Iridium(III) complexes as therapeutic and bioimaging reagents for cellular applications
Kenneth Kam-Wing
Lo
* and
Kenneth Yin
Zhang
Institute of Molecular Functional Materials (Areas of Excellence Scheme, University Grants Committee (Hong Kong)) and Department of Biology and Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, P. R. China. E-mail: bhkenlo@cityu.edu.hk; Fax: (852) 3442 0522
First published on 17th September 2012
Abstract
There is an emerging interest in applying inorganic and organometallic transition metal complexes to biomolecular and cellular studies. The cytotoxic effects of these complexes on various cancer and normal cells have been examined. Many of these complexes display intense, long-lived, and environment-sensitive emission, rendering them useful live-cell imaging reagents. Of particular interest are iridium(III) complexes, which possess a diversity of molecular structures with intriguing biological activity and photophysical properties. In this review article, we summarize recent work using iridium(III) complexes as anticancer drugs and cellular imaging reagents. We focus on the cytotoxic activity, cellular uptake efficiency and mechanisms, and intracellular distribution properties of these complexes. Additionally, we describe the applications of luminescent iridium(III) complexes in intracellular sensing for ions and small molecules, gene-delivery, and cancer cell detection.
1. Introduction
Despite the inertness of the iridium center, many efficient synthetic methods have been developed to allow coordination of a wide range of ligands.1 The resulting structures usually lead to novel and diverse photophysical and photochemical properties, which can be utilized in different areas such as light-emitting diodes, photocatalysis, and dye-sensitized solar cells.2 In the past decade, there has been an increasing interest in understanding the interactions of iridium(III) complexes with biomolecules as well as the development of these complexes as reagents for biological applications. Studies have focused on the biomolecular-binding interactions with biomolecules such as nucleic acids, amino acids, peptides, and proteins.3 Recently, there has also been a rapidly emerging interest in the cellular uptake properties and cytotoxic activity of iridium(III) complexes toward cancer cells, thus inspiring the design of anticancer drugs.4 The interesting emission properties of many of these complexes have also been utilized in the development of cellular imaging reagents as well as sensors for intracellular analytes such as ions and small molecules.5Iridium(III) complexes possess many advantageous characteristics for their use as biological reagents; for example, the possible utilization of a wide range of ligands enables fine tuning of their physical and chemical properties. Similar to other transition metal-based reagents, the cellular uptake of these complexes can be readily quantitated by atomic absorption spectroscopy (AAS) and inductively-coupled plasma mass spectrometry (ICP-MS). Many of these complexes, especially those with polypyridine ligands, show intense, long-lived, and environment-sensitive emission, which would allow the complexes to serve as reporters of their local surroundings and intracellular biological events.6 The intracellular trafficking and distribution of these complexes can also be followed conveniently by fluorescence methods such as laser-scanning confocal microscopy. The long-lived phosphorescence of these complexes facilitates the use of fluorescence lifetime imaging microscopy (FLIM), which offers very high sensitivity. Additionally, complexes with an ion- or molecule-recognition unit can be applied to detect intracellular ions and small molecules. As there are recent reviews on related topics such as the use of luminescent transition metal complexes as biological labels and probes,5 in this article we will focus concerning iridium(III) and summarize recent work on the biological applications of different types of complexes of this metal center. We will focus on the use of these complexes as anticancer drugs and cellular imaging reagents for organelle-staining, intracellular sensing, gene-delivery, and cancer cell detection.
2. Anticancer reagents
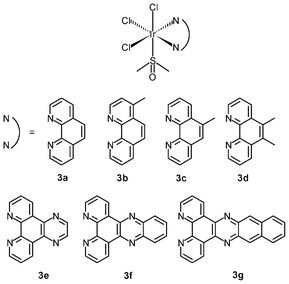
The success of cisplatin as an anticancer agent has led to much interest in the development of inorganic cytostatic drugs. In the early search for new metal-based anticancer drugs, iridium(III) complexes received less attention due to their prevailing chemical inertness, which would account for a lack of cytotoxic activity against cancer cells; for example, whilst the ruthenium(III) bis(imidazole) tetrachloride complex [Ru(Im)2Cl4]− and its monodimethylsulfoxide counterpart [Ru(Im)(DMSO)Cl4]− show very promising anticancer and antimetastatic properties associated with the hydrolysis of the chloride ligands,7 the iridium(III) analogues [Ir(Im)2Cl4]− (1) and [Ir(Im)(DMSO)Cl4]− (2) do not exhibit biological activity.8 Neither complex covalently modifies DNA or proteins nor inhibits the proliferation of representative human cancer cell lines. Despite the inertness, other iridium(III) complexes that exhibit relevant biological activity have been designed; for example, the diimine complexes fac-[Ir(NˆN)(DMSO)Cl3] (3a–3d) show antiproliferative properties toward the human cancer cell lines MCF-7 and HT-29.9a Since the order of activity of the complexes (3a < 3b, 3c < 3d) is in accordance with that of the free diimine ligands (phen < Me-phen < Me2-phen) (Table 1), the enhanced cytotoxic activity of these diimine complexes is believed to originate from the lipophilicity of the ligands, which plays a role in cellular uptake efficiency. The related complexes fac-[Ir(NˆN)(DMSO)Cl3] (3e–3g) also display similar ligand-dependent antiproliferative activity (3e < 3f < 3g) toward MCF-7 and HT-29 cells.9b Their cellular uptake efficiency has been analyzed by graphite furnace atomic absorption spectroscopy (GF-AAS) and the results show that the higher activity of complex 3g is related to its more efficient uptake. In addition to the diimine ligand, the solvent and halide ligands, as well as the stereostructure, all affect the activity of the complexes [Ir(NˆN)(solvent)X3];9c for example, the IC50 values of the aqua complexes are larger than those of their DMSO counterparts, whilst those of the chloride complexes are larger than those of the bromide analogues, which may be a consequence of the nature of the displaced ligands. Also, the meridional isomers of the complexes [Ir(phen)(DMSO)X3] (X = Cl, Br) are significantly less cytotoxic than their facial counterparts.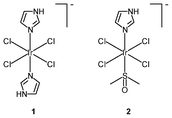
| Complex or ligand | IC50/μM | |
|---|---|---|
| MCF-7 | HT-29 | |
| 3a | 4.6 ± 0.5 | 4.6 ± 0.2 |
| 3b | 1.79 ± 0.14 | 2.21 ± 0.14 |
| 3c | 1.06 ± 0.04 | 1.88 ± 0.41 |
| 3d | 0.60 ± 0.27 | 0.86 ± 0.07 |
| phen | 3.5 ± 0.2 | 2.7 ± 0.5 |
| 4-Me-phen | 0.61 ± 0.24 | 0.80 ± 0.09 |
| 5-Me-phen | 0.72 ± 0.09 | 0.98 ± 0.03 |
| 5,6-Me2-phen | 0.34 ± 0.08 | 0.28 ± 0.16 |
| cisplatin | 2.0 ± 0.3 | 7.0 ± 2.0 |
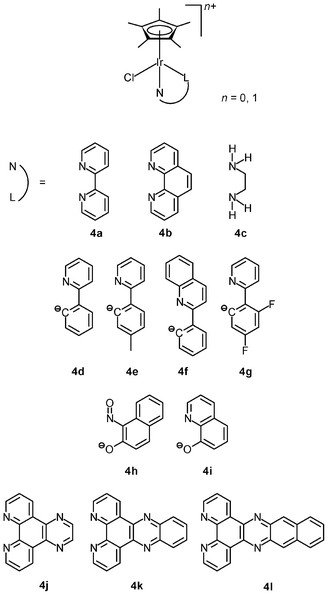
The lability of Ir–Cl bonds in octahedral iridium(III) complexes depends on the trans effect of the opposite ligand. Thus, the strong trans effect of pentamethylcyclopentadienyl (η5-C5Me5−) leads to rapid substitution of the chloride ligand by nucleophiles such as the nucleobase nitrogen atoms of nucleic acids; for example, the organometallic complexes [Ir(NˆN)(η5-C5Me5)Cl]+ (4a–4c) hydrolyze rapidly and readily form adducts with 9-ethylguanine.10a Although these complexes are not active or cytotoxic toward human ovarian cancer A2780 cells (IC50 > 100 μM), the introduction of a phenyl or biphenyl substituent to the cyclopentadienyl ring dramatically reduces the IC50 values of the complexes to as low as 0.57 μM (IC50 cisplatin = 1.22 μM). Thus, it is apparent that the anticancer potency of the complexes mainly results from the hydrophobicity and intercalative ability of the phenyl and biphenyl modified cyclopentadienyl ligands. Replacement of the neutral diimine and diamine ligands by a negatively charged ligand can switch on the anticancer activity. The cyclometalated complexes [Ir(NˆC)(η5-C5Me5)Cl] (4d–4g) show high activity toward A2780 cells with IC50 values ranging from 2.5 to 10.8 μM, which are comparable to that of cisplatin (1.2 μM).10b,c The NˆO complex [Ir(NˆO)(η5-C5Me5)Cl] (4h) exhibits much higher cytotoxicity toward HUVEC, HeLa, and HL60 cancer cell lines compared to cisplatin.10d The remarkable cytotoxicity of this complex has been attributed to necrotic cell death. The NˆO complex [Ir(NˆO)(η5-C5Me5)Cl] (4i) is not only cytotoxic toward SK-Mel, SNB-19, and C-32 cancer cells, but also active against Gram-positive bacteria including M. luteus, S. aureus, E. faecalis, and S. epidermidis.10e The use of extended planar diimine ligands allows the organometallic complexes [Ir(NˆN)(η5-C5Me5)Cl]+ (4j–4l) to initially intercalate into the base pairs of double-stranded DNA, which is followed by substitution of the labile chloride ligands by the N7 atom of purine bases.10f,g The dipyridophenazine (dppz) complex 4k exhibits high cytotoxicity toward MCF-7 and HT-29 cells; the IC50 values (2.3 and 7.4 μM, respectively) are similar to those of cisplatin (2.3 and 7.0 μM, respectively).
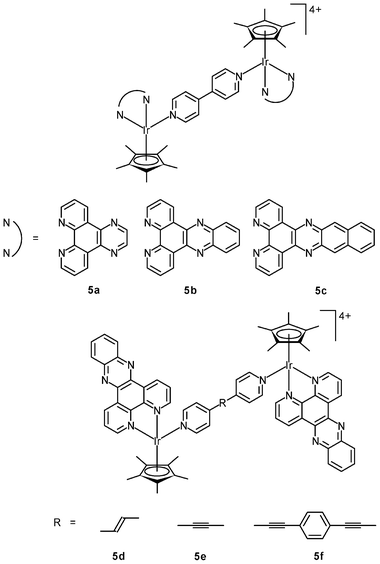
Related studies have been extended to multinuclear systems; for example, the DNA-binding properties and in vitro cytotoxicity of the dinuclear complexes [{Ir(NˆN)(η5-C5Me5)}2L]4+ (5a–5c) have been studied.11a The dppz complex 5b binds to DNA through a bis-intercalation mode involving the insertion of both dppz ligands into the base pairs. In contrast, complexes 5a and 5c with the smaller dipyridoquinoxaline (dpq) and larger benzodipyridophenazine (dppn) ligands do not intercalate into the base pairs of DNA. Thus, the size of the potentially intercalating diimine ligands is of great importance to the construction of effective bis-intercalators. The effects of the length of the rigid bridging ligands on the DNA-binding mode have been investigated; for example, the distance between the effectively parallel dppz ligands in complex 5b is about 10.2 Å, which is the ideal distance (3 × 3.4 Å) for sandwiching two base pairs. Interestingly, complexes 5d and 5e bind to DNA only in a mono-intercalation mode, though their Ir–Ir distances (13.1 and 13.5 Å, respectively) are close to the ideal distance for sandwiching three base pairs (13.6 Å).11b Upon addition of DNA, complex 5f (Ir–Ir distance = 20.6 Å) rapidly forms a kinetically-favored mono-intercalated adduct, which is slowly converted to a thermodynamically more stable intertwined bis-intercalation mode. Whilst complex 5f possesses photo-induced nuclease activity, both complexes 5d and 5e cleave DNA in the dark. All of these dinuclear complexes are more cytotoxic toward MCF-7 cells than their mononuclear counterparts. The IC50 values of the nuclease complexes 5d and 5e (0.61 and 0.49 μM) are smaller than those of the bis-intercalators 5b and 5f (3.1 and 2.2 μM) by one order of magnitude.
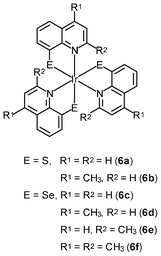
Neutral metal complexes with the 8-quinolinethiolate ligand display different anticancer activity. Most of them are very cytotoxic toward human fibrosarcoma HT-1080 and mouse hepatoma MG-22A cells. Among the 8-quinolinethiolate complexes [M(NˆS)n] (M = Cu(II), Cd(II), Pd(II), Pt(II), n = 2; M = In(III), Sb(III), Bi(III), Ru(III), Rh(III), Os(III), Ir(III), n = 3), the iridium(III) complex [Ir(NˆS)3] (6a) displays the highest activity against HT-1080 cells.12a However, this complex is also highly cytotoxic toward NIH 3T3 normal mouse embryo fibroblasts, limiting its potential as an anticancer drug. Interestingly, the introduction of a methyl group to the 4-position of the quinoline ring of these complexes significantly increases the selectivity of their cytotoxic activity; for example, the iridium(III) 4-methyl-8-quinolinethiolate complex [Ir(NˆS)3] (6b) is about 40 times more cytotoxic toward HT-1080 and MG-22A cancer cells than to 3T3 normal cells.12b,c Systematic studies of the complexes with methyl- and methoxy-substituted 8-quinolinethiolate ligands reveal that complexes with high cytotoxic activity substantially induce nitric oxide formation in the cells; for example, the iridium(III) 8-quinolineselenolate complexes [Ir(NˆSe)3] (6c–6f) show similar substituent-dependent cytotoxic activity and selectivity.12d,e The cytotoxicity of the complexes is also found to be closely related to their ability to generate nitric oxide.
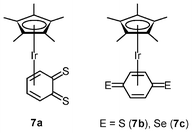
Quinones play an important role in electron transport in the respiratory chain and photosynthesis.13 Their potential uses as anticancer drugs and central components of antibiotics have been investigated; for example, 2-methyl-1,4-napthoquinone (menadione) displays in vitro and in vivo anticancer activity toward various tumor types, the mechanism of which involves activation by cytochrome P450 reductase to generate a semiquinone radical.14 The radical can reduce dioxygen to the superoxide anion, causing oxidative stress, depletion of glutathione, and induction of DNA single-strand breaks and eventually apoptosis. In contrast to benzoquinone, only a few quinones with heavier chalcogen elements have been reported, due to their inherent instability. To increase the stability, the [Ir(η5-C5Me5)] fragment has been introduced to the reactive dithio- and diseleno-benzoquinone species, leading to the isolation of the iridium(I) complexes [Ir(η4-C6H4E2)(η5-C5Me5)] (7a–7c).15 Subsequent protonation of the complexes yields iridium(III) hydroquinone complexes. The dithiobenzoquinone complexes 7a and 7b show moderate cytotoxic activity toward A2780 cells, whereas the diselenobenzoquinone complex 7c (IC50 = 5 μM) is as active as cisplatin (IC50 = 3 μM). Contrary to other iridium(III) anticancer drugs described above, which are hypothesized to act by DNA targeting, the benzoquinone complexes act in a mechanism similar to that of menadione.
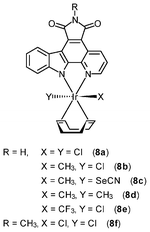
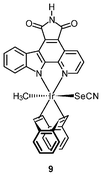
Chemically inert noncytotoxic metal complexes are promising scaffolds for targeting enzyme active sites, which may allow them to serve as enzyme inhibitors. The pyridocarbazole complexes [Ir(pcz)(cod)(X)(Y)] (8a–8e) are noncytotoxic and have been shown to inhibit the receptor tyrosine kinase Flt4, which plays an essential role in the maintenance of lymphatic vessels as well as in the development of the embryonic cardiovascular system.16 The Flt4 inhibition properties are strongly dependent on the ligands X and Y. The dichloro complex 8a shows the highest activity (IC50 = 123 nM), and is much more potent than the dimethyl complex 8d (IC50 = 1007 nM). Also, complex 8a displays high selectivity for Flt4 among 229 protein kinases. This complex is found to inhibit both angiogenesis in developing zebrafish embryos and cancer cell-induced angiogenesis. It strongly inhibits vessel formation of the zebrafish embryos being affected 3 days postfertilization at a concentration of 5 μM (Fig. 1). This bioactivity has been ascribed to Flt4 inhibition, because complex 8f lacking the protein inhibition ability does not show any effect on the zebrafish assays. Further studies show that the antiangiogenicity of complex 9 is independent of light.17 Interestingly, exposure to visible light for 1 h increases its cytotoxicity toward HeLa cells by ca. 34 fold and leads to significant apoptosis. The investigation of photoreactivity reveals that, in the presence of chloride, visible light induces the substitution of the selenocyanate ligand with a chloride ligand. However, the reason why such ligand substitution leads to efficient cellular apoptosis is unknown.
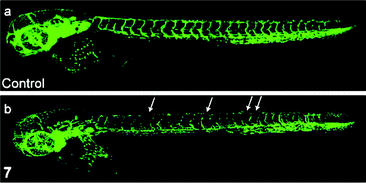 | ||
| Fig. 1 Laser scanning confocal microscopy images of 3 days postfertilization of zebrafish embryos treated with (a) DMSO or (b) complex 8a at 5 μM. Vessel defects are marked with white arrows. Taken from ref. 16. | ||
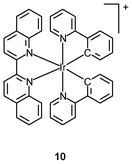
Tumor necrosis factor-α (TNF-α) is a cytokine involved in haematopoiesis, immunity, and systemic inflammation.18 It causes many of the clinical problems associated with autoimmune disorders, which are sometimes treated by using a TNF inhibitor that disrupts the TNF-α–TNFR (TNF receptor) interaction.19 Most of the small molecular inhibitors work by suppressing the expression of TNF-α.20 Small molecules capable of antagonizing TNF-α directly are very limited, and examples include the polysulfonated naphthylurea suramin and its analogues, the indole-linked chromone SPD304, and the isoindolo[2,1-a]quinazoline derivatives.21 The octahedral geometry of iridium(III) complexes provides a large structural complexity conformational flexibility, which may make access to the TNF-α binding site easier. Indeed, molecular docking shows that the iridium(III) biquinoline complex [Ir(ppy)2(piq)]+ (10) binds to the same binding pocket as chromone SPD304,22 which is one of the strongest small-molecule inhibitors of TNF-α. More importantly, both the Λ- and Δ-enantiomers of complex 10 show stronger activity against cellular TNF-α-induced NF-κB signaling than SPD304.
3. Bioimaging reagents
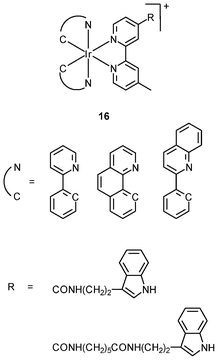
Many iridium(III) polypyridine complexes display intense and long-lived emission with large Stokes shifts in the visible region and the emission energy can be controlled using various ligands.6 The most common emissive states have been identified as being triplet metal-to-ligand charge-transfer (3MLCT), intraligand (3IL), and, in some systems, ligand-to-ligand charge-transfer (3LLCT), sigma-bond-to-ligand charge-transfer (3SBLCT), and aggregation-induced metal-to-ligand–ligand charge-transfer (3MLLCT), depending on the nature of the ligands.23 The intriguing emission properties have been exploited to design luminescent sensors for a range of analytes.3,5,24 In the past few years, there is also a rapidly growing interest in the use of these complexes for cellular imaging, intracellular sensing, gene-delivery, and cancer cell detection.3.1 Uptake efficiency
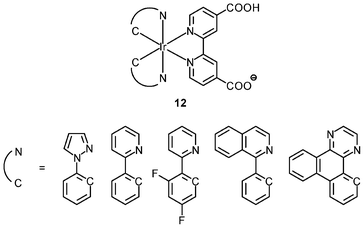
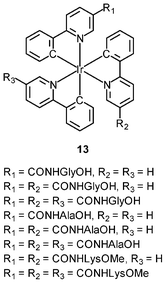
An important requirement for an intracellular imaging reagent is efficient uptake by cells, preferably in a controlled manner. Since the cellular uptake of iridium(III) complexes is known to be closely related to lipophilicity (vide supra), cyclometalated iridium(III) polypyridine complexes show more efficient cellular uptake compared to their chloro counterparts, as a result of the increased lipophilicity caused by the cyclometalating and polypyridine ligands; for example, HeLa cells loaded with the strongly emitting complexes [Ir(dfppy)2(NˆN)]+ (11) display exclusive staining in the cytoplasm and the uptake is dependent on their lipophilicity (Fig. 2).25 In a related study on the zwitterionic complexes [Ir(NˆC)2(Hdcbpy)] (12), KB cells treated with the complexes exhibit emission from the cytoplasm, and the intensity also increases with the lipophilicity of the complexes.26 Similarly, a series of iridium(III) mono-, bis-, and tris-amino acid complexes 13 shows variable cellular uptake; the monosubstituted complexes display a remarkably high cellular uptake than the bis- and tris-substituted complexes and the lysine complexes show a higher uptake than the glycine and alanine analogues.27 These differences are believed to be due to the lipophilicity of the complexes. The lipophilicity of the complexes [Ir(NˆC)2(bpy-CnH2n+1)]+ (n = 18, 10, 2) (14) bearing an aliphatic alkyl chain ranges from −0.34 to 9.89.28 Although efficient internalization of the complexes is supposed to be assisted by high lipophilicity, the most lipophilic complex of this family [Ir(pq)2(bpy-C18H37)]+ is taken up the least efficiently by cells. This could be a consequence of it having the largest molecular size and/or undergoing a higher degree of self-aggregation in aqueous solution.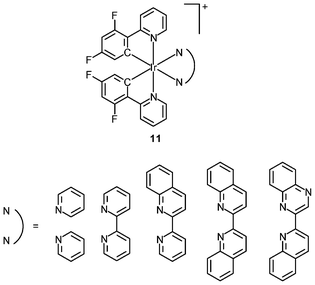
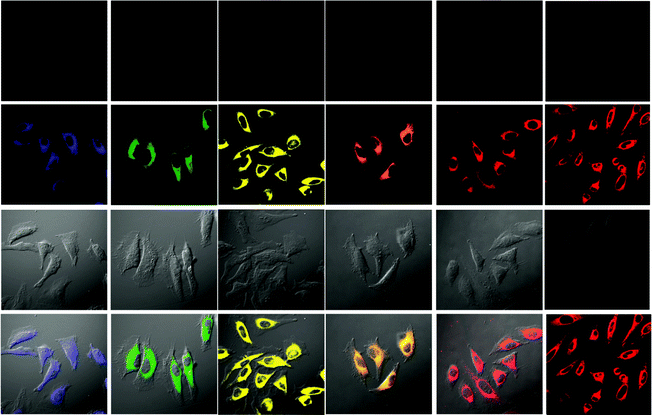 | ||
| Fig. 2 Microscopy images of HeLa cells: first row: cells without staining with complexes 11; second row: cells incubated solely with the complexes (20 μM) for 10 min at 25 °C; third row: DIC images of cells shown in the second row; last row: overlay image of the second and third rows. Taken from ref. 25. | ||
3.2 Uptake mechanisms
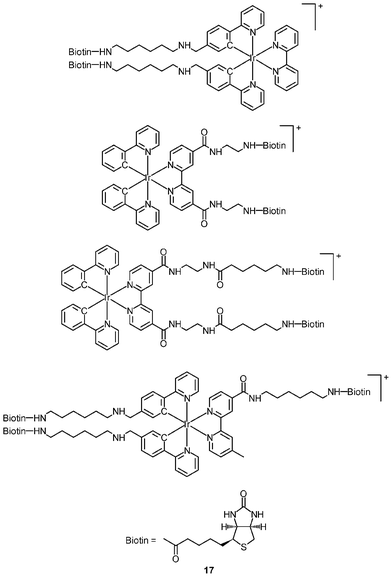
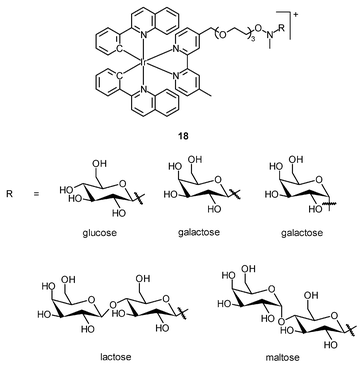
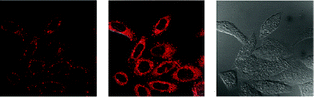
The cellular uptake mechanisms of iridium(III) polypyridine complexes have been investigated by studying uptake efficiency at different temperatures and by using various uptake inhibitors. The complex [Ir(ppy)2(bpy-(C4)2)]+ (15) forms a diffuse emissive background with cytoplasmic granules upon internalization into HeLa cells at 37 °C (Fig. 3).29 The disappearance of the cytoplasmic foci upon incubation of HeLa cells with this complex, either at 4 °C or in the presence of endocytic inhibitors, suggests that the complex enters cells by both passive diffusion and endocytosis pathways. The attachment of biological substrates to iridium(III) polypyridine complexes is found to affect their intracellular distribution and cellular uptake mechanisms; for example, the internalization of the indole (16)30 and biotin (17)29 complexes is via an energy-requiring process such as endocytosis. The sugar complexes [Ir(pq)2(bpy-sugar)]+ (18) appended with a D-glucose, D-galactose, D-lactose, and D-maltose moieties show similar photophysical properties but very different cellular uptake behaviors.31 Although HeLa cells treated with these complexes at 37 °C all exhibit emission from the perinuclear region, the emission intensity of the cells stained with the glucose complex is much higher than those treated with the other complexes (Fig. 4). Upon lowering the incubation temperature to 4 °C, the emission intensity of the cells loaded with the glucose complex is reduced and those with the non-glucose complexes become non-emissive. ICP-MS analysis further confirms that the glucose complex exhibits more efficient uptake compared to the non-glucose complexes. All of these differences between the glucose and non-glucose complexes have been ascribed to the glucose transporters (GLUTs) in the cell membrane that specifically transports glucose and its analogues.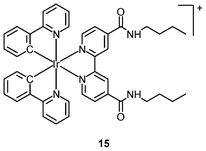
 | ||
| Fig. 3 Fluorescence (left), overlaid (middle), and bright-field (right) laser-scanning confocal microscopy images of HeLa cells incubated with complex 15 (5 μM) at 37 °C for 3 h. Taken from ref. 29. | ||
 | ||
| Fig. 4 Laser-scanning confocal microscopy images of HeLa cells incubated with complexes 18 (5 μM) (from left to right: complexes appended with β-glucose, α-galactose, β-galactose, β-lactose, and β-maltose) for 2 h at 37 °C. Taken from ref. 31. | ||
3.3 Cytotoxicity
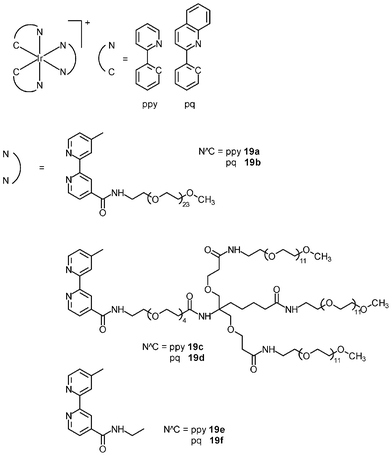
The cytotoxicity of the bioimaging reagents derived from iridium(III) polypyridine complexes with non-labile ligands has been studied. In most cases, a direct correlation between cellular uptake efficiency and cytotoxicity is observed; for example, among the alkyl complexes [Ir(NˆC)2(bpy-CnH2n+1)]+ (14), the bpy-C10H21 complexes exhibit both the highest uptake efficiency and cytotoxicity;28 for the indole complexes (16), the pq complexes enter HeLa cells more efficiently than the ppy complexes, with IC50 values of 1.1–2.4 and 6.0–6.3 μM, respectively;30 the biotin complexes 17 are much less cytotoxic (IC50 > 400 μM) toward HeLa cells compared to their biotin-free counterpart 15 (IC50 = 3.2 μM) because the latter has a much lower uptake efficiency, as revealed by ICP-MS measurements;29 among the sugar complexes 18, since the glucose complex enters cells through an additional GLUTs-mediated pathway, its cytotoxicity (IC50 = 4.0 μM) is higher than that of the non-glucose complexes (IC50 = 9.8–12.0 μM).31Many iridium(III) complexes suffer from high cytotoxicity, which limits their use as live-cell imaging reagents. Although decreasing the lipophilicity of these complexes can efficiently reduce their cytotoxicity through lowering their uptake efficiency, the detection sensitivity is also reduced accordingly. This problem of high cytotoxicity has been circumvented by the incorporation of poly(ethylene glycol) (PEG) into complexes; for example, the cellular uptake efficiency of the PEG complexes [Ir(NˆC)2(bpy-PEG)]+ (19a–d) toward HeLa cells is comparable to that of their PEG-free counterparts (19e and 19f) based on the results of ICP-MS measurements.32 Also, HeLa cells treated with the PEG and PEG-free complexes display emission of similar intensity from the cytoplasm. However, the cytotoxicity of the PEG complexes (IC50 = 286.5 to 1180.0 μM) is far lower than that of their PEG-free counterparts (IC50 = 4.1 to 14.6 μM) and cisplatin (10.3 μM) (Table 2). It is likely that the long PEG chains protect the complexes from interacting nonspecifically with intracellular biomolecules and/or organelles. Cells remain viable after prolonged exposure to the PEG complexes at a high concentration (200 μM), and the internalization of the PEG complexes does not adversely affect normal cellular processes such as cell growth and division. Thus, the high biocompatibility of the PEG complexes is a remarkable advantage toward the design of intracellular imaging reagents and sensors.
| Complex | IC50/μM |
|---|---|
| 19a | 830.4 ± 54.5 |
| 19b | 565.9 ± 49.4 |
| 19c | 1180.0 ± 70.5 |
| 19d | 286.5 ± 35.2 |
| 19e | 14.6 ± 1.5 |
| 19f | 4.1 ± 0.4 |
| cisplatin | 10.3 ± 0.7 |
3.4 Organelle targeting
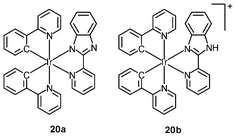
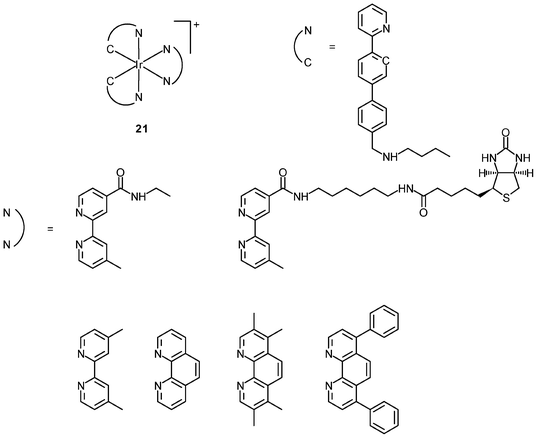
Organelle-specific probes are valuable tools that can monitor morphological changes of cellular compartments and offer insight into intracellular dynamic processes. Probes with controllable cytotoxicity can also be exploited as drugs for the treatment of organelle-related diseases. In the past few years, a number of iridium(III) complexes have been found to be capable of staining organelles in a specific manner; for example, CHO cells incubated with either the complex [Ir(ppy)2(pybz)] (20a) or [Ir(ppy)2(Hpybz)]+ (20b) display emission from discrete vesicles dispersed throughout the cytoplasm.33 The resemblance of the staining pattern to that observed with the dye LysoTracker Red confirms that the complexes are localized in the lysosomes. The long emission lifetimes of the complexes allow selective detection in co-staining experiments. CHO cells co-stained with complex 20a and the nuclear dye Hoeschst 33342 show bright emission from the nuclei. In contrast, time-gated imaging in a 10-ns delay reveals cytoplasmic staining and the emission of the Hoeschst dye becomes undetectable due to its much shorter fluorescence lifetime.Schemokine receptor 4 (CXCR4) is a G protein-coupled membrane receptor, which is most likely expressed in the cell membrane.34 It can bind several peptidic probes such as the Ac-TZ14011 peptide. The Ac-TZ14011 peptide-conjugated iridium(III) complexes specifically stain the cell membrane of schemokine receptor 4 (CXCR4), overexpressing MDA-MB-231 cells.35 A better signal-to-background ratio is obtained using the long luminescence lifetime of the iridium(III) complexes by FLIM. The intracellular localization of the bis(butylaminomethyl)-substituted complexes [Ir(pppy-C4)2(NˆN)]+ (21) is highly dependent on the polarity of the diimine ligands.36 Complexes with polar amide groups are well co-localized with the CellMask plasma membrane stain, indicative of membrane-targeting properties. In contrast, the lipophilic Ph2-phen complex is localized in the perinuclear region, and co-staining experiments with MitoTracker reveal a colocalization coefficient as large as 97%. Complexes with other diimine ligands are found to stain both the cell membrane and the mitochondria. These results illustrate that the use of ligands and substituents of various polarities would allow for control of the intracellular localization properties of these luminescent complexes.
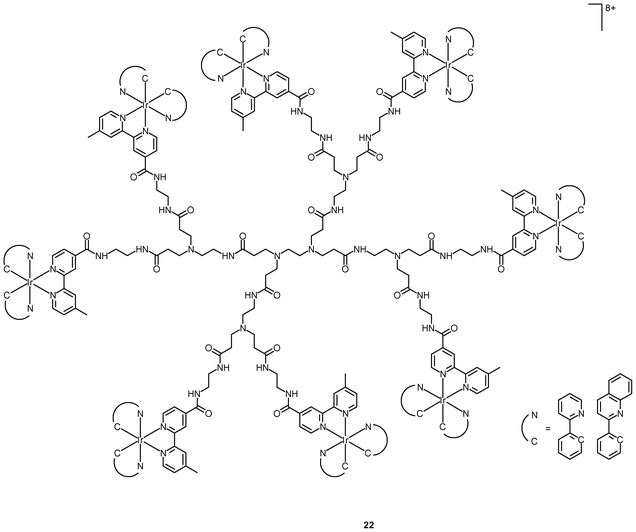
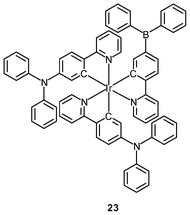
Polynuclear iridium(III) complexes display cellular uptake and intracellular staining behavior that can be very different from that of their mononuclear counterparts. Similar to other related iridium(III) complexes, the mononuclear complexes [Ir(NˆC)2(bpy-C2H5)]+ are localized in the perinuclear region of HeLa cells upon internalization. It is interesting to note that the octanuclear complexes [{Ir(NˆC)2}8(bpy-8)]8+ (22), with monomers as a building block, bind specifically to the Golgi apparatus (Fig. 5).37 The neutral borane-containing complex [Ir(ppy-N)2(ppy-B)] (23) also shows specific affinity toward the Golgi apparatus of the HeLa and A549 cells and stays in the organelle for more than 6 h.38 This complex also displays intense emission with a lifetime in the sub-microsecond scale upon two-photon excitation at 700 nm.
![Laser-scanning confocal microscopy images of HeLa cells treated successively with complexes 22 ([Ir] = 2 M) at 37 °C for 2 h, PBS containing 3% paraformaldehyde, anti-golgin-97 (human) mouse IgG1 (1 μg mL−1, 1 h), and Alexa 635 goat anti-mouse IgG (H+L) (10 μg mL−1, 30 min). Taken from ref. 37.](/image/article/2012/RA/c2ra20967e/c2ra20967e-f5.gif) | ||
| Fig. 5 Laser-scanning confocal microscopy images of HeLa cells treated successively with complexes 22 ([Ir] = 2 M) at 37 °C for 2 h, PBS containing 3% paraformaldehyde, anti-golgin-97 (human) mouse IgG1 (1 μg mL−1, 1 h), and Alexa 635 goat anti-mouse IgG (H+L) (10 μg mL−1, 30 min). Taken from ref. 37. | ||
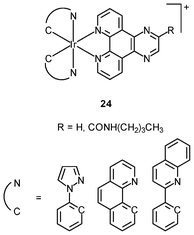
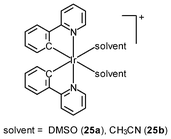
Generally speaking, it is very difficult to achieve nuclear staining using simple transition metal complexes, because of the very complicated intracellular environment and the presence of a wide range of biomolecules that the probes may bind to. However, with a judicious choice of ligands, complexes with appropriate formal charge, lipophilicity, and molecular shape may be able to traverse the nuclear envelope and reside inside the nucleus. MDCK cells incubated with the DNA intercalators [Ir(NˆC)2(NˆN)]+ (24) show very strong emission in the nucleoli, whilst the nucleoplasm emits weakly (Fig. 6).39 Results of emission titration and SDS-PAGE analysis indicate that the nucleolar staining originates from the binding of the complexes to hydrophobic pockets of nucleolar proteins. The non-emissive complexes [Ir(ppy)2(solvent)2]+ (25a and 25b) undergo substitution of the weak-field solvent ligands with the strong-field π-accepting imidazole of histidine (His) and His-containing proteins, resulting in significant emission enhancement.40,41 HeLa cells treated with complex 25a exhibit intense emission from their nuclei, indicative of the intracellular substitution of the ligands.41 Complex 25b has been used to label a His-modified mitochondrial targeting peptide.42 As expected, HeLa cells incubated with this conjugate reveal mitochondrial staining.
 (5 μM, 30 min). Taken from ref. 39.](/image/article/2012/RA/c2ra20967e/c2ra20967e-f6.gif) | ||
| Fig. 6 Fluorescence laser-scanning confocal microscopy images of fixed MDCK cells treated successively with fibrillarin antibody (20 μL mL−1, 1 h), Alexa 633 anti-rabbit IgG antibody (20 μg mL−1, 30 min), and one of complexes 24 [Ir(ppz)2(dpq)](PF6) (5 μM, 30 min). Taken from ref. 39. | ||
3.5 Other imaging reagents
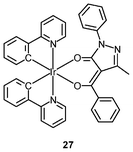
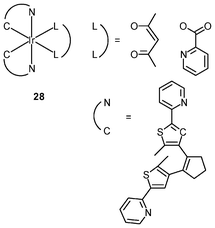
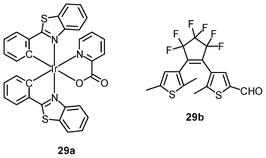
Luminescent iridium(III) complexes with functional ligands have been used as covalent labeling and imaging reagents. HeLa cells incubated with the complex [Ir(qba)2(Ph2-phen)]+ (26), which has two amine-specific aldehyde groups, show staining patterns that are similar to those treated with the aldehyde-free complex [Ir(pq)2(Ph2-phen)]+.43 However, upon fixation of the complex-treated cells with methanol or paraformaldehyde, only the qba complex retains its emission from the stained cells, whilst the emission intensity of the pq complex decreases significantly (Fig. 7). This indicates covalent attachment of the qba complex to the intracellular structures. Some iridium(III) complexes exhibit aggregation-induced luminescence, which has been utilized in cell imaging; for example, the complex [Ir(ppy)2(OˆO)] (27) emits only in the aggregated state, and is embedded in water-dispersible polymer nanoparticles prior to cellular imaging studies.44 The cytoplasm of KB cells incubated with the nanoparticles displays intense emission. The photoswitchable diarylethene complexes [Ir(py-bte)2(LˆL)] (28) switch from the open form to the closed form upon excitation at 488 nm, reversed by excitation at 440 nm.45 KB cells treated with the complexes reveal cytoplasmic staining, and the photon-induced switch between the open and closed forms can be controlled by the light sources of the confocal microscope at 458 and 488 nm, respectively. In another study, luminescent polymer nanoparticles have been constructed by using emulsion polymerization to trap the complex [Ir(bt)2(pic)] (29a) and the photochrome diarylethene (29b).46 The reversible photoswitching has been demonstrated in live KB cells.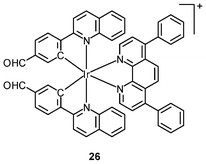
 was used as the staining reagent. Taken from ref. 43.](/image/article/2012/RA/c2ra20967e/c2ra20967e-f7.gif) | ||
| Fig. 7 Fluorescence images of HeLa cells incubated with complex 26 (5 μM) at 37 °C for 1 h (a) before and (b) after subsequent fixation with MeOH, or (c) fixation with paraformaldehyde, followed by extensive washing with MeOH; (d)–(f): fluorescence images of HeLa cells treated and fixed with the same procedures, except that [Ir(pq)2(Ph2-phen)](PF6) was used as the staining reagent. Taken from ref. 43. | ||
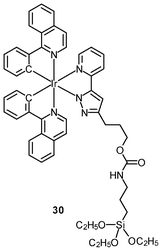
Multimodal probes have received increasing attention in recent years. Luminescent iridium(III) complexes have been coupled to magnetically active materials for luminescent and magnetic resonance bifunctional imaging applications; for example, the nanoparticles Fe3O4/SiO2(Ir) have been prepared from the luminescent complex [Ir(piq)2(ppTES)] (30) and the magnetic Fe3O4 core.47 These nanoparticles are non-cytotoxic toward HeLa cells, and bind to the cytoskeleton. Cells treated with the nanoparticles display a dose responsive signal change in magnetic resonance imaging (MRI) with a detection limit of about 104 cells. Irradiation of the nanoparticle-treated cells causes 1O2-induced apoptosis, highlighting the photocytotoxic nature of the material. In another study, HeLa cells incubated with polymeric nanoparticles prepared from the luminescent complex [Ir(ppy)2(H2dcbpy)]+ (31) and the paramagnetic gadolinium(III) ion display cytoplasm staining.48 Results of the MRI experiments indicate a linear relationship between 1/T1, 1/T2 and gadolinium(III) concentration.
3.6 Intracellular sensing
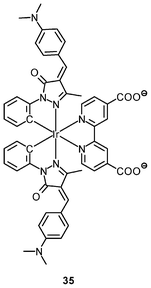
Luminescent cyclometalated iridium(III) polypyridine complexes have been widely used as sensors for proton, ions, oxygen, volatile organic compounds, and biological macromolecules such as proteins and nucleic acids.3,5,24 Recently, there has also been much interest in the incorporation of ion or molecular recognition groups into membrane-permeable complexes to afford new cellular sensors for ions and molecules; for example, a series of iridium(III) pyridylbenzaldehyde complexes has been designed to label amino acids and proteins through reductive amination.49a Since the aldehyde groups of the complexes can also react with cysteine (Cys)/homocysteine (Hcy), yielding cyclic thiazolidine and thiazinane derivatives, one of the complexes [Ir(pba)2(bpy)]+ (32) has been used to recognize intracellular Cys and Hcy.49b Introduction of an ammonium group to the diimine ligand renders the sensor soluble in pure water, which facilitates the intracellular Cys/Hcy detection.50 The complex [Ir(bt)2(acac)] (33) has been used as an intracellular ratiometric Hg2+ sensor based on the soft–soft interaction between the Hg and S atoms.51 The DPA complexes [Ir(NˆC)2(phen-DPA)](PF6) (34a) display Zn2+-induced emission enhancement in CH3CN due to the binding of the ion to the DPA moiety (Kd values in the order of 10−5 M). HeLa cells incubated with these complexes displayed intense emission, indicative of efficient cellular internalization.52 The related complex [Ir(dfppy)2(phen-DPA)]+ (34b) displays intracellular Zn2+ sensing capability.53 Adenocarcinoma human alveolar basal epithelial A549 cells loaded with complex 34b show weak emission (Fig. 8). Further incubation with ZnCl2/NaPT (sodium pyrithione) leads to intense and long-lived emission from the cytoplasm. Subsequent treatment with the cell-permeable Zn2+-specific chelator, TPEN, reduces the emission intensity. The complex [Ir(ppy)2(btp-DPA)] (34c) shows dual emission in the green and red regions, and binding of intracellular Cu2+ ions to the DPA unit of the complex selectively quenches the red emission, resulting in a significant change of emission profile.54 The complex [Ir(NˆC)2(dcbpy)]− (35) specifically reacts with CN− through a 1,4-addition reaction on the cyclometalating ligands (Scheme 1).55 An intracellular CN− sensing material (Ir-UCNPs) has been prepared from this complex and upconversion NaYF4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20%Yb, 1.6%Er, 0.4%Tm nanocrystals (UCNPs). The material exhibits green upconversion emission at 514–560 nm, with intensity that increases linearly with concentration of CN−. Incubation of CN−-pretreated HeLa cells with Ir-UCNPs displays much more intense upconversion emission compared to the untreated cells.
:20%Yb, 1.6%Er, 0.4%Tm nanocrystals (UCNPs). The material exhibits green upconversion emission at 514–560 nm, with intensity that increases linearly with concentration of CN−. Incubation of CN−-pretreated HeLa cells with Ir-UCNPs displays much more intense upconversion emission compared to the untreated cells.
 | ||
| Fig. 8 Microscopy images of live A549 cells incubated with complex 34b (5 μM, 30 min; left columns), then with ZnCl2/NaPT (1:1, v/v, 50 μM, 15 min; middle columns), and finally with TPEN (100 μM, 15 min; right columns). Left panels, bright-field images; right panels, phosphorescence images. Scale bar corresponds to 50 μm. Identical intensity scales have been applied. Images were acquired from four independent experiments (fields 1–4). Taken from ref. 53. | ||
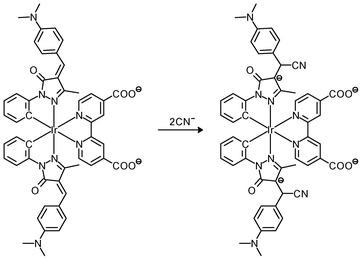 | ||
| Scheme 1 Proposed recognition mechanism of complex 35 toward CN−. | ||
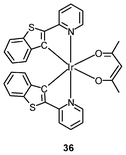
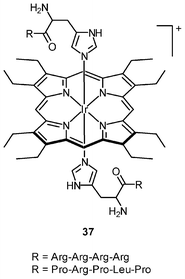
Iridium(III) complexes with intense and long-lived phosphorescence have been commonly employed in in vitro oxygen-sensing applications due to their efficient oxygen-quenching properties.5,24 These sensors have recently been applied in intracellular environments; for example, the strongly emissive complex [Ir(btp)2(acac)] (36) has been used for hypoxic tissue imaging. Cells from the CHO, HeLa, mouse oral squamous carcinoma SCC-7, and human glioma U251 lines cultured at both 20% and 5% oxygen concentrations have been incubated with complex 36.56 Whilst cells in 20% oxygen culture conditions do not exhibit notable emission, those in a 5% oxygen culture display bright red emission upon excitation (Fig. 9). This oxygen-sensing property has been extended to in vivo applications: five culture cell lines, SCC-7, human glioma U87, human lymphoma-derived RAMOS, human colon carcinoma HT-29, and mouse lung cancer LL-2, have been transplanted into the lower thigh of nude mice. With an oxygen monitoring probe, the pO2 of tumors has been determined to be 18–21 mmHg, which is lower than that of end-capillary blood (45–50 mmHg). All five tumors exhibit emission in 5 min after injection of complex 36 and the minimal image recognition size is about 2 mm in diameter. In another study, two iridium(III) porphyrin complexes 37 display low energy red emission at around 650 nm.57 Both complexes stain endoplasmic reticulum of a number of mammalian cell lines. Intracellular oxygen sensing by the complexes has been demonstrated using the rapid lifetime determination method.
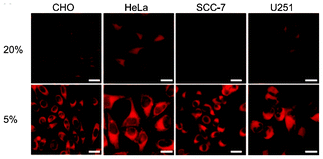 | ||
| Fig. 9 Microscopy images of CHO, HeLa, SCC-7, and U251 cells treated with complex 36. The cells were cultured at 20% O2 concentration (top) and 5% O2 concentration (bottom). Scale bars: 10 μm. Taken from ref. 56. | ||
3.7 Gene-delivery agents
Recently, a series of water-soluble iridium(III) poly(ethyleneimine) (PEI) complexes 38 have been synthesized and used as transfection reagents.58 These complexes retain the luminescence properties of iridium(III) complexes and the DNA retardation ability of the PEI unit. Formation of polyplexes from the iridium(III) complexes and plasmid DNA has been confirmed by zeta-potential measurements and particle size determination. One of the complexes has been grafted with PEG, yielding new copolymers. Various experiments indicated that the copolymers not only retained the photophysical properties of the complex, but also exhibited enhanced in vitro gene delivery capability, due to increased water solubility and biocompatibility.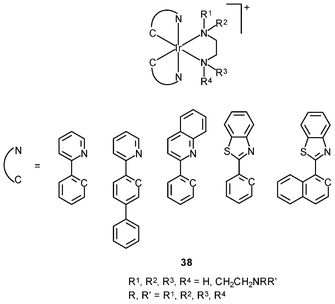
3.8 Cancer cell detection
The complex [Ir(ppy)2(H2dcbpy)]+ (31) has been used as a labeling reagent for DNA and cancer cell detection.59 The sensing procedure involves gold nanoparticles (AuNP) labeled with this complex and a signal probe DNA (Scheme 2). The target DNA (red) hybridizes through its two ends an electrode-immobilized capture DNA (blue) and the signal probe DNA on AuNP (green), respectively. The concentration of target DNA is quantified by the electrochemiluminescence (ECL) signal of the complex-labeled AuNP, and the detection limit is determined to be 8.6 × 10−16 M. The sensitivity has been improved by DNA polymerization and nicking the endonuclease assisted circular amplification process; the detection limit can be lowered to 8.2 × 10−17 M. Cancer cell detection has been achieved based on the high specificity and affinity of cell aptamers toward unique molecular signatures on the cell surface. The smallest number of Ramos cells that can be detected is 8.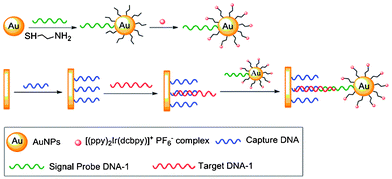 | ||
| Scheme 2 Process of the detection of target DNA. Taken from ref. 59. | ||
4. Conclusion
The use of iridium(III) complexes as anticancer drugs and live-cell imaging reagents has been summarized in this review. Although many iridium(III) complexes are chemically inert, their cytotoxic activity can be activated by increasing the lability of iridium–halide or iridium–solvent bonds or increasing their lipophilicity, which will enhance their cellular uptake. Various studies have illustrated that the cytotoxicity of these complexes is highly dependent on their uptake efficiency and, in some cases, on their ability to generate nitric oxide. Interestingly, the incorporation of a PEG moiety into the complexes can effectively reduce their cytotoxicity, while maintaining efficient cellular uptake. Many studies have also indicated that strongly-emissive iridium(III) polypyridine complexes can be used as live-cell imaging reagents. Different cellular uptake mechanisms, depending on the structures of the complexes, have been identified using various methods. Also, the introduction of specific substrates to the ligands has been found to enable receptor-mediated uptake, which may increase cellular uptake selectivity. Additionally, through functionalization of ligands, new reagents have been designed for targeting organelles such as cell membranes, lysosomes, mitochondria, Golgi apparatus, nuclei, and sub-nuclear structures. Luminescent iridium(III) complexes equipped with a recognition unit have also been exploited as probes for intracellular molecules and ions. In conclusion, the interesting structural and emissive properties of iridium(III) complexes has and will continue to contribute to the development of reagents with a wide range of biological applications.Acknowledgements
We thank the Hong Kong University Grants Committee (Areas of Excellence Scheme AoE/P-03/08) and the Hong Kong Research Grants Council (Project Nos. CityU 102410 and CityU 102311) for financial support.References
-
(a) J. A. Broomhead and W. Grumley, Inorg. Chem., 1971, 10, 2002 CrossRef CAS
; (b) C. M. Flynn Jr. and J. N. Demas, J. Am. Chem. Soc., 1974, 96, 1959 CrossRef
-
(a) H. Yersin, Top. Curr. Chem., 2004, 241, 1 CAS
-
(a) K. K.-W. Lo, D. C.-M. Ng and C.-K. Chung, Organometallics, 2001, 20, 4999 CrossRef CAS
-
(a) N. P. E. Barry and P. J. Sadler, Chem. Soc. Rev., 2012, 41, 3264 RSC
-
(a) C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418 CrossRef CAS
-
(a) I. M. Dixon, J. P. Collin, J.-P. Sauvage, L. Flamigni, S. Encinas and F. Barigelletti, Chem. Soc. Rev., 2000, 29, 385 RSC
-
(a)
Metal Complexes in Cancer Chemotherapy, ed. B. K. Keppler, Wiley-VCH, Weinheim, 1993 Search PubMed
-
(a) G. Marcon, A. Casini, P. Mura, L. Messori, A. Bergamo and P. Orioli, Met.-Based Drugs, 2000, 7, 195 Search PubMed
-
(a) Y. Geldmacher, I. Kitanovic, H. Alborzinia, K. Bergerhoff, R. Rubbiani, P. Wefelmeier, A. Prokop, R. Gust, I. Ott, S. Wölfl and W. S. Sheldrick, ChemMedChem, 2011, 6, 429 Search PubMed
-
(a) Z. Liu, A. Habtemariam, A. M. Pizarro, S. A. Fletcher, A. Kisova, O. Vrana, L. Salassa, P. C. A. Bruijninicx, G. J. Clarkson, V. Brabec and P. J. Sadler, J. Med. Chem., 2011, 54, 3011 CrossRef CAS
-
(a) M. A. Nazif, J.-A. Bangert, I. Ott, R. Gust, R. Stoll and W. S. Sheldrick, J. Inorg. Biochem., 2009, 103, 1405 CrossRef CAS
-
(a) E. Lukevics, I. Shestakova, I. Domracheva, A. Nesterova, D. Zaruma and J. Ashaks, Chem. Heterocycl. Compd., 2006, 42, 761 Search PubMed
-
(a)
The Chemistry of the Quinonoid Compounds, ed. S. Patai and Z. Rappoport, vol. 2, plts. 1 and 2, Wiley, New York, 1988, p. 1711 Search PubMed
- E. Misaka and K. Nakanishi, J. Biochem., 1965, 58, 1 Search PubMed
-
(a) H. Amouri, J. Moussa, A. K. Renfrew, P. J. Dyson, M. N. Rager and L.-M. Chamoreau, Angew. Chem., Int. Ed., 2010, 49, 7530 CrossRef CAS
- A. Wilbuer, D. H. Vlecken, D. J. Schmitz, K. Kräling, K. Harms, C. P. Bagowski and E. Meggers, Angew. Chem., Int. Ed., 2010, 49, 3839 CrossRef CAS
- A. Kastl, A. Wilbuer, A. L. Merkel, L. Feng, P. Di Fazio, M. Ocker and E. Meggers, Chem. Commun., 2012, 48, 1863 RSC
- H. Wajant, K. Pfizenmaier and P. Scheurich, Cell Death Differ., 2003, 10, 45 CrossRef CAS
-
(a) B. B. Aggarwal, Nat. Rev. Immunol., 2003, 3, 745 CrossRef CAS
-
(a) R. Alzani, A. Corti, L. Grazioli, E. Cozzi, P. Ghezzi and F. Marcucci, J. Biol. Chem., 1993, 268, 12526 Search PubMed
-
(a) F. Mancini, C. M. Toro, M. Mabilia, M. Giannangeli, M. Pinza and C. Milanese, Biochem. Pharmacol., 1999, 58, 851 Search PubMed
- C.-H. Leung, H.-J. Zhong, H. Yang, Z. Cheng, D. S.-H. Chan, V. P.-Y. Ma, R. Abagyan, C.-Y. Wong and D.-L. Ma, Angew. Chem. Int. Ed. DOI:10.1002/amie.201202937
-
(a) Y. Ohsawa, S. Sprouse, K. A. King, M. K. DeArmond, K. W. Hanck and R. J. Watts, J. Phys. Chem., 1987, 91, 1047 CrossRef CAS
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC
-
(a) M. Yu, Q. Zhao, L. Shi, F. Li, Z. Zhou, H. Yang, T. Yi and C. Huang, Chem. Commun., 2008, 2115 RSC
- W. Jiang, Y. Gao, Y. Sun, F. Ding, Y. Xu, Z. Bian, F. Li, J. Bian and C. Huang, Inorg. Chem., 2010, 49, 3252 CrossRef CAS
- P. Steunenberg, A. Ruggi, N. S. van den Berg, T. Buckle, J. Kuil, F. W. B. van Leeuwen and A. H. Helders, Inorg. Chem., 2012, 51, 2105 Search PubMed
- K. K.-W. Lo, P.-K. Lee and J. S.-Y. Lau, Organometallics, 2008, 27, 2998 CrossRef CAS
- K. Y. Zhang and K. K.-W. Lo, Inorg. Chem., 2009, 48, 6011 CrossRef CAS
- J. S.-Y. Lau, P.-K. Lee, K. H.-K. Tsang, C. H.-C. Ng, Y.-W. Lam, S.-H. Cheng and K. K.-W. Lo, Inorg. Chem., 2009, 48, 708 CrossRef CAS
- H.-W. Liu, K. Y. Zhang, W. H.-T. Law and K. K.-W. Lo, Organometallics, 2010, 29, 3474 CrossRef
- S. P.-Y. Li, H.-W. Liu, K. Y. Zhang and K. K.-W. Lo, Chem.–Eur. J., 2010, 16, 8329 CrossRef CAS
- L. Murphy, A. Congreve, L.-O. Pålsson and J. A. G. Williams, Chem. Commun., 2010, 46, 8743 RSC
-
(a) B. Furusato, A. Mohamed, M. Uhlen and J. S. Rhim, Pathol. Int., 2010, 60, 497 CrossRef CAS
- J. Kuil, P. Steunenberg, P. T. K. Chin, J. Oldenburg, K. Jalink, A. H. Velders and F. W. B. van Leeuwen, ChemBioChem, 2011, 12, 1897 CrossRef CAS
-
K. Y. Zhang, H.-W. Liu, A. W.-T. Choi, M.-C. Tang, N. Zhu, X.-G. Wei, K.-C. Lau and K. K.-W. Lo, unpublished work
- K. Y. Zhang, H.-W. Liu, T. T.-H. Fong, X.-G. Chen and K. K.-W. Lo, Inorg. Chem., 2010, 49, 5432 CrossRef CAS
- C.-L. Ho, K.-L. Wong, H.-K. Kong, Y.-M. Ho, C. T.-L. Chan, W.-M. Kwok, K. S.-Y. Leung, H.-L. Tam, M. H.-W. Lam, X.-F. Ren, A.-M. Ren, J.-K. Feng and W.-Y. Wong, Chem. Commun., 2012, 48, 2525 RSC
- K. Y. Zhang, S. P.-Y. Li, N. Zhu, I. W.-S. Or, M. S.-H. Cheung, Y.-W. Lam and K. K.-W. Lo, Inorg. Chem., 2010, 49, 2530 CrossRef CAS
- D.-L. Ma, W.-L. Wong, W.-H. Chung, F.-Y. Chan, P.-K. So, T.-S. Lai, Z.-Y. Zhou, Y.-C. Leung and K.-Y. Wong, Angew. Chem., Int. Ed., 2008, 47, 3735 CrossRef CAS
- C. Li, M. Yu, Y. Sun, Y. Wu, C. Huang and F. Li, J. Am. Chem. Soc., 2011, 133, 11231 CrossRef CAS
- X. Wang, J. Jia, Z. Huang, M. Zhou and H. Fei, Chem.–Eur. J., 2011, 17, 8028 CrossRef CAS
- P.-K. Lee, H.-W. Liu, S.-M. Yiu, M.-W. Louie and K. K.-W. Lo, Dalton Trans., 2011, 40, 2180 RSC
- H. Wu, T. Yang, Q. Zhao, J. Zhou, C. Li and F. Li, Dalton Trans., 2011, 40, 1969 RSC
- W. Tan, J. Zhou, F. Li, T. Yi and H. Tian, Chem.–Asian J., 2011, 6, 1263 CrossRef CAS
- T. Yang, Q. Liu, S. Pu, Z. Dong, C. Huang and F. Li, Nano Res., 2012, 5, 494 Search PubMed
- C.-W. Lai, Y.-H. Wang, C.-H. Lai, M.-J. Yang, C.-Y. Chen, P.-T. Chou, C.-S. Chan, Y. Chi, Y.-C. Chen and J.-K. Hsiao, Small, 2008, 4, 218 CrossRef CAS
- Z. Zhou, D. Li, H. Yang, Y. Zhu and S. Yang, Dalton Trans., 2011, 40, 11941 RSC
-
(a) K. K.-W. Lo, C.-K. Chung and N. Zhu, Chem. Eur. J., 2003, 9, 475 CrossRef CAS
- Y. Ma, S. Liu, H. Yang, Y. Wu, C. Yang, X. Liu, Q. Zhao, H. Wu, J. Liang, F. Li and W. Huang, J. Mater. Chem., 2011, 21, 18974 RSC
- Y. Wu, H. Jing, Z. Dong, Q. Zhao, H. Wu and F. Li, Inorg. Chem., 2011, 50, 7412 CrossRef CAS
- P.-K. Lee, W. H.-T. Law, H.-W. Liu and K. K.-W. Lo, Inorg. Chem., 2011, 50, 8570 Search PubMed
- Y. You, S. Lee, T. Kim, K. Ohkubo, W.-S. Chae, S. Fukuzumi, G.-J. Jhon, W. Nam and S. J. Lippard, J. Am. Chem. Soc., 2011, 133, 18328 CrossRef CAS
- Y. You, Y. Han, Y.-M. Lee, S. Y. Park, W. Nam and S. J. Lippard, J. Am. Chem. Soc., 2011, 133, 11488 CrossRef CAS
- J. Liu, Y. Liu, Q. Liu, C. Li, L. Sun and F. Li, J. Am. Chem. Soc., 2011, 133, 15276 CrossRef CAS
- S. Zhang, M. Hosaka, T. Yoshihara, K. Negishi, Y. Iida, S. Tobita and T. Takeuchi, Cancer Res., 2010, 70, 4490 CrossRef CAS
- K. Koren, R. I. Dmitriev, S. M. Borisov, D. B. Papkovsky and I. Klimant, ChemBioChem, 2012, 13, 1184 Search PubMed
- S. P.-Y. Li, T. S.-M. Tang, K. S.-M. Yiu and K. K.-W. Lo, Chem. Eur. J., 2012 DOI:10.1002/chem.201200979
- C. Li, J. Lin, Y. Guo and S. Zhang, Chem. Commun., 2011, 47, 4442 RSC
| This journal is © The Royal Society of Chemistry 2012 |