and 2.1 Hz, 4-H), 6.53 (2H, br d, J = 7.8 Hz, 3′′-H), 6.63 (1H, d, J = 10.2 Hz, 5-H), 6.68 (1H, dd, J = 2.1 and 0.96 Hz, 2-H), 6.89-6.97 (5H, m, 4′-H, 4′′′-H and 5′′-H), 7.12 (2H, dt, J = 7.8 and 1.5 Hz, 4′′-H), 7.21 (2H, dd, J = 7.4 and 1.5 Hz, 6′′-H), 7.23 (2H, d, J = 8.1 Hz, 3′,5′-H), 7.28 (4H, d, J = 8.0 Hz, 3′′′,5′′′-H), 8.22 (1H, d, J = 0.96 Hz, CHO2) ppm. δC (75 MHz; CDCl3) 38.0 (CH2), 85.4 (CHO2), 118.9 (C-3′′), 122.5 (C-5′′), 123.76 (C-1′′), 123.78 (C-4′′′), 123.9 (C-2′,6′), 125.9 (C-4′), 128.1 (C-3′,5′), 128.3 (C-4′′), 128.7 (C-3′′′,5′′′), 128.8 (C-5), 129.3 (C-2′′′,6′′′), 130.8 (C-2), 130.9 (C-6′′), 133.3 (C-4), 137.8 (C-1′′′), 142.7 (C-2′′), 143.4 (C-1 and C-1′), 158.4 (C-6), 169.3 (CO2CH2-), 186.3 (C-3) ppm. HRMS-ESI-TOF mz calcd for C41H27Cl6N3NaO5 [M+Na]+ 873.9974, found 873.9965.
The available crystals of 6 and 7 for the structural analysis were composed of very thin plates or blocks (the maximum dimension was of 0.09 mm – see Table 4) which systematically diffracted very poorly at high angles, in particular for resolutions higher than 1.0 Å, even when long X-ray exposures for the individual frames were employed. In addition, the large unit cell axes lengths (see, for example, the parameters for compound 6) in combination with the sole presence of very light atoms, proved to be detrimental factors concerning reflection intensity at high angles. As a consequence, even though data integration up to a resolution of about 0.83 Å produced complete data sets for both structures (data completeness of 99.6 and 98.3% for 6 and 7, respectively), the overall mean I > σ(I) was less than to 2.2, hence the high Rint values for both structures. Nevertheless, both structures could be unequivocally solved using the integrated and scaled data set and by employing the direct methods implemented in SHELXS-97.53,54 This strategy allowed the immediate location of the vast majority of the atoms. All the remaining non-hydrogen atoms were directly located from difference Fourier maps calculated from successive full-matrix least squares refinement cycles on F2 using SHELXL-97.54,55 All non-hydrogen atoms have been successfully refined using anisotropic displacement parameters.
| 6 | 7 | |
|---|---|---|
a
R1 = Σ||Fo| − |Fc||/Σ|Fo|
b
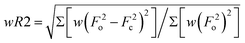 c
w = 1/[σ2(F2o) + (mP)2 + nP] where P = (F2o + 2F2c)/3
c
w = 1/[σ2(F2o) + (mP)2 + nP] where P = (F2o + 2F2c)/3
|
||
| Formula | C27H18Cl4N2O3 | C17H13Cl2NO5 |
| Formula weight | 560.23 | 382.18 |
| T/K | 180(2) | 150(2) |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21/n | Pī |
| a/Å | 12.923(4) | 10.851(2) |
| b/Å | 8.679(3) | 10.951(2) |
| c/Å | 23.217(7) | 14.929(3) |
| α (°) | — | 87.827(12) |
| β (°) | 102.312(11) | 84.530(11) |
| γ (°) | — | 80.818(11) |
| Volume/Å3 | 2544.0(13) | 1742.6(6) |
| Z | 4 | 4 |
| D c/g cm−3 | 1.463 | 1.457 |
| μ(Mo-Kα)/mm−1 | 0.498 | 0.400 |
| F(000) | 1144 | 784 |
| Crystal size/mm | 0.09 × 0.05 × 0.01 | 0.08 × 0.06 × 0.03 |
| Crystal type | Orange plates | Colourless blocks |
| θ range | 3.55 to 25.35 | 3.52 to 25.35 |
| Index ranges | −15 ≤ h ≤ 15 | −13 ≤ h ≤ 12 |
| −9 ≤ k ≤ 10 | −13 ≤ k ≤ 12 | |
| −27 ≤ l ≤ 27 | −17 ≤ l ≤ 17 | |
| Reflections collected | 25989 |
21445 |
| Independent reflections | 4645 (Rint = 0.1442) | 6267 (Rint = 0.1184) |
| Data completeness | 99.6% | 98.3% |
| Data/parameters | 4645/325 | 6267/455 |
| Final R indices [I > 2σ(I)]ab | R1 = 0.0583 | R1 = 0.0956 |
| wR2 = 0.1032 | wR2 = 0.2254 | |
| Final R indices (all data)ab | R1 = 0.1766 | R1 = 0.1683 |
| wR2 = 0.1374 | wR2 = 0.2686 | |
| Weighting schemec | m = 0.0498 | m = 0.1436 |
| n = 0 | n = 0.0 | |
| Largest diff. peak and hole | 0.256 and −0.381 eÅ−3 | 0.681 and −0.865 eÅ−3 |
Hydrogen atoms bound to nitrogen and carbon were placed at their idealised positions using appropriate HFIX instructions in SHELXL: 43 for the aromatic carbon atoms and N–H moieties, 13 for the tertiary carbons, 23 for the methylene moieties (–CH2–) and 137 for the terminal methyl groups (–CH3). These atoms have been included in subsequent refinement cycles in riding-motion approximation with isotropic thermal displacements parameters (Uiso) fixed at 1.2 (for the three former families of hydrogen atoms) or 1.5 × Ueq (only for the methyl groups) of the nitrogen or carbon atom to which they are attached, respectively.
The last difference Fourier map synthesis showed: for 6, the highest peak (0.256 eÅ−3) and deepest hole (−0.381 eÅ−3) located at 2.07 Å and 0.88 Å from H3 and Cl2, respectively; for 7, the highest peak (0.681 eÅ−3) and deepest hole (−0.865 eÅ−3) located at 1.09 Å and 0.84 Å from Cl3 and Cl4, respectively. Information concerning crystallographic data collection and structure refinement details is summarized in Table 4. Geometrical details concerning the various supramolecular contacts present in the crystal structures of 6 and 7, namely, D–H⋯A weak and strong hydrogen bonding interactions, Cl⋯π and π⋯π contacts are summarized in Tables 2 and 3, respectively. Structural drawings have been created using the software package Crystal Impact Diamond.56
Crystallographic data (including structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication Nos. CCDC-862153 and -862154 for 6 and 7, respectively. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 2EZ, UK. FAX: (+44) 1223 336033. E-mail: deposit@ccdc.cam.ac.uk.
Conclusions
The in vitro formation of several new diclofenac derivatives, initially resulting from oxidative decarboxylation, similar to what happens in vivo, is revealed. Manganese(III) porphyrins, namely [Mn(TDCPP)Cl] and [Mn(TPFPP)Cl], were tested at room temperature in the presence of hydrogen peroxide, a safe and environmentally friendly oxidant. Two potential mechanistic proposals for the formation of diclofenac derivatives 2–9 are presented. The formation of monoesters 5 and 6 and geminal diesters 7–9 suggest that alcohol 12 and geminal diol 14 are key intermediates in the oxidation process. The new diclofenac derivatives obtained are fully characterized, three of them being characterized in the solid state using X-ray diffraction.Acknowledgements
Thanks are due to Fundação para a Ciência e a Tecnologia (FCT/FEDER) for funding QOPNA (Project PEst-C/QUI/UI0062/2011) and CICECO (Project PEst-C/CTM/LA0011/2011) and for specific funding towards the purchase of the single-crystal X-ray diffractometer. Authors also acknowledge the Portuguese National NMR Network supported with funds from FCT.References
- I. Wiesenberg-Boettcher, J. Pfeilschifter, A. Schweizer, A. Sallmann and P. Wenk, Agents Actions, 1991, 34, 135–137 CrossRef CAS.
- R. J. Sawchuk, J. A. Maloney, L. L. Cartier, R. J. Rackley, K. K. H. Chan and H. S. L. Lau, Pharm. Res., 1995, 12, 756–762 CrossRef CAS.
- W. Blum, J. W. Faigle, U. Pfaar and A. Sallmann, J. Chromatogr., Biomed. Appl., 1996, 685, 251–263 CrossRef CAS.
- M. P. Grillo, J. Ma, Y. Teffera and D. J. Waldon, Drug Metab. Dispos., 2008, 36, 1740–1744 CrossRef CAS.
- P. E. Stackelber, E. T. Furlong, M. T. Meyer, S. D. Zaugg, A. K. Henderson and D. B. Reissman, Sci. Total Environ., 2004, 329, 99–113 CrossRef.
- M. S. Díaz-Cruz, M. J. García-Galán, P. Guerra, A. Jelic, C. Postigo, E. Eljarrat, M. Farré, M. J. López de Alda, M. Petrovic and D. Barceló, TrAC, Trends Anal. Chem., 2009, 28, 1263–1275 CrossRef.
- L. H. M. L. M. Santos, A. N. Araujo, A. Fachini, A. Pena, C. Delerue-Matos and M. C. B. S. M. Montenegro, J. Hazard. Mater., 2010, 175, 45–95 CrossRef CAS.
- J. Hofmann, U. Freier, M. Wecks and S. Hohmann, Appl. Catal., B, 2007, 70, 447–451 CrossRef CAS.
- L. A. Pérez-Estrada, S. Malato, W. Gernjak, A. Agüera, E. M. Thurman, I. Ferrer and A. R. Fernández-Alba, Environ. Sci. Technol., 2005, 39, 8300–8306 CrossRef.
- I. Forrez, M. Carballa, K. Verbeken, L. Vanhaeke, M. Schulüsener, T. Ternes, N. Boon and W. Verstraete, Environ. Sci. Technol., 2010, 44, 3449–3454 CrossRef CAS.
- J. Bernadou and B. Meunier, Adv. Synth. Catal., 2004, 346, 171–184 CrossRef CAS.
- D. Mansuy, C. R. Chim., 2007, 10, 392–413 CrossRef CAS.
- W. Lohmann and U. Karst, Anal. Bioanal. Chem., 2007, 391, 79–96 CrossRef.
- G. T. Balogh and G. M. Keserű, Arkivoc, 2004,(vii), 124–139 CAS.
- C. Bochot, J.-F. Bartoli, Y. Frapart, P. M. Dansette, D. Mansuy and P. Battioni, J. Mol. Catal. A: Chem., 2007, 263, 200–205 CrossRef CAS.
- S. Othman, V. Mansuy-Mouries, C. Bensoussan, P. Battioni and D. Mansuy, C. R. Acad. Sci. II C, 2000, 3, 751–755 CAS.
- M. Komuro, T. Higuchi and M. Hirobe, Bioorg. Med. Chem., 1995, 3, 55–65 CrossRef CAS.
- S. M. S. Chauhan, P. P. Mohapatra, B. Kalra, T. S. Kohli and S. Satapathy, J. Mol. Catal. A: Chem., 1996, 113, 239–247 CrossRef CAS.
- T. Higuchi and M. Hirobe, J. Mol. Catal. A: Chem., 1996, 113, 403–422 CrossRef CAS.
- S. M. S. Chauhan and B. B. Sahoo, Bioorg. Med. Chem., 1999, 7, 2629–2634 CrossRef CAS.
- V. Mirkhani, S. Tangestaninejad, M. Moghadam and Z. Karimiana, Bioorg. Med. Chem. Lett., 2003, 13, 3433–3435 CrossRef CAS.
- G. R. Karimipour, B. Karami, M. Montazerozohori and S. Zakavi, Chin. J. Catal., 2007, 28, 940–946 CrossRef CAS.
- M. Moghadam, S. Tangestaninejad, V. Mirkhani, I. Mohammadpoor-baltork, N. Sirjanian and S. Parand, Bioorg. Med. Chem., 2009, 17, 3394–3398 CrossRef CAS.
- R. R. L. Martins, M. G. P. M. S. Neves, A. J. D. Silvestre, M. M. Q. Simões, A. M. S. Silva, A. C. Tomé, J. A. S. Cavaleiro, P. Tagliatesta and C. Crestini, J. Mol. Catal. A: Chem., 2001, 172, 33–42 CrossRef CAS.
- S. L. H. Rebelo, M. M. Q. Simões, M. G. P. M. S. Neves, A. M. S. Silva and J. A. S. Cavaleiro, Chem. Commun., 2004, 608–609 RSC.
- S. L. H. Rebelo, M. M. Pereira, M. M. Q. Simões, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Catal., 2005, 234, 76–87 CrossRef CAS.
- S. L. H. Rebelo, M. M. Q. Simões, M. G. P. M. S. Neves, A. M. S. Silva, J. A. S. Cavaleiro, A. F. Peixoto, M. M. Pereira, M. R. Silva, J. A. Paixão and A. M. Beja, Eur. J. Org. Chem., 2004, 4778–4787 CrossRef CAS.
- S. L. H. Rebelo, M. M. Q. Simões, M. G. P. M. S. Neves, A. M. S. Silva, P. Tagliatesta and J. A. S. Cavaleiro, J. Mol. Catal. A: Chem., 2005, 232, 135–142 CrossRef CAS.
- S. L. H. Rebelo, A. R. Gonçalves, M. M. Pereira, M. M. Q. Simões, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Mol. Catal. A: Chem., 2006, 256, 321–323 CrossRef CAS.
- R. De Paula, M. M. Q. Simões, M. G. P. M. S. Neves and J.A.S. Cavaleiro, Catal. Commun., 2008, 10, 57–60 CrossRef CAS.
- S. M. G. Pires, R. D Paula, M. M. Q. Simões, M. G. P. M. S. Neves, I. C. M. S. Santos, A. C. Tomé and J. A. S. Cavaleiro, Catal. Commun., 2009, 11, 24–28 CrossRef CAS.
- C. M. B. Neves, M. M. Q. Simões, I. C. M. S. Santos, F. M. J. Domingues, M. G. P. M. S. Neves, F. A. A. Paz and J. A. S. Cavaleiro, Tetrahedron Lett., 2011, 52, 2898–2902 CrossRef CAS.
- R. D Paula, M. M. Q. Simões, M. G. P. M. S. Neves and J.A.S. Cavaleiro, J. Mol. Catal. A: Chem., 2011, 345, 1–11 CrossRef.
- S. M. G. Pires, R. D Paula, M. M. Q. Simões, A. M. S. Silva, M. R. M. Domingues, I. C. M. S. Santos, M. D. Vargas, V. F. Ferreira, M. G. P. M. S. Neves and J. A. S. Cavaleiro, RSC Adv., 2011, 1, 1195–1199 RSC.
- C. M. B. Neves, J. A. Fernandes, M. M. Q. Simões, M. G. P. M. S. Neves, J. A. S. Cavaleiro and F. A. A. Paz, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o3022 Search PubMed.
- S. L. H. Rebelo, M. M. Q. Simões, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Mol. Catal. A: Chem., 2003, 201, 9–22 CrossRef CAS.
- I. D. Cunningham, T. N. Danks, J. N. Hay, I. Hamerton and S. Gunathilagan, Tetrahedron, 2001, 57, 6847–6853 CrossRef CAS.
- I. D. Cunningham, T. N. Danks, J. N. Hay, I. Hamerton, S. Gunathilagan and C. Janczak, J. Mol. Catal. A: Chem., 2002, 185, 25–31 CrossRef CAS.
- E. Bolzonella, S. Campestrini, F. Di Furia and P. Ghiotti, J. Phys. Org. Chem., 1996, 9, 539–544 CrossRef CAS.
- J. H. Han, S.-K. Yoo, J. S. Seo, S. J. Hong, S. K. Kim and C. Kim, Dalton Trans., 2005, 402–406 RSC.
- V. T. Kamble, V. S. Jamode, N. S. Joshi, A. V. Biradara and R. Y. Deshmukh, Tetrahedron Lett., 2006, 47, 5573–5576 CrossRef CAS.
- N. Sumida, K. Nishioka and T. Sato, Synlett, 2001, 1921–1922 CrossRef CAS.
- M. M. Oliveira, L. H. M. Carvalho, F. P. Peixoto, A. M. F. Oliveira-Campos, A. Reis, P. Domingues and M. R. M. Domingues, Rapid Commun. Mass Spectrom., 2010, 24, 2171–2174 CrossRef.
- P. Bartels and W. Tümpling Jr., Sci. Total Environ., 2007, 374, 143–155 CrossRef CAS.
- J. Grell, J. Bernstein and G. Tinhofer, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 1030–1043 CrossRef.
- C. S. Raska, C. E. Parker, C. Huang, J. Han, G. L. Glish, M. Pope and C. H. Borchers, J. Am. Soc. Mass Spectrom., 2002, 13, 1034–1041 CrossRef CAS.
- D. J. Waldon, Y. Teffera, A. E. Colletti, J. Liu, D. Zurcher, K. W. Copeland and Z. Zhao, Chem. Res. Toxicol., 2010, 23, 1947–1953 CrossRef CAS.
- T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef.
- APEX2, Data Collection Software Version 2.1-RC13, Bruker AXS, Delft, The Netherlands, 2006 Search PubMed.
- Cryopad, Remote monitoring and control, Version 1.451, Oxford Cryosystems, Oxford, United Kingdom, 2006 Search PubMed.
- SAINT+, Data Integration Engine v. 7.23a ©Bruker AXS, Madison, Wisconsin, USA, 1997–2005 Search PubMed.
- G. M Sheldrick SADABS v.2.01, Bruker/Siemens Area Detector Absorption Correction Program, Bruker AXS, Madison, Wisconsin, USA, 1998 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, University of Göttingen, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, 1997 Search PubMed.
- K. Brandenburg, DIAMOND, Version 3.2f, Crystal Impact GbR, Bonn, Germany, 1997–2010, Search PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 862153 and 862154 for compounds 6 and 7, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20801f |
| This journal is © The Royal Society of Chemistry 2012 |