Catalytic conversion of lactide to optically pure heterocycles†
Shinji
Tsunoi
,
Hiroki
Takahashi
,
Yugo
Takano
,
Aritomo
Okamura
and
Ikuya
Shibata
*
Research Center for Environmental Preservation, Osaka University, 2–4 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: shibata@epc.osaka-u.ac.jp; Fax: (+81)-6-6879-8978; Tel: (+81)-6-6879-8975
First published on 25th June 2012
Abstract
The catalytic preparation of optically pure nitrogen heterocycles was developed via cycloaddition using lactide catalyzed by tin alkoxide.
Glycolide and lactide, the related cyclic dimers of glycolic and lactic acid, are plant-derived substrates. Of special interest is the fact that lactide is an easily available chiral source. The major utility of these substrates is in the application to biodegradable aliphatic polyesters such as poly(glycolide) (PGA) and poly(lactide) (PLA).1 As far as we could ascertain, however, their direct transformation to monomeric fine chemicals such as heterocycles has not been reported.2 Oxazolidine-2,4-diones are used in the areas of medicinal,3 agricultural4 and synthetic applications.5 We have recently reported the equimolar tin-mediated synthesis of heterocycles (Scheme 1).6 The in situ formed tin alkoxide was added to an isocyanate followed by cyclization of the remaining functional group. Thus α-stannoxy-ester A and stannyl amine B were the reactive intermediates. We present here the catalytic synthesis of oxazolidine-2,4-diones from glycolide and lactide by using a tin alkoxide catalyst. In the catalytic reaction, A and B worked well as active catalytic species.7,8 In particular, optically pure oxazolidine-2,4-diones 4 were obtained directly from easily available chiral sources.
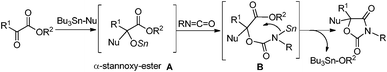 | ||
| Scheme 1 The equimolar tin-mediated reaction. | ||
Initially, methyl glycolate (1a) was used as the substrate as we thought that it could be a good precursor of α-stannoxy-ester A. Table 1 shows the results of the catalytic effect in the reaction with BuN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO.9 Without a catalyst, the reaction proceeded in a low yield even with heating where no cyclization proceeded, and linear adduct 2a′ was obtained in a 10% yield (entry 1). In the presence of a tin catalyst (10 mol%) such as di-n-butyltin oxide (Bu2SnO) or the tin(II) bis(2-ethylhexanoate) system [Sn(Oct)2–MeOH],101a reacted with BuNCO well at rt for 3 h, however, only linear adduct 2a′ was obtained (entries 2 and 3).11 On the other hand, using tri-n-butyltin methoxide (Bu3SnOMe) as the catalyst afforded a cyclic product, 1,3-oxazolidin-2,4-dione 2a (entry 4). Further, di-n-butyltin dimethoxide [Bu2Sn(OMe)2] was employed as an effective catalyst for the preparation of 2a (entry 5). Microwave irradiation gave the desired 2a in only 10 min (entry 6).
CO.9 Without a catalyst, the reaction proceeded in a low yield even with heating where no cyclization proceeded, and linear adduct 2a′ was obtained in a 10% yield (entry 1). In the presence of a tin catalyst (10 mol%) such as di-n-butyltin oxide (Bu2SnO) or the tin(II) bis(2-ethylhexanoate) system [Sn(Oct)2–MeOH],101a reacted with BuNCO well at rt for 3 h, however, only linear adduct 2a′ was obtained (entries 2 and 3).11 On the other hand, using tri-n-butyltin methoxide (Bu3SnOMe) as the catalyst afforded a cyclic product, 1,3-oxazolidin-2,4-dione 2a (entry 4). Further, di-n-butyltin dimethoxide [Bu2Sn(OMe)2] was employed as an effective catalyst for the preparation of 2a (entry 5). Microwave irradiation gave the desired 2a in only 10 min (entry 6).
COa
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Conditionsb | 2a [%]c | 2a′ [%] |
|
a
1a 1 mmol, BuNCO 1 mmol, cat. 0.1 mmol, MeCN 1 mL.
b rt = room temperature; MW = microwave.
c tr = trace.
|
||||
| 1 | None | 80 °C, 3 h | tr | 10 |
| 2 | Bu2SnO |
rt, 3 h | tr | 72 |
| 3 | Sn(Oct)2–2 MeOH | rt, 3 h | tr | 99 |
| 4 | Bu3SnOMe | rt, 3 h | 19 | 60 |
| 5 | Bu2Sn(OMe)2 | rt, 3 h | 70 | 6 |
| 6 | Bu2Sn(OMe)2 | MW, 90 °C (30 W), 10 min | 80 | 4 |

A plausible catalytic cycle is shown in Scheme 2. The Sn–O and Sn–N bonds bear high nucleophilicity.10 Initially, tin methoxide reacts with 1a to give α-stannoxy-ester A. The Sn–O bond of A reacts with isocyanate to form stannyl-carbamate B.12 The resultant Sn–N bond of B reacts with the remaining ester moiety. The Sn–O bond of cyclized product C reacts with the starting 1a. As a result, 1,3-oxazolidine-2,4-dione 2a is obtained and the catalytic active species A is regenerated.13 On the other hand, linear adduct 2a′ is obtained by the protonolysis of B with 1a. Bu2Sn(OMe)2 forms a dimeric structure in which apical Sn–OMe bonds of 5-coordinated tin have high nucleophilicity.14 Thus, the highly nucleophilic Bu2Sn(OMe)2 is employed as an effective catalyst rather than the Sn(Oct)2–MeOH-system.15
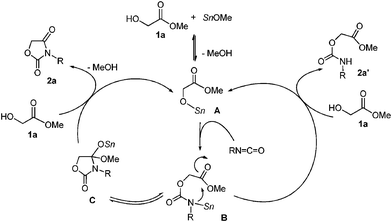 | ||
| Scheme 2 A plausible catalytic cycle in the reaction using 1a. | ||
Thus, protonation occurs instead of cyclization. In fact, when glycolic acid was used as a starting material, no desired products were obtained when using Bu2Sn(OMe)2 as the catalyst.
As shown in Table 2, we then tried to use the related cyclic dimer, glycolide 1b, as a substrate. This substrate has no proton source. When the reaction with BuNCO was carried out under the optimized conditions in Table 1, entry 6, the reaction scarcely proceeded (entry 1). However, the reaction using higher MW power increased the yield of 2a (entries 2 and 3), and the reaction at 120 °C for 20 min gave a quantitative yield (entry 4). The reaction with a smaller amount of catalyst decreased the yield (entry 5). The polyester, PGA, can be obtained by the ring-opening polymerization of glycolide 1b.1 The most widely used catalyst for industrial preparation is the tin(II) bis(2-ethylhexanoate) system [Sn(Oct)2–MeOH]. This catalytic system did not give 2a from 1a as shown in Table 1, entry 3, however, using glycolide 1b as a substrate enabled the preparation of 2a (entry 6). Thus, the absence of proton sources in the reaction of 1b allowed the use of Sn(Oct)2 as the catalyst. The reaction using 1b was highly atom-economical and no side products were obtained. In place of aliphatic isocyanate, aromatic isocyanates were reactive giving oxazolidine-2,4-diones, 2b and 2c (entries 7–10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Catalyst | Conditions | Product 2 [%] | |
|
a
1b 0.5 mmol, isocyanate 1 mmol, tin catalyst 0.1 mmol, MeCN 1 mL.
b Sn(Oct)2Sn(OCOC7H17)2.
|
|||||
| 1 | Bu | Bu2Sn(OMe)2 | MW 80 °C (30 W), 5 min | 2a | trace |
| 2 | Bu2Sn(OMe)2 | MW 95 °C (50 W), 10 min | 11 | ||
| 3 | Bu2Sn(OMe)2 | MW 110 °C (100 W), 10 min | 78 | ||
| 4 | Bu2Sn(OMe)2 | MW 120 °C (100 W), 20 min | 99 | ||
| 5 | Bu2Sn(OMe)2 (0.1 equiv) | MW 120 °C (100 W), 20 min | 44 | ||
| 6 | Sn(Oct)2–2 MeOHb | MW 120 °C (100 W), 20 min | 74 | ||
| 7 | PMP | Bu2Sn(OMe)2 | MW 120 °C (100 W), 20 min | 2b | 99 |
| 8 | Sn(Oct)2–2 MeOH | MW 120 °C (100 W), 20 min | 71 | ||
| 9 | p-IC6H4 | Bu2Sn(OMe)2 | MW 120 °C (100 W), 20 min | 2c | 79 |
| 10 | Sn(Oct)2–2 MeOH | MW 120 °C (100 W), 20 min | 86 | ||

Hence, organotin alkoxides could be good candidates to catalyze the cycloaddition of 1b in which the high nucleophilicity of the Sn–O bond to the lactone ring is an important factor.16 A plausible catalytic cycle is shown in Scheme 3. Initially, tin methoxide cleaves the acyl–oxygen bond of 1b. The resultant Sn–O bond of A′ reacts with an isocyanate to form stannyl-carbamate B′. The resultant Sn–N bond reacts with the remaining carbonyl moiety intramolecularly to give 1,3-oxazolidine-2,4-dione 2. The subsequent addition–cyclization through A and B also gave product 2, regenerating tin methoxide.
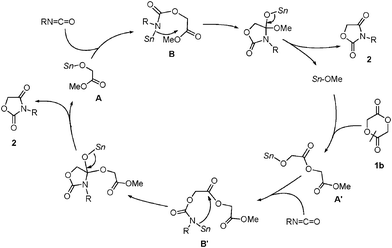 | ||
| Scheme 3 A plausible catalytic cycle in the reaction using 1b. | ||
As shown in Table 3, instead of isocyanates, when isothiocyanate (BuNCS) was used, 1,3-oxazolidine-2-thione 3a, was obtained catalyzed by Bu2Sn(OMe)2 (entry 1). On the other hand, the Sn(Oct)2–2MeOH catalytic system gave no satisfactory results (entry 2). Using other isothiocyanates also gave the corresponding adducts 3b, 3c, and 3d that were effectively catalyzed by Bu2Sn(OMe)2 (entries 3–5), whereas, the Sn(Oct)2–2MeOH system basically showed less catalytic activity (entries 3–5, parentheses). It is known that a Sn–OR bond adds across the CS group of an isothiocyanate (R′NCS) to give a Sn–S–C(NR′)OR adduct.17 Because of the high affinity of Sn toward the S atom,18 the cyclization step accompanying the cleavage of the Sn–S bond would be difficult compared with the reaction using isocyanates (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | R | Catalyst | Product 3 [%] | |
| a 1b 0.5 mmol, isothiocyanate 1 mmol, tin catalyst 0.1 mmol, MeCN 1 mL. b Yields from the Sn(Oct)2–2MeOH system-catalyzed reactions are in parentheses. | ||||
| 1 | Bu | Bu2Sn(OMe)2 | 3a | 91 |
| 2 | Sn(Oct)2–2MeOH | 12 | ||
| 3 | Ph | Bu2Sn(OMe)2 | 3b | > 99 (tr)b |
| 4 | PMP | Bu2Sn(OMe)2 | 3c | 90 (tr)b |
| 5 | p-NO2C6H4 | Bu2Sn(OMe)2 | 3d | 90 (25)b |

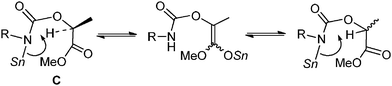
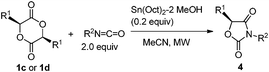
Lactide 1c is also a naturally occurring compound like glycolide 1b, and is an easily available chiral source.1 As shown in Scheme 4, the reaction of 1c with p-methoxyphenyl isocyanate proceeded quantitatively catalyzed by Bu2Sn(OMe)2 at 130 °C for 10 min (Scheme 4, (1)). However, after product 4a was isolated, its enantiopurity decreased to 56% ee despite the fact that optically pure 1c was used. Namely, racemization occurred during the reaction. It has already been reported that tin amide (Bu3Sn–NR2) provides tin enolate from reaction with α-alkoxyketone.19 Hence, as shown in Scheme 5, intermediate stannyl-carbamate C had enough basicity to abstract acidic hydrogen to cause undesirable racemization. Fortunately, when the Sn(Oct)2–MeOH system was used as the catalyst, 4a was obtained with no loss of enantiopurity (Scheme 4, (2)). As described, the basicity of the Sn–N bond generated in the Sn(Oct)2–MeOH system was lower than that of Bu2Sn(OMe)2. So abstraction of the acidic hydrogens and the inducement of racemization was suppressed.
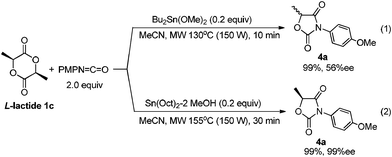 | ||
| Scheme 4 Reaction of lactide with isocyanate. | ||
 | ||
| Scheme 5 A plausible racemization mechanism. | ||
Table 4 shows the results of the reactions of lactide 1c with various isocyanates catalyzed by the Sn(Oct)2–MeOH system. Thus, optically pure products, 4a–4c, were obtained (entries 1–3). The reaction under MW irradiation was essential because reflux conditions did not give the desired product 4a (entry 1, parenthesis). Phenyl lactide 1d is also easily available in an optically pure form.20 Thus 5-benzyl-1,3-ozazolidine-2,4-diones 4d–4f were obtained with no loss of enantiopurity (entries 4–6).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2b | Conditions | Product 4 [%] | ee [%] | ||
| a 1c or 1d 0.5 mmol, isocyanate 1 mmol, Sn(Oct)2 0.1 mmol, MeOH 0.2 mmol, MeCN 1 mL. b PMP = p-MeOC6H4; Ts = tosyl. c 80 °C, 3 h. | |||||||
| 1 | Me | 1c | PMP | MW 155 °C (150 W), 30 min | 4a | 99 | 99 (trace)c |
| 2 | Me | 1c | p-IC6H4 | MW 155 °C (150 W), 30 min | 4b | 96 | 98 |
| 3 | Me | 1c | Ts | MW 160 °C (150 W), 30 min | 4c | 89 | 98 |
| 4 | Bn | 1d | PMP | MW 155 °C (150 W), 30 min | 4d | 99 | 94 |
| 5 | Bn | 1d | p-IC6H4 | MW 158 °C (150 W), 30 min | 4e | 90 | 97 |
| 6 | Bn | 1d | Ts | MW 160 °C (150 W), 30 min | 4f | 79 | 92 |
Conclusions
In conclusion, the first synthetic use of plant-derived substrates, glycolide and lactide, to give heterocycles was achieved by using tin alkoxide catalysts. In particular, optically pure products were obtained from easily available chiral sources.References
- For reviews see: (a) O. D. Cabaret, B. M.-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef; (b) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- Alcoholysis: (a) A. Grala, J. Ejfler, L. B. Jerzykiewicz and P. Sobota, Dalton Trans., 2011, 40, 4042–4044 RSC; (b) K. Phomphrai, S. Pracha, P. Phonjanthuek and M. Pohmakotr, Dalton Trans., 2008, 3048–3050 RSC; Aminolysis: (c) G. A. Brine, K. G. Boldt, D. Prakash, D. J. Kotchmar, V. C. Bondeson, D. J. Bradley, P. Singh and F. I. Carroll, J. Chem. Soc., Perkin Trans. 1, 1991, 1809–1814 RSC; (d) Bromination: F. Jing and M. A. Hillmyer, J. Am. Chem. Soc., 2008, 130, 13826–13827 CrossRef CAS; (e) Alkylation: K. C. Nicolaou, R. Jautelat, G. Vassilikogiannakis, P. S. Baran and K. B. Simonsen, Chem.–Eur. J., 1999, 5, 3651–3665 CrossRef CAS; (f) Reduction: E. Balaraman, E. Fogler and D. Milstein, Chem. Commun., 2012, 48, 1111–1113 RSC; (g) R. J. Wehmschulte and L. Wojtas, Inorg. Chem., 2011, 50, 11300–11302 CrossRef CAS.
- Hypoglycemic agents: (a) R. L. Dow, B. M. Bechle, T. T. Chou, D. A. Clark, B. Hulin and R. W. Stevenson, J. Med. Chem., 1991, 34, 1538–1544 CrossRef CAS; (b) NK1 antagonists: R. T. Lewis, A. M. Macleod, K. J. Merchant, F. Kelleher, I. Sanderson, R. H. Herbert, M. A. Cascieri, S. Sadowski, R. G. Ball and K. Hoogsteen, J. Med. Chem., 1995, 38, 923–933 CrossRef CAS.
- Fungicide: (a) J. A Sternberg, D. Geffken, J. B Adams Jr, R. Pöstages, C. G Sternberg, C. L Campbell and W. K Moberg, Pest Manage. Sci., 2001, 57, 143–152 CrossRef; (b) Y.-J. Zheng, R. Shapiro, W. J Marshall and D. B Jordan, Bioorg. Med. Chem. Lett., 2000, 10, 1059–1062 CrossRef CAS; (c) D. B Jordan, R. S Livingston, J. J. Bisaha, K. E. Duncan, S. O. Pember, M. A. Picollelli, R. S. Schwartz, J. A Sternberg and X.-S. Tang, Pestic. Sci., 1999, 55, 105–118 CrossRef.
- (a) J. W. Clark-Lewis, Chem. Rev., 1958, 58, 63–99 CrossRef CAS; (b) G. Galliani, B. Rindone and F. Saliu, Tetrahedron Lett., 2009, 50, 5123–5125 CrossRef CAS.
- I. Shibata, R. Kojima, S. Tsunoi, T. Nozaki, T. Watanabe, A. Ninomiya, M. Yasuda and A. Baba, Org. Biomol. Chem., 2010, 8, 2009–2011 CAS.
- Synthetic uses of organotin compounds, see: (a) A. Baba, I. Shibata and M. Yasuda, in Comprehensive Organometallic Chemistry III, ed. P. Knochel, Elsevier, Oxford, 2007, vol. 9, ch. 8, pp. 341–380. Search PubMed; (b) M. Pereyre, P. J. Quintard and A. Rahm, in Tin in Organic Synthesis, Butterworth, London, 1987, pp. 261–258 Search PubMed; (c) A. G. Davies, in Organotin Chemistry, VCH, Weinheim, 1997, pp. 166–193. Search PubMed.
- Sn–O and Sn–N bonds bear high nucleophilicity. In some cases, their nucleophilicity is higher than that of the corresponding free alcohols and amines: E. W. Abel, D. A. Armitage and D. B. Brady, Trans. Faraday Soc., 1966, 62, 3459–3462 RSC.
- We have recently reported the reaction of α-hydroxy ketones with isocyanate catalyzed by Bu2Sn(OMe)2: R. Kojima, S. Sawamoto, A. Okamura, H. Takahashi, S. Tsunoi and I. Shibata, Eur. J. Org. Chem., 2011, 7255–7258 CrossRef CAS.
- (a) H. R. Kricheldorf, I. Kreiser-Saunders and A. Stricker, Macromolecules, 2000, 33, 702–709 CrossRef CAS; (b) A. Kowalski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 7359–7370 CrossRef CAS.
- Tin methoxide catalyzed urethane formation: J. Otera, T. Yano and R. Okawara, Chem. Lett., 1985, 14, 901–904 CrossRef.
- A. G. Davies, T. N. Mitchell and W. R. Symes, J. Chem. Soc. (C), 1966, 1311–1315 RSC.
- Tin-catalyzed reaction using epoxides: I. Shibata, A. Baba, H. Iwasaki and H. Matsuda, J. Org. Chem., 1986, 51, 2177–2184 CrossRef CAS.
- A. G. Davies and P. G. Harrison, J. Chem. Soc. (C), 1967, 298–300 RSC.
- In the Sn(Oct)2–MeOH-system, the catalytic species form 4-coordinated tin,1a and has no alkyl substituents on tin. Hence compared with the Bu2Sn(OMe)2 system, the Sn–N bonds in B would not bear enough nucleophilicity toward an ester moiety beyond protonolysis.
- Cleavage of lactide by tin alkoxide: M. H. Chisholm and E. E. Delbridge, New J. Chem., 2003, 27, 1167–1176 RSC.
- I. Shibata, A. Baba, H. Kashiwagi and H. Matsuda, Bull. Chem. Soc. Jpn., 1986, 59, 341–343 CrossRef.
- (a) K. Itoh, I. K. Lee, I. Matsuda, S. Sakai and Y. Ishii, Tetrahedron Lett., 1967, 8, 2667–2670 CrossRef; (b) K. Itoh, Y. Fukumoto and Y. Ishii, Tetrahedron Lett., 1968, 9, 3199–3202 CrossRef.
- I. Shibata, K. Yasuda, Y. Tanaka, M. Yasuda and A. Baba, J. Org. Chem., 1998, 63, 1334–1336 CrossRef CAS.
- T. L. Simmons and G. L. Baker, Biomacromolecules, 2001, 2, 658–663 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20831h/ |
| This journal is © The Royal Society of Chemistry 2012 |