Recent developments in the production of liquid fuels via catalytic conversion of microalgae: experiments and simulations
Fan
Shi
*ab,
Ping
Wang
a,
Yuhua
Duan
*a,
Dirk
Link
a and
Bryan
Morreale
a
aNational Energy Technology Laboratory, United States Department of Energy, Pittsburgh, Pennsylvania 15236, USA. E-mail: Yuhua.duan@netl.doe.gov; Fax: +1-412-386-5920; Tel: +1-412-386-5771
bURS Corporation, South Park, Pennsylvania 15219, USA. E-mail: Fan.shi@netl.doe.gov; Fax: +1-412-386-4542; Tel: +1-412-386-7350
First published on 2nd August 2012
Abstract
Due to continuing high demand, depletion of non-renewable resources and increasing concerns about climate change, the use of fossil fuel-derived transportation fuels faces relentless challenges both from a world markets and an environmental perspective. The production of renewable transportation fuel from microalgae continues to attract much attention because of its potential for fast growth rates, high oil content, ability to grow in unconventional scenarios, and inherent carbon neutrality. Moreover, the use of microalgae would minimize “food versus fuel” concerns associated with several biomass strategies, as microalgae do not compete with food crops in the food chain. This paper reviews the progress of recent research on the production of transportation fuels via homogeneous and heterogeneous catalytic conversions of microalgae. This review also describes the development of tools that may allow for a more fundamental understanding of catalyst selection and conversion processes using computational modelling. The catalytic conversion reaction pathways that have been investigated are fully discussed based on both experimental and theoretical approaches. Finally, this work makes several projections for the potential of various thermocatalytic pathways to produce alternative transportation fuels from algae, and identifies key areas where the authors feel that computational modelling should be directed to elucidate key information to optimize the process.
 Fan Shi | Dr Fan Shi is a senior materials scientist of URS Corporation working at the Department of Energy's (DOE) National Energy Technology Laboratory (NETL). Dr Shi has been actively working on sustainable energy production and greenhouse gas separation for more than 10 years. He is also working on two books focusing on sustainable energy research. Dr Shi obtained his BS and MS in chemical engineering from the Nanjing University of Technology, China. He holds a Ph.D. in chemical engineering from the University of Pittsburgh. |
 Ping Wang | Dr Ping Wang is a chemical and biological engineer at the U.S.A. DOE-NETL in Pittsburgh, PA. Her research interests are in the areas of biomass and coal from their preparation and characterization to their conversion to gaseous/liquid fuels. Prior to joining NETL, she had many years experience with grain based liquid fuel process development at the University of Illinois at Urbana-Champaign (UIUC) in Urbana, IL. She earned her Ph.D. in Agricultural and Biological Engineering from UIUC. |
 Yuhua Duan | Dr Yuhua Duan is a physical scientist at the U.S. DOE-NETL. He obtained his BS in Chemistry, a MS in Chemical Physics, Ph.D. in Condensed Matter of Physics from the University of Science and Technology of China, and another MS in Computer Engineering from the University of Minnesota. Dr Duan has authored and co-authored more than 60 publications. His research interests include developing technologies for CO2 capture and energetic materials applied to batteries and solar cells, exploring the mechanisms of biological enzyme catalysis and high-temperature gas sensors, and multi-scale modelling of energy systems. |
 Dirk Link | Dr Dirk Link received his Ph.D. in Analytical Chemistry from Duquesne University. His initial research at U.S. DOE was in trace analysis of sulfur species in liquid fuels, and structure–performance relationships for sulfur species. His experience broadened to other polar species in fuels, as well as biological sources for alternative fuels, primarily microalgae. Dr Link has developed protocols to accurately assess the lipid content of fuels using a combination of extraction and conversion, and has shown that gravimetric lipid determination can dramatically overstate the lipid content of algae. This, in turn, affects the projections of potential yield of lipid-based fuels. |
 Bryan Morreale | Dr Bryan Morreale is the acting Materials Science and Engineering focus area lead within the Office of Research and Development at the DOE-NETL. Dr Morreale leads activities across a diverse research portfolio related to both structural and functional materials for advanced energy conversion applications, specifically focused on advanced gasification, advanced combustion and minimizing environmental impacts. Dr Morreale's tenure at NETL began in 1999, where he supports the NETL Office of Research and Development as well as the NETL Strategic Center for Coal. |
1. Introduction
With the rapid development of modern civilization, national energy security, availability and affordability, as well as environmental impact are major drivers for developing renewable energy sources, such as biofuels, bioproducts and biopower.1 During recent decades, sustainable energy sources have been widely researched to meet such challenges. Among these efforts, liquid biofuels from renewable bio-resources are one of the most attractive alternatives, with the features of sustainability and the ability to utilize current infrastructure (i.e. pipelines, vehicle engines, etc.). 1–6In the United States, petroleum accounts for approximately 90% of the energy consumed within the transportation sector, and this usage reflects nearly 70% of the oil processed nationally.1 In addition, the large consumption of petroleum-based transportation fuels accounts for roughly one-third of CO2 emissions, which is thought to be a major contributor to global climate change.1 With increasing demand but limited fossil fuel resources, as well as growing concerns about national energy security and climate change, the production of transportation fuels via alternative renewable routes, especially from relatively inexpensive and plentiful photosynthetic organisms as feedstocks, is becoming more important for addressing future energy needs. Biomass, especially algae based biomass, has been considered as a potential alternative feedstock for the production of traditional petroleum-based commodities, including transportation fuels.7–11 In addition, diversifying the energy portfolio to include more renewable energy resources can assist in overcoming impacts associated with increased greenhouse gas concentrations in the atmosphere.1,12,13
Biomass, including agricultural residues, forest resources, energy crops, wastes and algae, is an opportunistic renewable resource that can be converted to liquid transportation fuels in the near-term.1 Current trends show that the United States biofuel production for transportation will continually increase over the next few decades, as shown in Fig. 1.1 In the United States, the Energy Independence and Security Act (EISA) of 2007 requires production of 36 billion gallons per year (bgy) of renewable transportation fuels by 2022.1 The United States Environmental Protection Agency's (EPA) final rule projects that these renewable fuels could come from starch-based ethanol and advanced biofuels including cellulosic ethanol, biobutanol, hydrocarbons from algae and biomass-based hydrocarbon fuels (renewable gasoline, diesel, jet fuel).1,7,11
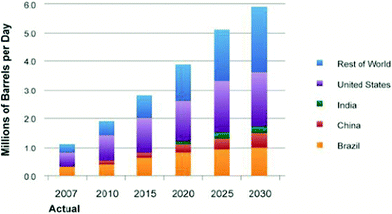 | ||
| Fig. 1 Biofuels production worldwide.1 | ||
Algae, including microalgae and macroalgae (such as seaweed), is currently commercially used in food and other products and has potential as a carbon-neutral feedstock for biofuel production.11,14–21 Among renewable biomass resources for advanced biofuels, microalgae continues to attract attention because of its potential for fast growth rates, high oil yield (1000–6500 gal acre−1 yr−1vs. soybean 48 gal acre−1 yr−1), the use of non-arable land for algae cultivation, growth in a variety of water sources and the benefits associated with large-scale CO2 mitigation.3,6,11,14–19 Furthermore, microalgae-based biofuels do not compete with food crop production, unlike conventional biofuels, which typically use fertile land and edible oils in their production cycle. Research supported by the U.S. Department of Energy is studying the potential use of microalgae to produce biodiesel with CO2 captured from point sources, including coal fired power plants.22–27 Microalgae are able to produce more than 50% dry weight of bio-crude oil with the potential to yield 100-times higher oil production than conventional crops.28 The compositions of typical microalgal oils include higher molecular weight species ranging from C14 to C26, and often contain carboxylic acids. Overall, the bio-oil has a chemical nature and energy density comparable to that of petroleum-based diesel, making algae oil-based biofuels a target for diesel replacement.29 Microalgae-based oil also typically contains from 20% to 50% free fatty acids.30,31
The three major components of microalgae are carbohydrates, lipids and proteins.15 The lipid content of dry biomass of various microalgae species varies from 4% (such as in spirulina) to as high as 80% (such as in botyococcus braunii).18 Depending on the species of microalgae, three major pathways can be used to convert it to transportation fuel, including whole algae conversions (thermochemical conversion), lipid (algal oil) extraction followed by catalytic reactions (thermochemical conversion), and hydrolysis of carbohydrates (biochemical conversion), as shown in Fig. 2.1,14,32 Because the aim of this work is to focus on the thermocatalytic conversion of microalgae, the hydrolysis of carbohydrates and fermentation of sugar to ethanol will not be discussed further.
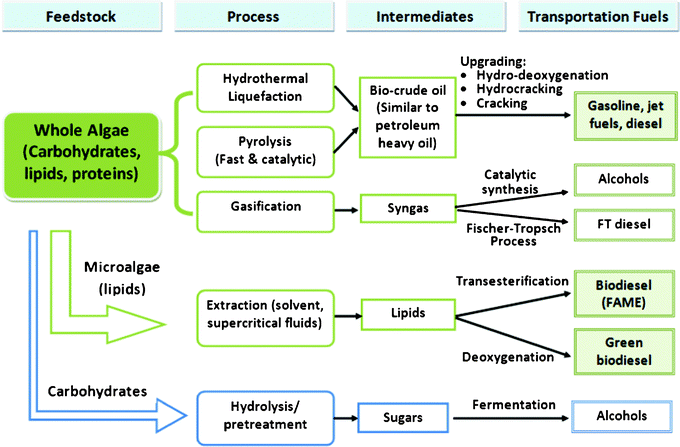 | ||
| Fig. 2 Pathways for converting microalgae to biofuels.1,14 | ||
Whole microalgae can be processed into fuels directly via gasification, pyrolysis, and hydrothermal liquefaction (HTL).14,32–35 The gasification or partial oxidation of microalgae in the presence of steam produces an intermediate synthesis gas (H2 and CO) that can be further converted via thermocatalytic pathways to liquid fuels and chemicals.1,14,34,36,37 Microalgae can also be converted thermochemcially in the absence of oxygen, where the pyrolysis (fast pyrolysis, 450–550 °C and 103–104 °C s−1) generates bio-oil with less oxygen content and higher heating values than lignocellulosic based bio-oil,38,39 which is upgraded to transportation fuels by thermocatalytic processes.14,34,37,40–42 High water content (as high as 92%)43 in harvested microalgae makes HTL processes more economically attractive than pyrolysis, gasification or extraction methods, all of which require an energy-intensive pre-drying procedure.34,44,45 The resulting bio-crude oil is subsequently fed into the refinery stream and upgraded via conventional petroleum catalytic processes to transportation fuel through cracking, hydrocracking and hydro-deoxygenation using commercial catalysts.
For the microalgae extraction pathway, 45% lipid content is estimated as the “optimistic case”, based on energy needed from microalgae production to biodiesel.33 The residues remaining after lipid extraction could potentially be converted into biofuel by carbohydrate hydrolysis and fermentation to obtain an optimized yield, productivity and process efficiency.1,14 Selective conversion of triacylglycerides and free fatty acids (FFAs) to transportation fuels from lipids produced by microalgae has received extensive attention due to environmental concerns and high demand for transportation fuels. Traditional biodiesel fuels, i.e., fatty acid methyl esters (FAME), from renewable resources including microalgae, are generally produced via transesterification reactions in solvents using homogeneous catalysts.36,37,40–42,46 However, due to undesirable oxygen groups present in biofuels compared to petroleum-derived diesel fuels, these conventional biodiesels suffer from the propensity to have lower energy densities and poor low-temperature properties, and they absorb water, and are also susceptible to microbial fouling.31,47,48 In addition, a large quantity of the by-product from the transesterification reactions, specifically the glycerol, is rapidly being considered as a waste material. However, a new generation of green diesels produced from microalgae49–51 are alkane hydrocarbons similar to those found in conventional fossil fuel-derived fuels, thus making them fully compatible with existing engines and infrastructure. This green diesel from microalgae also produces fewer by-products, such as the waste glycerol produced from transesterification. While many examples of the growth and extraction of the resultant microalgae are available in the literature,4,21,31,52–56 instances discussing the upgrade or catalytic conversion of microalgal lipids to alkane-based transportation fuels are relatively sparse and not discussed extensively.
Among the major stages for the production of algal biofuel, as shown in Fig. 2, are several factors that need to be considered and optimized, including energy and material inputs. Computational modelling can be applied to each of these stages to identify the optimizing conditions for algal biofuel technology development. However, of the relatively few simulations found in the literature, many focused only on the microalgae growth, specifications, and harvesting process, 3,4,6,12,15,21,31,52–61 and very few discussed the catalytic conversion of algal oils to fuel, especially using the computational modelling approach. Most simulation studies were done for model compounds, which are only indirectly related to the catalytic conversion of a complex biological mixture like algal oils.
In this review, we summarize both the experimental studies of the catalytic conversion of microalgae to transportation fuels and the studies that provide a fundamental understanding of the processes using computational simulation. To address the importance of computational modelling in the field of biofuel production, we also summarize the modelling progress in hydrocracking, decarboxylation, and hydrodeoxygenation of model compounds. This review is organized as follows: section two summarizes the progress in HTL of microalgae to bio crude oil; section three describes progress on transesterification of algae to FAME; section four summarizes the current status of the catalytic upgrading of algae feedstock to liquid fuels and the model compounds typically used as surrogates for lipid-rich microalgae; and finally, some perspectives and concluding remarks are given in section five.
2. HTL of microalgae to bio-crude oil
HTL generally converts biomass to bio-crude oil in subcritical water (below the water critical point of 374 °C and 22 MPa).44,62 Bio-crude oil is much like a petroleum-derived product but with a higher viscosity and oxygen content, typically 10–20 wt% compared to <1% in conventional petroleum.44 Therefore, bio-crude-oil requires upgrading by removing oxygenates and reducing molecular weight to produce transportation fuel.63 Many studies37,44,64–73 report use of the HTL process for conversion of biomass to fuels. In the 1970s, pioneering work on the HTL of cellulosic biomass using a sodium carbonate catalyst to produce a heavy oil was conducted by researchers at the Pittsburgh Energy Research Center.64 A liquefaction pilot plant capable of treating 3 tons per day of biomass was demonstrated in Albany, Oregon.66 One of the pioneering works on HTL for microalgae (Botryococcus braunii) with high lipid content was done by Dote et al. in 1994.74 Since then, HTL has been applied to a wide range of lipid contents of microalgae (<1 to 50% of total microalgae),74–81 microalgae residues (e.g. from post-lipid extraction)82–83 and macroalgae.73,84–85 Sawayama et al.86 studied energy consumption for the HTL of various microalgae and presented it as an energy consumption ratio (ECR) that represents energy for liquefaction and the lowest heating value of the oil production. They found that ECRs were low, less than 1.0, which means that HTL of microalgae could produce net energy.86 Therefore, HTL has been considered as a promising technology to generate liquid fuel from wet biomass. In this section, we summarize the current progress on HTL for converting whole algae to bio-crude-oil.2.1 Microalgae composition and conversions in HTL conditions
Microalgae are single-celled organisms that include both eukaryotic cells and prokaryotic cells (cyanobacteria, also called blue-green algae).15,56 The enormous number of microalgae species is divided into groups, with the most important including green algae (Chlophyceae), red algae (Rhodophyceae), and diatoms (Bacillariophyta).15,18 Microalgae have three major components: carbohydrates, lipids and proteins.15Table 1 lists the biochemical and chemical compostions of selected species of microalgae reported in the literature. Microalgae can be classified as having low (15% of total mass), medium (25%) and high (50%) lipid contents.15| Microalgae | Biochemical composition (daf %) | Proximate | Ultimate (daf %) | High heating value (MJ kg−1) | Refs. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipid/Crude fat | Crude protein | Carbohydrate | Ash (db%)a | C | H | N | Ob | S | |||
| a Organic content = 100 – ash content b By difference c On dry basis for biochemical composition and ultimate analysis d Residues after recovering β-carotene as a bioactive compound | |||||||||||
| Botryococcus braunii | 50 | — | — | 2 | 63.1 | 11.7 | 2.8 | 22.4 | — | — | 74 |
| Nannochloropsis oculata | 32 | 57 | 8 | 26.4 | 57.8 | 8 | 8.6 | 25.7 | — | 17.9 | 76 |
| Nannochloropsis sp. c | 28 | 52 | 12 | — | 43.3 | 6 | 25.1 | 6.4 | 0.5 | — | 77 |
| Chlorella vulgaris | 25 | 55 | 9 | 7 | 52.6 | 7.1 | 8.2 | 32.2 | 0.5 | 23.2 | 76 |
| Dunaliella tertiolecta | 20.5 | 63.6 | 14.7 | 23.6 | 53.3 | 5.2 | 9.8 | 31.7 | — | — | 78 |
| Dunaliella tertiolecta | 22.17 | 32.13 | 20.16 | 12.12 | 40.28 | 5.41 | 9.22 | 47.36 | — | 19.68 | 62 |
| Microcystis viridis | — | — | — | — | 46 | 7.3 | 9.5 | — | — | — | 80 |
| Desmodesmus sp. | 10–14 | 38–44 | 13–20 | — | 51.96 | 7.31 | 6.86 | 33.87 | — | 23.44 | 88 |
| Spirulina platensis | 13.1 | 65.38 | — | 6.6 | 46.87 | 6.98 | 10.75 | 34.86 | 0.54 | 20.52 | 89 |
| Scenedesmus c | 13 | 56 | 25 | 6 | 52.1 | 7.4 | 8.8 | 31.1 | 0.48 | 22.6 | 82 |
| Porphyridium cruentum | 8 | 43 | 40 | 24.4 | 51.3 | 7.6 | 8 | 33.1 | — | 14.7 | 76 |
| Cyanobacteria (Spirulina) | 5 | 65 | 20 | 7.6 | 55.7 | 6.8 | 11.2 | 26.4 | 0.8 | 21.2 | 76 |
| Cyanobacteria (Spirulina)c | 5 | 64 | 20 | 11 | 45.2 | 6.4 | 9.8 | 37.8 | 0.8 | 17.7 | 82 |
| Cyanobacteria (Spirulina)c | 12 | 57.5 | <0.5 | — | 46.1 | 7.4 | 4.8 | 41.3 | 0.4 | — | 90 |
| Defatted scenedesmus | <1 | 72 | 21 | 7 | 49.9 | 7.1 | 9.9 | 32.1 | 0.96 | 21.3 | 82 |
| Dunaliella tertiolecta cake d | 2.87 | 61.32 | 21.69 | 86.46 | 39 | 5.37 | 1.99 | 53.02 | — | 20.08 | 83 |
Carbohydrates are monosaccharide polymers with an elemental composition C1H1.67O0.83, 17.3 MJ kg−1 of calculated calorific value and 10–50% of typical cell content.15 Sugars (monosaccharide and disaccharide) and starches (polysaccharides in the form of amylose and amylopectin) are all carbohydrates. Different polysaccharides belong to different groups of microalgae, e.g. starch consisting of amylose and amlopectin is in green algae as an energy store and floridean starch consisting mostly of amylose is in red algae. In the HTL conditions (subcritical water system), starch is rapidly hydrolyzed to monosaccharides with glucose as one of the main products. As shown in Fig. 3, the glucose is readily converted to fructose, an isomer of glucose.87 This fructose subsequently undergoes degradation with fragmenation procucts e.g. glycolaldehyde and glyceraldehyde. Further fragmentaion and dehydrations produce complex products e.g. acetic acid, formic acid and aromatic compounds.44,87
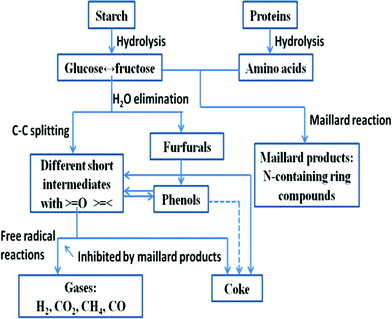 | ||
| Fig. 3 Simplified reaction pathways for hydrothermal carbohydrate and proteins, adapted from reference 87. | ||
Most proteins consist of linear polymers of amino acids with an elemental composition of C1H1.56O0.3N0.26S0.006, 23.9 MJ kg−1 of calculated calorific value, and 20–60% of typical cell content.15 They have both structural and metabolic functions. The peptide C–N bond links the amino acids together between the carboxyl and amine groups. The bond will be hydrolyzed under hydrolysis conditions and results in production of amino acids with optimal yields at ∼250 °C.44 The peptide bonds of proteins are more stable than the glycosidic bonds in starch, and peptide bond hydrolysis occurs slowly below 230 °C.87 The amino acids rapidly undergo decarboxylation and deamination and produce mainly hydrocarbons, amines, aldehydes and acids.44,87 Some of these products are the same as those from hydrolysis of carbohydrate. Thus, similar condensation may take place in protein degradation alone or together with carbohydrate degradation, in the case of HTL of algae as shown in Fig. 3.87 In addition, a considerable fraction of the nitrogen in protein will be incorporated in the bio-oil during the HTL process. The nitrogen in oil causes NOx emission when it is combusted.
Algal lipids mainly consist of simple fatty acid triglycerides (TAG), glycolipids and a phospholipid with elemental compositions of C1H1.83O0.17N0.0031P0.006S0.0014, 36.3 MJ kg−1 of calculated calorific value and 15–60% of typical cell content.15 The lipids can function as the structural membranes of the cells. The prokaryotic algae (cyanobacteria) has less lipid content compared to eukaryotic algae, possibly due to the lack of internal membranes in prokaryotes with fast growth.15 High contents of unsaturated fatty acids, 50% of those with a carbon number less than C18, are typical of algal lipids and may need to be hydrogenated to improve their potential fuel properties.15 In the HTL conditions, TAGs are hydrolyzed to fatty acids and glycerol, which are not converted to an oil phase but rather to water-soluble soluble compounds in HTL. The free fatty acids are relatively stable, but partially degrade to produce long-chain hydrocarbons for transportation.44,87 Adding NaOH or KOH can increase the decomposition of fatty acids.44 In addition, hydrocarbons and lipids may be formed from small organic materials by Fisher–Tropsch reactions with water acting as the hydrogen source in the HTL conditions.44
2.2 Effect of catalysts and HTL conditions on bio-crude oil properties and yields
In general, HTL of microalgae is operated in a temperature range between 200 and 370 °C, at pressures from 12 to 20 MPa, for 5 to 60 min in subcritical water with or without catalysts (alkaline or metal catalysts) under CO and H2 or inert gases, such as N2 and He. Selected HTL processes of microalgae with low to high lipid content,74,76–82 microalgae residues after extracting lipid or β-carotene,82,83 and macroalgae73,84,85 are summarized in Table 2. Table 2 lists the key parameters of temperature, time and catalyst used in HTL processes, the optimum conditions where maximum bio-oil yield was obtained, maximum bio-oil yields, and heating values. The table shows that feedstocks strongly affect bio-oil yield and properties, as optimum conditions are different for different feedstocks. Microalgae with high lipid content tend to generate a high oil yield. HTL also can convert low-lipid microalgae and its residues to bio-oil. Utilization of the low lipid algae may have potential for large-scale production, as well as potential environmental advantages because high-lipid algae usually have lower growth productivity compared to low-lipid algae. Algae that are typically used for wastewater treatment, and also these species that form algal blooms in nature usually have low lipid contents.75,81,82,89| Feedstocks | Lipid (wt%) | Catalyst/ Organic acid | T (°C) | Time (min) | Optimal condition | Max. oil-yield (wt %)b | Oil heating value (MJ Kg−1) | Refs. |
|---|---|---|---|---|---|---|---|---|
| a Weight percentage of catalyst to dry solid in algal cells b Maximum oil-yield on a dry organic in algal cells (dry ash free basic, daf) = (weight of oil/dry weight of organic in algal cells)*100 c Maximum oil-yield on a dry matter in algal cells (dry basis, db) = (weight of oil/dry weight in algal cells)*100 d Grown in open pond with waste water e Residues after recovering β-carotene as a bioactive compound | ||||||||
| Microalgae | ||||||||
| Botryococcus braunii | 50 | 0, 5 wt% Na2CO3a | 200, 300, 340 | 60 | 300 °C 60 min 5% Na2CO3 | 64 | 49 | 43,74 |
| Dunaliella tertiolecta | 20.5 | 0, 5 wt% Na2CO3 | 250,300, 340 | 5, 60 | 300 °C 5 min | 43.8 | 34 | 78 |
| Spirulina | 12 | 0, Fe(CO)5–S | 350 | 60 | 350 °C 60 min | 61 | 26 | 90 |
| Microcystis viridis | 0, 5 wt% Na2CO3 | 300, 340 | 30, 60 | 340 °C 30 min 5% Na2CO3 | 33 | 31 | 80 | |
| Spirulina, | 13.1 | 0, 5 wt% Na2CO3 NiO | 350 | 30–60 | 350 °C 60 min 5% Na2CO3 | 45.06 | 29.32 | 89 |
| platensis mixed algae d | 6.7 | 17.46 | 35.18 | |||||
| Dunaliella tertiolecta | 22.17 | NA | 300–380 | 10–90 | 360 °C 30 min | 36.9c | 26.62 | 62 |
| Nannochloropsis sp. | 28 | NA | 200–550 | 60 | 350 °C 60 min | 43c | 39 | 77 |
| Chlorella vulgaris, | 25 | 1M Na2CO3, 1M KOH, 1M CH3COOH, 1M HCOOH | 300, 350 | 60 | 350 °C 60 min 1M Na2CO3 | 27.3 | 37.1 | 79 |
| Spirulina | 5 | 20 | 34.8 | |||||
| Dunaliella tertiolecta cake e | 2.87 | 5 wt% Na2CO3 | 280–380 | 10–90 | 360 °C 50 min 5% Na2CO3 | 25.8 | 30.74 | 83 |
| Chlorella vulgaris, | 25 | 0, 1M Na2CO3, 1M HCOOH | 350 | 60 | 350 °C 60 min | 35.8 | 35.1 | 76 |
| Nannochloropsis oculata, | 32 | 34.3 | 34.5 | |||||
| Porphyridium cruentum, | 8 | 20 | 35.7 | |||||
| Spirulina | 5 | 29 | 36.8 | |||||
| Chlorella pyrenoidosa | <1 | NA | 100–300 | 10–120 | 280 °C 120 min | 39.4c | 35.42 | 81,91 |
| Desmodesmus sp. | 10–14 | NA | 175–450 | 5–60 | 375 °C 5 min | 49c | 35.4 | 75,88 |
| Scenedesmus, | 13 | NA | 300 | 30 | — | 45 | 35.5 | 82,92 |
| Spirulina, | 5 | 36 | 35.8 | |||||
| Defatted scenedesmus | <1 | 31 | 35.3 | |||||
| Nannochloropsis sp. | 28 | 0, Pd(Pt, Ru)/C Ni/SiO2-Al2O3, CoMo/γ-Al2O3, zeolite | 350 | 60 | 350 °C 60 min Pd/C | 57 | 38 | 93 |
| Macroalgae | ||||||||
| Laminaria saccharina | — | 0–100% KOH/algae | 250–370 | 15–120 | 350 °C 15 min | 19.3 | 36.5 | 84 |
| S.patens C. Agardh | — | NA | 320–380 | 15–90 | 340 °C 15 min | 32.1 | 27.1 | 85 |
| Enteromorpha prolifera | — | 0, 5 wt% Na2CO3 | 220–320 | 5–60 | 300 °C 30 min 5% Na2CO3 | 23d | 28.74 | 73 |
The effect of catalyst loading along with temperature and reaction time on yield and properties of produced oil including viscosity and ultimate analysis were studied by Minowa et al.78 In their study, Dunaliella tertiolecta (microalgae) with 78.4% moisture content in common algae loading of about 10–20% 75,89 was converted in a 100 ml autoclave. The process conditions were temperatures of 250, 300 and 340 °C, residence times of 5 and 60 min with high pressure (about 10 MPa), and Na2CO3 catalyst loadings of 0 and 5 wt % of the dry algae. The products of HTL included a bio-oil fraction (extracted by dichloromethane), a water soluble fraction, gases and solid residues. The oil yields on an organic basis were 31–44 wt% (average 37 wt%), while the microalgae had 76.4 wt% dry basis non-ash organic content (daf). The oil obtained at 340 °C for 60 min had heating values of 36 MJ kg−1 and a viscosity of 150–330 mPa s, which is comparable to fuel oil (about 40 MJ kg−1, 50–1000 mPa s). The gas yield was about 10% on an organic basis and the gas contained mainly CO2. The solid residue was negligible and the inorganic matter in the microalgae was mostly soluble in the aqueous phase. The catalyst, temperature, and reaction time affected the oil properties, but they did not significantly affect the oil yield. At the maximum oil yield obtained at 300 °C for 5 min, the viscosity of the oil with the catalyst was 500 mPa s which is much lower than 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mPa s without the catalyst. As the temperature increased, the viscosity of the oil decreased, its heating value increased slightly, the carbon and hydrogen contents increased and the oxygen content decreased.78 HTL of the Microcystis viridis strain was investigated using a similar method.80 The maximum oil yield was obtained at 340 °C for 30 min with 5 wt% Na2CO3 and the oil had the maximum energy yield (the ratio of the weight of C and H in oil products after liquefaction to the weight of C and H in feedstock).80 In addition, the higher temperature increased both the oil yield and nitrogen content in the oil of Desmodesmus sp.75 and Chlorella pyrenoidosa.81
000 mPa s without the catalyst. As the temperature increased, the viscosity of the oil decreased, its heating value increased slightly, the carbon and hydrogen contents increased and the oxygen content decreased.78 HTL of the Microcystis viridis strain was investigated using a similar method.80 The maximum oil yield was obtained at 340 °C for 30 min with 5 wt% Na2CO3 and the oil had the maximum energy yield (the ratio of the weight of C and H in oil products after liquefaction to the weight of C and H in feedstock).80 In addition, the higher temperature increased both the oil yield and nitrogen content in the oil of Desmodesmus sp.75 and Chlorella pyrenoidosa.81
Researchers also expanded the temperatures to 550 °C (above the water critical temperature of 374 °C) to determine the effect on maximum oil yields. It was found that the maximum oil yields were obtained in the subcritical condition (Table 2).75,77,88 Brown et al. conducted tests using the microalgae Nannochloropsis at a processing temperature range of 200 to 500 °C and a reaction time of 60 min.77 The highest bio-oil yield (dry basis) was 43 wt% at 350 °C in subcritical water liquefaction. The oil yields decreased modestly from 400 °C to 450 °C, and at 500 °C the yield was nearly half that achieved in supercritical water. In addition, the chemical composition of the oil and gas fractions changed with varying temperatures. The oils generated at lower temperatures consisted of fatty acids, alkenes, sterol-related compounds and heterocyclic N-containing compounds, with CO2 being the main compound in the gas phase. As the temperature increased, the oils showed more polycyclic aromatic hydrocarbons (PAHs), and the gas phase contained some lighter hydrocarbons (such as CH4, C2H4 and C2H6) in addition to CO2.
Recently, HTL research has moved toward providing a more detailed characterization of products, and determining the relationship between composition and HTL process conditions.75,80,88,92,94 The understanding of bio-oil chemical composition is required for improving the upgrading pathways for bio-oil to achieve higher quality products. Yang et al.80 studied HTL conditions for the Microcystis viridis strain using a method similar to that used by Monowa (Table 2).78 The liquefied oil was separated into four fractions using a thin layer chromatography and flame ionization detector (TLC-FID).80 The fractions included saturated compounds, aromatic compounds, resin and asphalt. Furthermore, a gas chromatography with mass spectrometer (GC-MS) was used to analyze low molecular weight saturated and aromatic hydrocarbon compounds. The saturated compounds in the oil were mainly n-alkanes of the C17–C18 hydrocarbon range, and the aromatic compounds contained n-naphthalene and n-dibenzothiophene, which are typical components in petroleum-based heavy oil. Therefore, the liquefied oil is similar to heavy oil. The gas consisted primarily of CO2 and methane. The solid residues decreased with increasing temperature and sodium carbonate addition. The aqueous phase had a total nitrogen concentration ranging from 998 to 1157 mg l−1 and a total phosphate concentration from 2.47 to 5.38 mg l−1. Half of the total nitrogen was detected as ammonia (NH3–N). Therefore, authors suggested that the final process water will require further treatment to remove nitrogen and phosphate prior to disposal. On the other hand, microalgae growth requires nitrogen and phosphate. So, the nitrogen and phosphate in the water could potentially be recycled as nutrients for microalgae growth.75,81,95 At the same time, this approach would have an environment benefit. Microalgae grown in wastewater from secondary treated sewage or from the carpet industry was converted to bio-crude oil using HTL.43,89 Nitrogen distributions in the products of HTL of microalgae and macroalgae were evaluated for future nitrogen control and recycling studies.75,84
Ross et al.79 investigated the effect of alkali catalysts and organic acids along with temperature on yield and properties of produced oil. The alkali included potassium hydroxide and sodium carbonate. The organic acids as hydrogen donors were acetic acid and formic acid. The microalgae Chlorella vulgaris, which was found to have a low lipid content and high protein content, was selected as the feedstock. The HTL was performed in a batch reactor at 300 and 350 °C for 1 h. The oil yield based on organic content followed the trend Na2CO3 > CH3COOH > KOH > HCOOH. The oil yield on a dry basis followed the trend CH3COOH > HCOOH > KOH > Na2CO3. The bio-crude oils using the alkali catalysts had higher heating values, but the oils generated from HTL using organic acids had lower boiling points, as measured using thermal gravimetric analysis (TGA), and had better flow properties. The oils, analyzed using GC-MS, contained aromatic hydrocarbons, substituted phenols, nitrogen heterocycles, fatty acids and fatty acid amides. Elemental analysis of the algae oil showed that it had significantly lower oxygen content compared with the biomass pyrolysis oil. The overall elemental balance of nitrogen indicated that a large portion of the algae nitrogen (up to 50%) is transferred to the aqueous phase in the form of ammonium (NH4−). The nitrogen content of the oils typically was 4 to 6%. Such high nitrogen content in the oil is undesirable because it increases NOx emission as the fuel is combusted, and because of the technical challenges for upgrading bio-oil to remove nitrogen.75,82,84
Alkalis such as sodium carbonate have been widely used in the HTL of algae. Formates also have been found to have some amount of catalytic activity.64 The weak organic acids as hydrogen donors positively affect bio-crude oil yield and boiling point distribution of the oils, but may either decompose to gaseous products (such as CO and H2) or react with the microalgae degradation compounds in the HTL process.79 For considering homogeneous catalyst recovery and reuse issues following the HTL process, researchers applied metal catalysts to the HTL process (Table 2) as discussed in section 4.1.89,90,93 The catalysts affected the oil yield and oil properties. The HTL of Nannochloropsis sp. with Pd/C had higher bio-crude oil yield and lower O/C ratio in the oil than the oil from the uncatalyzed process.93 The metal catalyst function and catalytic mechanism are not clearly understood and more research is needed.
2.3 Pathway of the hydrothermal liquefaction process
The mechanisms for hydrothermal liquefaction reactions of microalgae to crude oil are not well understood. The proposed pathway is for the macromolecules of microalgae (lipids, proteins and carbohydrates) to first be hydrolyzed into small fragments (fatty acids, amino acids and glucose). Subsequently, the small fragments can be converted to smaller compounds, e.g. the amino acids undergo decarboxylation and deamination and produce mainly hydrocarbons, amines, aldehydes and acids. The generated compounds (intermediates) are unstable, and rearrange via condensation, cyclization and polymerization reactions to larger compounds.37,44,75,81,87 The complex interactions are not clearly understood and further study is needed. The properties of the product obtained depend on the components of the microalgae and the reaction conditions. An experimental study of the HTL of microalgae Desmodesus sp. at temperatures between 175 and 450 °C showed that hydrolysis dominates from 175 °C to 225–250 °C, as shown by an increase in water-soluble organics, and low yields of oil were obtained. In the temperature range 250–375 °C, re-polymerization becomes predominant, with increasing oil yields and decreasing organic content in water.75In the HTL of raw microalgae having an oxygen content of 31.7 wt% on a dry organic basis,78 the oil derived from the microalgae had an oxygen content of 8.4 to 13.7 wt%. Oxygen was removed during the HTL process in the form of water by dehydration or carbon dioxide by decarboxylation.44 The raw microalgae had an initial crude lipid content of 20.5 wt% on an organic basis, and produced 37 wt% of oil yield on an organic basis, which was higher than the microalgae crude lipid content. This means that not only lipids but also proteins and carbohydrates were converted to oil in the process. In addition, the maximum oil yield was 64% from HTL of the microalgae Botryococcus braunii while the microalgae starting material had crude lipid content of 50%.74 So, the higher the lipid content, the higher the bio-oil yield. Biller and Ross 76 further confirmed this using model compounds of proteins (albumin, soya protein, and amino acids of asparagines and glutamine), carbohydrates (starch and glucose) and lipids (triglyceride from sunflower oil) and microalgae strains with high (Chlorella vulgaris, Nannochloropsis occulata) and low (Porphyridium cruentum and the cyanobacteria Spirulina) lipid contents. Fig. 4 shows the yields of products of the model compounds and microalgae based on a dry, ash-free basis in sodium carbonate at 350 °C for 60 min. Bio-crude oil was generated from each biochemical component. This is a unique advantage of HTL compared with conventional lipid extraction and conversion methods, and speaks to the conversion efficiencies and yields that are possible with the HTL process. Furthermore, the results of studies using model compounds and microalgae oil also showed that the relative oil contributions were lipids > proteins > carbohydrates.
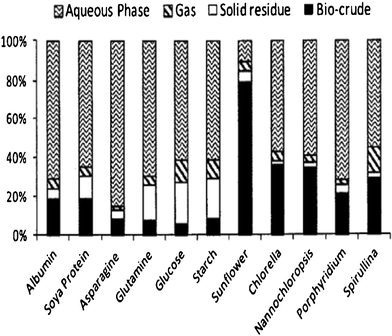 | ||
| Fig. 4 Yields of products from hydrothermal processing of microalgae and model compounds at 350 °C for 60 min.76 | ||
Water plays an important role in the HTL processes. It serves as a solvent (vehicle) for the reactions, supplies hydrogen for addition to the substrate, and hydrolyzes high molecular weight reactants such as carbohydrates and proteins.44,64 As the water temperature increases, for example from 25 °C to 300 °C, the relative permittivity decreases from 78.85 to 19.66, indicating that ordinarily very polar water molecules become relatively non-polar, which allows the water to show greater affinity for the non-polar organic hydrocarbons.68 In addition, the dissociation of water dramatically increases with the increase of temperature. For example, the water dissociation constant at 300 °C is about 500 times higher than that at 25 °C, under atmospheric pressure.68 Water is split into H+ and OH− ions via hydrolysis or dissociation, which is reversible and rapid. In that way, the rate of both acid- and base-catalyzed reactions is increased.
3. Transesterification of algae feedstocks
Traditional biodiesel fuels, i.e., FAMEs, produced from various biomass feedstocks via transesterification reactions are commercially avaliable.36,37,40–42 Although the technique of transesterification has been around for over a century, it has only recently been used for obtaining fuel from algae oil. Transesterification of extracted algal lipids is another fuel production pathway, in addition to thermochemical conversion of the entire algal mass, such as via HTL as described previously.3.1 Transesterification
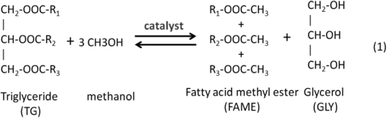
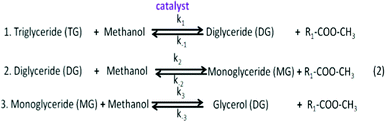
Many microalgae species can be induced to produce and accumulate substantial amounts of lipids (such as fatty acids and glycerides). Under certain conditions, lipid content in some species can reach up to 90% of dry weight.21,42 Beal et al.96 laid out a framework to report the production of renewable diesel from algae and analyzed several production pathways. Usually, triglycerides (TG) in algal lipids can be converted to traditional biodiesel (mixture of FAMEs) through the transesterification process. A general overview of the transesterification reaction of TG with methanol isin which one triglyceride and three alcohols (methanol) form three FAMEs and one by-product glycerol (GLY), in the presence of a catalyst.42,97–101 In the literature, computational modelling of transesterification focuses on the kinetics and the mechanism of the reactions and product design.102 Freedman et al.98 proposed a three-step reaction scheme for the overall transesterification reaction (1) that includes the reaction intermediates, monoglyceride (MG) and diglyceride (DG), as shown in the following:
First, the carbonyl carbon of the triglyceride molecule reacts with the alkoxide ion to form a tetrahedral intermediate. Then, this intermediate reacts with an alcohol to produce an alkoxide ion. Finally, the tetrahedral intermediate rearranges into an ester and a diglyceride. Similarly, this transesterification mechanism can be extended to di- and mono-glyceride intermediates. By examining the effects of the molar ratio of methanol to soybean oil, the type and amount of catalyst and the reaction temperature on rate constants and kinetic order, they found that the forward reactions appear to be pseudo-first order or second order depending upon conditions used, while the reverse reactions appear to be second order.103 Based on batch and loop reactor modelling, Tesser et al.101 simulated the FFAs esterification and found that with a packed bed loop reactor configuration with multi-steps operation, high FFA conversion can be obtained, and the methanol–water separation costs could be reduced by lowering the methanol/acid ratio.
Such transesterification reactions can be base-catalyzed, acid-catalyzed and enzyme-catalyzed. Traditional homogeneous catalysts, such as KOH, NaOH and H2SO4, have been reported for direct transesterification of algae oil to biodiesel.25,104,105 Usually, base-catalyzed transesterification requires shorter reaction times and lower temperatures, while acid-catalyzed transesterification requires more time as well as elevated temperatures to complete.106 For example, Freedman et al.106 reported that, when using the base catalyst NaOH, the transesterification of vegetable oil was 99% complete at 32 °C in 4 h, whereas at 60 °C the reaction was complete in one hour. Compared with the alkali catalyst, they also observed that the transesterification by acid catalyst was much slower. However, solid acid catalysts can carry out transesterification and esterification simultaneously using low-cost feedstocks, without multiple reaction and post-treatment steps, greatly improving the economics of biodiesel production.107 Because algal biomass contains lipids and other chemicals, a pre-treatment process, i.e., extraction, is typically involved before the transesterification reactions. D'Oca et al.108,109 investigated the extraction of lipids from the dry biomass of Chlorella pyrenoidosa and compared the transesterification of lipidic extracts with direct transesterification of dry biomass. They found that extraction with methanol followed by the transesterification process resulted in a higher FAMEs yield from biomass than the direct transesterification process using methanol. Miao and Wu110 reported the acidic transesterification of the extracted algae oil from Chlorella protothecoides using n-hexane as extraction solvent. The optimized operating conditions were reported as 56:1 molar ratio of methanol to oil at a temperature of 30 °C, with a reaction time of 4 h.
It is difficult to extract algae oil from their tough, rigid cell wall structures using conventional mechanical pressing and subsequent solvent extraction methods. Microwaves can directly penetrate through these cell walls to assist the oil extraction. Recently, a new microwave assisted process111 has been reported to simultaneously extract algae oil and transesterify it to FAME. A schematic of the microwave-assisted extractive-transesterification111 process to transform algal biomass (Inoculum Nanno-chloropsis sp.) to biodiesel is shown in Fig. 5. Wet algal biomass was first pre-dried in a vacuum oven at 50–60 °C overnight and was ground under liquid N2 conditions. The dry algal powder thus obtained was added to the premixed homogeneous solution of methanol and KOH catalyst to produce biodiesel. The FAME yield for this microwave-assisted simultaneous extraction and transesterification reaction process was greater than 70% at optimal conditions, having a dry algae to methanol (wt/vol) ratio of around 1:12, a KOH concentration of 2 wt.% and a reaction time of 4 min, at a reaction temperature of around 60–64 °C. The mechanism of base-catalyzed microwave transesterification is similar to traditional transesterification of vegetable oil98. It was also reported112 that the microwave assisted process could shorten FAME recovery time from reaction mixtures.
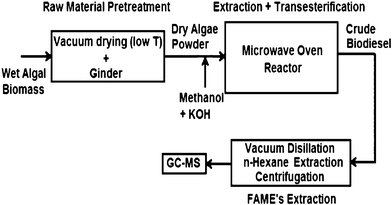 | ||
| Fig. 5 Single-step microwave transesterification process for dry algal biomass. Modified from Ref. 111 | ||
Direct conversion111,113–116 of algal biomass to bio-diesel is of high interest to researchers because of the economic and energy advantages of by-passing the time-intensive extraction and clean up steps. Direct transesterification enhanced by a solvent extraction process was recently reported 113 for the production of biodiesel from the algae Schizochytrium limacinum, as shown in Fig. 6. Under similar reaction conditions, using sulfuric acid at 90 °C for 40 min, direct transesterification of algae biomass resulted in a higher biodiesel yield and FAME content than the traditional two-step extraction-transesterification method. The sulfur content was also higher than the ASTM required level, probably due to the use of MgSO4 in the algal culture media. A one-step supercritical methanol transesterification process was reported116 for direct liquefaction and conversion of a wet algal biomass (Inoculum Nanno-chloropsis sp.), containing about 90% water, to biodiesel. The reaction conditions are milder than those required for pyrolysis and prevent the formation of by-products. More than 80% FAME yield can be reached under the optimal conditions, given as a wet algae to methanol ratio (wt/vol) of around 1:9, with a reaction pressure, temperature, and time of about 1200 psi, 255 °C and 25 min respectively.
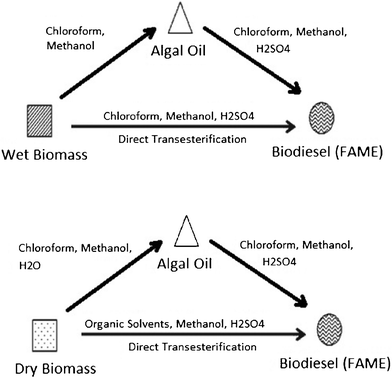 | ||
| Fig. 6 Methods used in the preparation of biodiesel from algal biomass: extraction–transesterification and direct transesterification. Modified from Ref. 113. | ||
Normally, these homogeneous transesterifications are performed in a batch operation. But because the catalysts are soluble in both reaction products, FAME and glycerol phases, after the reaction, the resulting biodiesel needs to be neutralized, washed with water, and then dried. The complicated post-treatment process and the huge amount of produced wastewater limit the homogeneous catalytic transesterification process. Because heterogeneous catalysts can be easily separated from the reaction mixture and regenerated through many cycles, the development of a continuous reaction system based on a heterogeneous catalyst can greatly reduce the process and catalyst consumption costs. Many attempts have been made to develop a heterogeneous catalyst with high tolerance to FFA and water in the feedstock.
Base heterogeneous catalysts, including alkaline earth metal oxides, zeolites and mixed oxides, have been widely used in biofuels production.117 A modified ZnO catalyst (Zn3La1) was used in the heterogeneous transesterification118 of crude algae oil with an additional 3% water and 5% FFA at 200 °C, 500 psi, and a methanol/oil ratio of 14:1–26:1 (mol/mol). The FAME yield was around 70%. In Zn3La1, lanthanum partially replaced zinc atoms in ZnO crystals, which weakened the neighbouring oxygen bonds, making them more reactive. In addition, the wurtzite ZnO crystal structure doped with La was stable under the reaction conditions, as verified by the similar XRD patterns for both fresh and spent catalysts.
Mixed metal oxides, Al2O3-supported CaO and MgO catalysts, have been investigated in the transesterification of lipid from the microalgae, Nannochloropsis oculata.119 Although CaO and MgO were solid basics, no transesterification activity was obvious for the microalgal lipid under the reaction conditions of 50 °C and a methanol/lipid molar ratio of 30. The support catalyst, i.e., 80% CaO/Al2O3, showed a very high biodiesel yield, over 97%, under these reaction conditions. Rather than the crystallite sizes of these alkaline earth metal oxides, the basic site density and basic strength played significant roles in the biodiesel yield. Mg–Zr solid base catalyst120 was used in the one-step transesterification of the green microalgae Nannochloropsis sp. at 65 °C. Compared to the biodiesel yield via the conventional extraction–transesterification method (22.2%), with a 10% Mg–Zr catalyst and a mixed solvent (methanol/methylene dichloride = 2:1, v/v) under the same reaction temperature of 65 °C for 4 h, the one-step method could give a higher yield of methyl ester (28.0%). Moreover, the solid catalyst could be easily separated from the microalgae residue, which could eventually reduce operating units as well as overall costs.
Hydroxide-modified Ti catalysts121,122 have been used for continuous catalyzed transesterifications of algae oil from species Dunaliella tertiolecta and Inoculum Nanno-chloropsis sp for fast production of biodiesel at elevated temperature and pressure. About 90% FAMEs yield was achieved within 60 s at 340–350 °C, and 3650 psi, or about 85% FAMEs yield within 30 s under supercritical methanol conditions, i.e., 340 °C and 2250 psi, with an algae lipids:methanol:hexane ratio of 1:3:96.
3.2 Saponification and hydrolysis
Typically, homogeneous catalysts are sensitive to water and FFAs in the feedstock. Compared to other catalytic reactions, reactants for transesterification must be free of water because in the presence of water, triglycerides hydrolyze and form the salts of the fatty acids and then transesterification stops. FFAs react with basic catalysts (NaOH and KOH) and form soaps, as shown in the following reactions: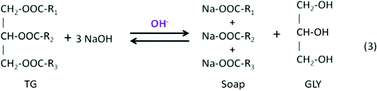
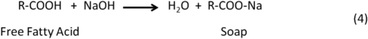
This complicates the glycerol separation and drastically reduces the methyl ester yield.
The saponification leads to lower production of biodiesel and a higher cost for the required purification process. Based on the work done by Freedman et al.,98,106 Komers et al.99 explored the kinetics and mechanism of the KOH-catalyzed methanolysis of rapeseed oil for biodiesel production using a model consisting of two sequences of consecutively competitive reactions (methanolysis and saponification). Their modelling results could accurately describe real cases with a probability of about 78% at given conditions. This model showed that all reaction rates were proportional to the amount of catalyst, which varies over time and is consumed in the competing saponification reactions. For base-catalyzed reactions, if the starting lipid contains more than 0.5 wt% of FFAs, a preliminary stage of acidity reduction is necessary by means of an esterification reaction of the FFAs, to avoid their saponification. Tesser et al.100 explored the kinetics of fatty acid esterification on acid exchange resins. As catalysts, the active site in acid ionic exchange resins is the sulfuric group, which exchanges its hydrogen ion with the components involved in the reaction after they are adsorbed on the resin surface. Their results showed that the rate-determining step is the surface reaction of protonated fatty acid with methanol that leads to the formation of a protonated methyl ester. This model can provide a good description of the kinetic behaviour, in particular for reactions performed at different catalyst concentrations. Meanwhile, in order to minimize problems associated with the homogeneous catalytic process, such as removal of catalyst after reaction, soap formation minimization, corrosion, and environmental issues, heterogeneous solid catalyst systems are favoured. For example, Kim et al.123 found that strongly basic anion-exchange resins could be used as a heterogeneous catalyst exhibiting synergetic effects for the transesterification reactions.
Hydrolysis and transesterification are two competing reactions that can occur during the synthesis of biodiesel, in which the catalysts play important roles.121 Cheirsilp et al.97 developed three kinetic models of the transesterification of palm oil fatty acids to ethanol using an immobilized lipase catalyst. Their results showed that the rate constants for alcoholysis of palm oil with ethanol are much higher than those for the hydrolysis reaction. The simulated results also showed that increasing the initial ethanol concentration increases the initial production rate and yield of fatty acid ethyl ester and lowers the final concentration of free fatty acid. Using a tungstated zirconia catalyst, Ngaosuwan et al.107 investigated the mechanistic pathways for hydrolysis (with water) and transesterification (with methanol) of tricaprylin and found that increasing the concentration of tricaprylin increased the reaction rates for both hydrolysis and transesterification under all conditions. However, water inhibited the reaction rate of hydrolysis by poisoning the active sites. With a low water-to-tricaprylin ratio, the rate-determining step for hydrolysis is the adsorption of tricaprylin, while the rate-determining step for transesterification is the surface reaction at a low methanol-to-tricaprylin ratio. Based on quantum mechanical calculations, Zhang et al.124 investigated the mechanisms of the conversion of methyl lactate (ML) over sodium tripolyphosphate. Their calculated results indicated that the conversion of ML over a catalyst is mainly through the direct decomposition of ML to methyl acrylate and methanol, and also through the decarbonylation of ML to acetaldehyde, methanol and CO via stepwise mechanisms. Based on ab initio quantum chemistry calculations, Turner et al.125,126 investigated the alkaline hydrolysis of esters with methyl acetate and methyl benzoate model systems. From the calculated reaction pathway for the first step in the alkaline hydrolysis of esters, they identified one transition state and one intermediate product.
Except for the catalytic hydrotreating or transesterification of TG to hydrocarbons as described above, the thermal cleavage of TG is another pathway with a unique feature to form cyclic hydrocarbons. Instead of via a Diels–Alder reaction, based on the experimental evidence, Kubatova et al.127 proposed that the cyclic hydrocarbons are produced through intramolecular radical cyclization. In general, although varying by engine type, ignition characteristics and specific compression ratios, the presence of 5–20 wt% cyclic hydrocarbons (including aromatics) in transportation fuels can improve the operational performance. Therefore, such attempts are meaningful and further discovery is highly desired.127
3.3 Glycerol reforming
As described in the previous sections (eqn (2)) glycerol (GLY) is a by-product formed in biodiesel production, and new efficient procedures for its transformation into valuable derivatives, such as via hydrodeoxygenation, are in high demand.4,128–132 Some promising derivatives include dihydroxyacetone (DHA), glyceric acid (GLS) and H2. Fig. 7(a) demonstrates a reaction network of Pt–Bi/C-catalyzed glycerol oxidation proposed by Wörz et al.130 The figure shows that the main products could be GLA, GLS and DHA. MOS, HBTS, and TS (their molecular structures are defined in Fig. 7(a)) are chelating agents and not the main products from their results. One major problem for technical realization is the deactivation of the catalyst, especially in acidic media. Based on kinetic modelling as shown by the schematic in Fig. 7(b), the produced GLS easily adsorbs on the catalyst active sites and therefore reduces both the conversion of glycerol and the selectivity toward DHA. Therefore, to increase the DHA yield and glycerol conversion, developing new methods of separating glyceric acid in situ from the reaction mixture is critical. With a ZrO2–FeOx catalyst, Yoshikawa et al.133 used an aqueous glycerol solution as a feedstock in a fixed-bed flow reactor at 623 K under atmospheric pressure to produce useful chemicals. Their products include propylene, allyl alcohol, carboxylic acids and ketones. | ||
| Fig. 7 (a) Typical reaction network of Pt–Bi/C-catalyzed glycerol oxidation. (b) Proposed mechanism of selective deactivation induced by glyceric acid.130 | ||
Another method for glycerol reforming is to produce renewable H2 and CO2 through the steam reforming process: C3H8O3 + 3H2O → 7H2 + 3CO2.128,129 Employing a COMSOL Multiphysics modelling package, Adhikari et al.128 explored the kinetics and reactor modelling of the glycerol steam reforming process over a Ni-based CeO2-supported catalyst. Based on a power model, the simulated activation energy and the reaction order for the above reaction were found to be 103.4 kJ mol−1 and 0.233, respectively. Wawrzetz et al.129 measured the rate for the formation of gaseous products from 30 wt% glycerol at T = 498 K and total pressure = 29 bar, with a 3 wt% Pt/Al2O3 catalyst, and found that the main products are H2 (with a rate of 3.53 min−1) and CO2 (2.32 min−1), with other minor products being CH4 (0.079 min−1), C2H6 (0.040 min−1), C3H8 (0.012 min−1), and CO (0.003 min−1). By photofermentation under optimal conditions, the reformation of biodiesel-derived crude glycerol to hydrogen could reach as high as 6.1–6.96 mol hydrogen/mole of crude glycerol, a yield of 96% of the theoretical maximum.134,135 Recent studies showed that by adding CaO into the reaction system for enhanced steam reforming, a high purity of hydrogen can be achieved from a single-reactor process.131 The esterification of glycerol with acetic acid was carried out over different solid acid catalysts in a slurry reactor by Zhou et al.136 Their results showed that at optimal operating conditions (molar ratio of acetic acid to glycerol = 9:1 at 110 °C), the predicted glycerol conversion could reach 98.47%.
The biological conversion of crude glycerol into high-value biochemicals may be one of the most promising alternatives available. Nitayavardhana and Khanal137 examined the potential for producing an edible fungus (animal feed) from biodiesel-derived crude glycerol. Such an alternative approach for glycerol utilization could provide a unique sustainable option for biodiesel refineries and an additional source of revenue from reaction products.
4. Heterogeneous catalytic conversion of microalgal lipids to liquid fuels
Most of the current technologies to convert microalgae-based feedstocks are still in their early stages.138 Due to undesirable oxygen groups in FAMEs, these conventional biodiesels suffer from high water content and poor low-temperature properties and are susceptible to microbial fouling.31,47,48 Besides, a large quantity of glycerol produced from FAMEs manufacturing is being considered a waste material. Therefore, catalytic heterogeneous conversions, including hydrothermal liquefaction, hydrocracking, hydrodeoxygenation and decarboxylation, of microalgae-based feedstocks, i.e. bio-crude and lipids, to transportation fuels have attracted lots of attention. While the use of commercial hydro-processing technology139–141 for the production of transportation fuels is less of a challenge in terms of material development and reactor design, it consumes a large amount of H2 at operating conditions. The catalytic deoxygenation of FFAs (the model compounds most often studied as surrogates for lipid-rich microalgae) to diesel-range hydrocarbons over heterogeneous catalysts has attracted much attention because it requires little or no H2 consumption. However, this process requires further understanding of the catalyst function and catalytic mechanism. In this section, we summarize the current progress on the conversion of microalgae to transportation fuels.4.1. Heterogeneous hydrothermal liquefaction of algae feedstock
Heterogeneous hydrothermal liquefaction of microalgae has recently been investigated by many researchers. Typically, the bio-oil yields were in a range of 24–45 wt%. However, bio-oil physico-chemical characteristics, including heating values and distribution of components, were highly influenced by conversion method and feedstock.89,90,93,142 The use of heterogeneous catalysts during HTL of microalgae was recently reported to produce higher quality bio-oil by lowering the oxygen content in products.Early in 1997, Matsui et al.90 investigated the liquefaction of micro-algae (spirulina) with an iron catalyst (Fe(CO)5–S) and obtained 66.9 wt% oil yield at 350 °C for 60 min in tetralin. Fig. 8 shows the effect of Fe(CO)5–S catalyst level on the product distribution. The oil yields increased linearly from 54.4 to 63.7 wt% as the amount of catalyst increased from 0 to 1.0 mmol, but the conversion and gas yield were nearly constant. They also found that liquefaction in toluene gave oil fractions of high carbon content and lower oxygen content, while liquefaction in water had a lower carbon content and a higher oxygen content with an oil yield of 61 wt% at 350 °C as shown in Table 2. The presence of Ni/Al2O3 catalysts was found 142 to convert the triglycerides to fatty acids then to alkanes at 350 °C, at pressures of 150–200 bar in water, and the de-oxygenation of bio-crude increased by up to 10%. At near-supercritical and supercritical conditions, the addition of water could enhance the mixing of reactants and therefore speed up the reactions. Water could also serve as solvent, and reactant.143–145 Fu et al.146,147 also reported the deoxygenation of model algae fatty acids to linear hydrocarbons in near-supercritical or supercritical water. Activated carbons, metal salts and high-surface-area supported metal catalysts were investigated for activity toward deoxygenation of palmitic acid. Reaction temperatures ranged from 290 (near-critical water) to 380 °C (supercritical water), and the pressure was the saturated or supercritical pressure of water at reaction temperatures, for example, 280 bar at 380 °C. On activated carbon, 5% platinum and 5% palladium were effective for hydrothermal deoxygenation of palmitic acid, with more than 90% selectivity toward pentadecane.146 Also for the first time activated carbons were shown to convert saturated and unsaturated fatty acids to fuel-range hydrocarbons in a hydrothermal environment.147 This approach has important practical value for the energy industry by offering a low-cost approach to convert triglyceride-derived fatty acids into hydrocarbons, eliminating the need for expensive precious metal catalysts. The kinetic study suggested that the reaction was first-order for palmitic acid over Pt/C146 and activated carbons,147 with a calculated Arrhenius activation energies of 79 kJ mol−1, and 125 kJ mol−1, respectively. Nevertheless, these results shed new light on the role of activated carbons in bio-fuel production. A preliminary regenerability study indicated that no significant activity loss occurred for regenerated catalysts.
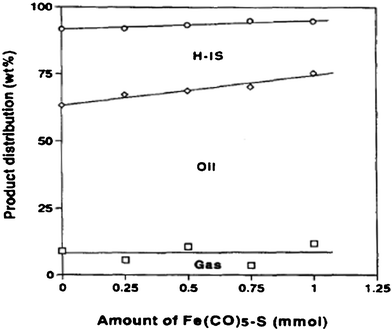 | ||
| Fig. 8 Effect of catalyst level on product distribution in liquefaction of Spirulina in 1-methylnaphthalene under hydrogen at 350 °C for 60 min.90 | ||
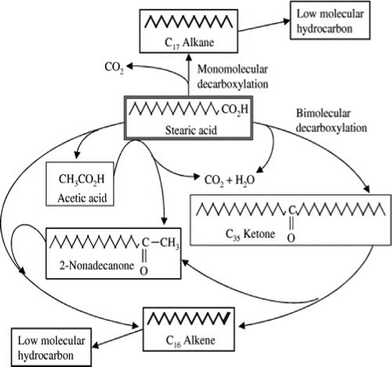
Possible pathways for the deoxygenation of stearic acid over heterogeneous catalysts at supercritical water conditions was investigated.148 In a batch reactor at 673 K, by adding basic catalysts, such as alkali hydroxide (NaOH and KOH), the monomolecular decarboxylation of stearic acid produced CO2 and C17 alkane. Different bimolecular decarboxylation pathways were proposed over metal oxide catalysts (CeO2, Y2O3 and ZrO2), as shown in Fig. 9. The resultant C16 alkene may be produced via the direct bimolecular decarboxylation of stearic acid, or via the decomposition of intermediate C35 ketone, and/or 2-nonadecanone, which were found in the products as well.
4.2 Hydroprocessing of bio-crude oils
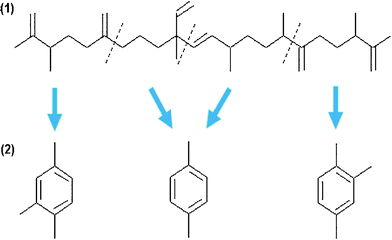
The chain-lengths of the hydrocarbons obtained from biological feedstocks are typically C11 and above (typically, C14–C22). In order to produce light hydrocarbons (such as the jet fuel JP-8) from heavy hydrocarbons, hydrocracking and hydroisomerization processes are necessary.Currently, hydrocracking is commonly practiced in the petroleum refinery industry to treat oil residua. Most frequently used hydrocracking catalysts for bio-crude oils were precious metal based catalysts, including Co and Ru, and zeolites. Hillen and coworkers140,141 reported the hydrocracking of botryococcus extracts using cobalt–molybdenum catalysts at temperatures of 400–440 °C. The resulting products consisted of more than 80% of low molecular weight, linear alkanes and isomers, which were in the gasoline and jet fuels range. About 15% of the cracking products were classified in the diesel fraction. Kitazato et al.149 studied aromatic hydrocarbons produced from the cracking of botryococcenes using H-zeolite as the catalysts. The proposed mechanism involved pathways of consecutive cracking of botryococcenes with subsequent aromatization, as shown in Fig. 10. The potential cracking sites were between a carbon with a methyl group and a carbon with an unsaturated double bond. However, Sharma and Bakhshi150 reported that large amounts of oxygenated compounds were formed at lower temperatures (370 °C) while low gasoline yields with catalyst coking occurred at higher temperatures (410 °C). Furthermore, these unselective catalytic crude bio-oil upgrading processes produced a wide variety of hydrocarbons, including light molecular weight hydrocarbons, which are of less interest.
 | ||
| Fig. 10 Pathways of production of aromatic hydrocarbons from a botryococcene: (1) cracking, and (2) aromatization. Modified from Ref. 149. | ||
To obtain high quality fuels with less oxygen contents, a new co-processing139 of hydrodeoxygenated (HDO) crude bio-oil with so-called “long residue” oil in a lab-scale fluid catalytic cracking (FCC) fluidized bed reactor has been reported. Under reaction conditions with temperature ranges from 230 to 340 °C and with total pressure constant at around 290 bar, 5 wt% of ruthenium supported on carbon powder was used as deoxygenation catalyst. Near oxygenate-free hydrocarbons with more than 40wt% of typical FCC gasoline and 20+ wt% of light cycle oil (LCO) and about 10% of liquefied petroleum gas (LPG) were obtained. However, compared to a pure FCC feed, excessive amounts of undesired coke and dry gas were found in co-processing experiments, as shown in Fig. 11.
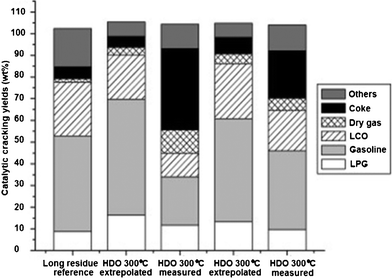 | ||
| Fig. 11 Product yields after catalytic cracking. Results for experiments denoted as “extrapolated” are theoretical yields obtained when extrapolating actual yields at 20 wt% mix HDO oil in long residue to 100 wt% HDO oil.139 | ||
In the literature, direct modeling work has not been published for hydrocracking of algal biofuels. Ancheyta et al.151 reviewed the kinetic modeling of hydrocracking of heavy oil fractions. The models were based on the lumping technique, continuous mixtures, structure-oriented lumping and single event, and were all fully discussed and compared in terms of the capacity to predict detailed product composition, difficulty for parameter estimation, dependency of rate coefficient with feed properties, and required experimental data. Recently, Martinez et al.152 reviewed the process aspects and modeling of an ebullated bed reactor for hydrocracking of heavy oils. Some key factors for applying these reactors to hydrocracking of heavy petroleum fractions, such as sediment formation, catalyst attrition and catalyst deactivation, were clearly discussed. Here we are summarizing progress in theoretical modelling of mechanisms of catalyzed hydrocracking of heavy compounds.
Based upon the elementary steps of carbenium-ion chemistry, Froment153 developed a detailed kinetic model of acid-catalyzed oil-refining processes. In this approach, the computer calculation of the number of events in an elementary step requires knowledge of the configuration of the transition state which could be determined through quantum chemical calculations. Krishna and Balasubramanian154 provided a general analytical solution for the full stoichiometry based discrete lumped kinetic equations in hydrocracking of the heavier petroleum fractions. Their results revealed that the full stoichiometry is the best performing model for hydrocracking of heavier petroleum fractions and that the product distribution in a hydrocracker is governed by ordinary differential equations. Thybaut and Marin155 investigated the kinetic modeling of the conversion of complex hydrocarbon feedstocks by acid catalysts and found that the number of elementary reaction families, and hence of the corresponding kinetic parameters, is rather limited, and the variations in acid strength have a significant effect on the activation energy of the acid-catalyzed rearrangement steps. Using the principles of the single-event microkinetic concept, Mitsios et al.156 investigated the hydrocracking and hydroisomerization of long-chain paraffin on an amorphous Pt/SiO2·Al2O3 catalyst. Their results showed that the invariant rate constants do not depend on the feedstock composition or the process configuration. From the experimental program involving liquid-phase pure n-hexadecane hydrocracking, a complete set of only nine rate constants for paraffin hydrocracking and hydroisomerization were obtained and gave a satisfactory fit of the experimental data.
Zeolite catalysts are often used in hydrocracking processes. According to the bifunctional reaction scheme, kinetic modeling for hydrocracking of alkanes on Pt/US-Y zeolites has been carried out by Martens et al.157 and Thybaut et al.158 The differences in both the number of acid sites and the average acid length determine the activities of the zeolites. On this catalyst, the reactivity of alkanes increased with the carbon number, which related to three phenomena: (1) the physical sorption of heavier molecules was more favorable; (2) the reaction pathway and the number of parallel reactions became larger with larger molecules; and (3) the stabilization of alkylcarbenium ions and, hence, their concentration, increased with the increasing size and electron-donating property of alkyl-substituents. In order to explore the mechanisms of hydrocarbon conversion in zeolites, several atomistic-level simulations have been done.159–162 Based on ab initio quantum chemical calculations, Kazansky et al.161 investigated the isobutene cracking on zeolites for three main elementary steps: (1) chain initiation, which resulted in formation of adsorbed carbenium ions by the protonation of olefins by protolytic cracking of paraffins and by protolytic dehydrogenation of paraffins; (2) chain termination, which represented the decomposition of the tert-butoxide group into isobutene and bridging hydroxyl; and (3) chain propagation, which can involve hydride transfer. For each elementary step, at least one transition state was identified. Their calculated heats of reaction and the activation energies of the main elementary steps are comparable with available experimental and other theoretical results. Using the ab initio method at the Hartree–Fock level with an SCF-3-21G/6-31G* basis set and MP2 perturbation theory, Ridby et al.162 further studied the mechanism of hydrocarbon conversion in zeolites and found that the intermediates are covalent alkoxide species. In their work, the influence of differences in the acid strength on the activation energies was explored. The variations in acid strength were found to only have minor effects on the alkoxide stability and little effect on the activation energies for the D/H exchange in methane and for olefin chemisorptions, which have covalent transition states. However, acid strength was found to have a significant effect on the activation energy of the acid-catalyzed rearrangement steps that have ionic transition states. In such cases, all of these activation energies are strongly reduced with increasing acidity. Based on first-principles density functional theory, Frash et al.159 explored the mechanism of the β-scission reaction in zeolites and found that the potential energy surface of this reaction is very complex. Three reaction paths were identified: (1) a one-step reaction via the “ringlike” transition state; (2) a two-step β-scission reaction via a “hydrogen-bonded” transition state and substituted cyclopropane; and (3) a one-step β-scission reaction via a “hydrogen-bonded” transition state. Among them, path 2 has the lowest activation barrier. Combining experimental measurements with molecular dynamic simulations, Isoda et al.160 explored the mechanism of hydrocracking of pyrenes (Py) over nickel-supported Y-zeolite catalysts. Their results showed that pyrene was cracked into 1- and 2-ring aromatics by partial hydrogenation of aromatic rings. Molecular dynamic simulation results indicated that diffusion of the polycyclic aromatic products into the zeolite pore is not easy and that these molecules are cracked on the surface of the zeolite rather than in the pore.
As described above, the first-principles model (FPM) strategies exist in the literature for modeling hydrocracking (HC) reactions with some success, but FPM strategies may not be adequate because of process complexity and common process variations, such as changes in feed composition, operating conditions, and catalyst deactivation.163 Obviously, multi-scale modeling approaches are needed to simulate such complex systems. Unfortunately, in the literature information on this kind of multi-scale modeling for hydrocracking is very limited. Bhutani et al.163 employed first principles, data-based and hybrid modeling strategies to simulate an industrial hydrocracking unit with comparative performance assessment and to optimize the operation of the industrial HC unit and thereby estimate the costs. Their results demonstrated that the application of data-based and hybrid models to represent the behavior of an industrial HC unit provides accurate and consistent predictions in the presence of common process variations and changing operating conditions.
4.3. Deoxygenation of microalgae feedstocks
After transesterification or hydrolysis, the obtained biodiesel (FAMEs, FFAs) may still contain unsaturated C–C bonds and carboxyl groups, depending on the algal lipid composition. These unsaturates will reduce the heat of combustion of the fuel. In some cases, further treatments may be desirable to upgrade the biodiesel by decarboxylation and hydrodeoxygenation processes.164 Deoxygenation of FFAs can be categorized into three major reactions: hydrogenation, decarboxylation and decarbonylation. In the presence of H2, the hydrogenation reaction involves the replacement of a hydroxyl group and an oxo group by hydrogen atoms without losing a carbon number, or by removal of the carboxyl group through the release of carbon monoxide to form hydrocarbons having one less carbon number. In the absence of H2, direct decarboxylation or decarbonylation removes the carboxyl group by releasing carbon monoxide or carbon dioxide and producing either a paraffinic hydrocarbon or an olefinic hydrocarbon. Catalytic carbonylations of alkenes and alkynes over transition metal catalysts, especially Pd based catalysts, have been commercially used for the preparation of C5–C18 carboxylic acids and esters.165 For the reverse decarbonylation and similar decarboxylation reactions, supported Pd catalysts have also been extensively studied.166–171The reaction pathways for deoxygenation of both saturated and unsaturated C18 free fatty acids, including stearic, oleic, and linoleic acids have been investigated by Immer et al.174 In the presence of inert gas, He, or diluted H2, the reactions were conducted over a 5 wt% Pd/C catalyst in organic solvents in a semi-batch reactor. The major products for catalytic deoxygenation of stearic acid (SA) under He were decarboxylation products, n-heptadecane and heptadecenes. Based on the analysis of CO2 and H2 evolution, they explained that at complete SA conversion, a 98% n-heptadecane yield could be reached because of the hydrogenation of heptadecenes via hydrogen transfer from the solvent. However, at low SA conversion, the hydrogenation of heptadecenes was not significant. Similarly, due to the formation of unsaturated products on the catalysts, the reaction requires much more time to reach full conversion under He than in the presence of H2. They also found that the pathway for deoxygenation of the unsaturated C18 free fatty acids, oleic and linoleic, were consecutively hydrogenated to saturated SA during the heating of the reaction mixture to reaction temperature, followed by subsequent decarboxylation.
Snåre et al.172 examined the possible reaction matrix for the deoxygenation of stearic acid in dodecane over metal-supported activated carbon catalysts at 300 °C and 600 KPa under an inert atmosphere (as shown in Fig. 12). Catalysts were found, under provided conditions, favourable for deoxygenation reactions (decarbonylation and decarboxylation, reactions 1–2), as well as hydrogenation reactions (reactions 3–4). Under hydrogen lean conditions, several side reactions (i.e., isomerization, dehydrogenation, and cyclization, reactions 5–9) were observed. The isomerized C17 cyclic hydrocarbons and aromatics are subsequent products of these side reactions. Also found in the reaction products were short-chain fatty acids and hydrocarbons, but no fatty alcohols. Therefore, cracking (reaction 10) rather than alcoholation occurred over heterogeneous catalysts under the inert environment, even though alcohol formation via acid hydrogenation is well-known.175 To a lesser extent, dimers and heavy products (products of reactions 11–13) were easier to form over nickel catalysts than either Pt- or Pd-supported catalysts. Free hydrogen atoms may be transferred from cracking products or solvents.
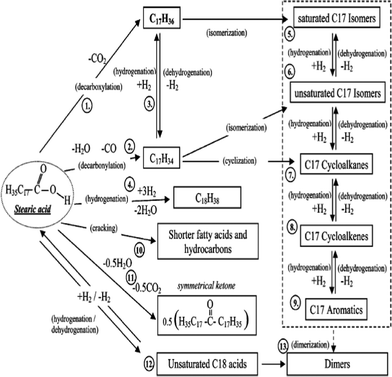 | ||
| Fig. 12 Tentative reaction routes for deoxygenation of stearic acid over a heterogeneous catalyst at 300 °C under inert atmosphere.172 | ||
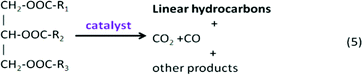
With a Ni catalyst, the conversion of triglyceride produced high yields of linear C5 to C17 alkanes and alkenes, while small amounts of light alkanes (C1–C4) and H2 were also formed. Compared to Ni, catalysts containing Pd or Pt supported on activated carbon showed lower activity for both triglyceride deoxygenation and for cracking of the fatty acid chains.176 They found that the selectivity towards the cracked and/or hydrogenated products increased with increasing unsaturation of the feed. As shown in Fig. 13, their experimental results suggested that one pathway for triglyceride deoxygenation to C17 alkanes involves the liberation of fatty acids via C–O bond scission, followed by a subsequent step of elimination of CO2 from the acids. Similar results were also reported by Murzin et al.,29,168 where stearic acid was formed from ethyl stearate as an intermediate, and then it subsequently decarboxylated to the C17 alkane. Alternate pathways for hydrocarbon formation, as both ethene and ethane were detected, involved the scission of the C–C bond to C17 and C15 hydrocarbons, respectively. This pathway may either correspond to alkanes or terminal alkenes depending on whether hydrogen abstraction occurs during bond scission.
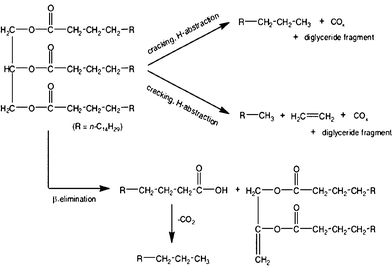 | ||
| Fig. 13 Simplified reaction scheme for deoxygenation of tristearin. Modified from Ref. 176. | ||
The deoxygenation of triglycerides over Ni, Mo and NiMo sulphide catalysts at 260–280 °C, 3.5 MPa, and in the presence of H2 in a fixed-bed reactor was investigated.177 Significantly different reaction pathways were exhibited over two monometallic supported catalysts, as shown in Fig. 14: the Ni/Al2O3 produced only decarboxylation hydrocarbon products, which were also reported by other groups,29,168,176 while Mo/Al2O3 yielded mainly hydrodeoxygenation of hydrocarbon products. For the bimetallic NiMo catalysts, a mixture of decarboxylation and hydrodeoxygenation of hydrocarbon products was found, apart from the various oxygenated product intermediates.
 | ||
| Fig. 14 Reaction pathways involved in conversion of triglycerides into hydrocarbons.177 | ||
Other liquid-phase catalytic deoxygenation reactions 29,169–170 were also been investigated for unsaturated renewables (oleic acid, linoleic acid, tristearine and methyl oleate) on a Pd/C catalyst and ethyl stearate to produce high carbon number hydrocarbons. With 5 wt% Pd/C catalyst, Immer et al.174 explored the catalytic reaction pathways in liquid-phase deoxygenation of C18 FFAs, such as stearic, oleic and linoleic acids, and found that the main liquid products are n-heptadecane and heptadecenes. FFA deoxygenation can occur via decarboxylation (C17H35COOH → CO2 + n-C17H36) and decarbonylation (C17H35COOH → CO + H2O + C17H34) pathways. Thermodynamics of both reactions are favourable at 300 °C: ΔGrxn = −83.5 kJ mol−1 for decarboxylation and −17 kJ mol−1 for decarbonylation.174 Snåre et al.170 studied the kinetics of ethyl stearate decarboxylation over a Pd/C catalyst. Their kinetic modelling and estimated parameters of temperature dependence (300–360 °C) of ethyl stearate decarboxylation fit the experimental results very well with a 99.28% degree of explanation. Recently, Lei et al.178 explored the effect of metal loading for the conversion of fatty acid methyl esters to monoethanolamides. By decreasing the amount of Al in the layers of the MgAl-layered double hydroxide (LDH) precursors, they found that the conversion of methyl stearate increased dramatically. Therefore, the study and optimizing of metal catalyst compositions will be able to improve and eventually provide effective and feasible routes for algae based bio-diesel production.
 | ||
| Fig. 15 Schematic of the intermediate stages in a decarboxylation of a model fatty acid C2H5COOH.179 Each of the four model sets in this study were geometry optimized from these initial configurations, with the relevant substitution of Si for Al and the addition of Na as described in the text. In the figure, oxygen is red, hydrogen is white, aluminium is pink, carbon is gray and silicon is yellow. | ||
Based on the use of methyl formate as the model for biodiesel, Metcalfe et al.181 investigated the chemical kinetics of methyl formate decomposition by ab intio calculations. Their results indicated that the decomposition is dominated by a single channel producing methanol and CO over all temperatures. The other two pathways—producing formaldehyde or producing methane and CO2— are unlikely to occur based on the calculated kinetic mechanism between 500 and 2500 K and for all pressures. Obviously, these results can be used as the basis to build more realistic models that eventually will provide information for optimizing the efficiency of the production of biofuels.
5. Perspectives and concluding remarks
Microalgae has potential as a renewable biomass resource for advanced biofuels and mitigating CO2 emissions because of its high productivity, high oil content, fast growth rates and minimal competition with food crops.14 As an alternative fuel, microalgal biodiesel must be competitive with petroleum-derived fuels that are, at present, the least expensive transportation fuels. This will depend mainly on the cost of producing the algal biomass.5,182,183 However, for microalgae to be a viable fuel resource it faces challenges of balancing energy inputs, economics and CO2 emission advantages.54,184 For 10 million gallons per year of algae lipid production (including algae growth and solvent lipid extraction) with 10% investment return, it costs $8.52/gal for an open ponds and $18.10/gal for closed tubular photobioreactors. Adding the algae lipid conversion to biodiesel through hydrotreating, the corresponding total costs increase to $9.84/gal and $20.53/gal based on techno-economic analysis.185 Therefore, the current cost of the algae-based biofuel production is much higher than petroleum as well as seed crop-based fuels, mainly due to the energy required for production and processing.184–186 The energy consumed primarily in drying algae and extracting lipids from algae comes directly and indirectly from fossil energy (electricity and heat).33,54 The high fossil energy input takes away a certain amount of microalgae's CO2 emission advantage from life cycle CO2 analysis.33,54,184 To reduce the energy input and lower the cost, one approach is to increase algae lipid content and growth rate.33,185 Similarly, developing low cost and low energy intensity harvesting and algae processing technologies are also highly sought.9 In addition, value-added co-products (such as animal feed) and co-services (such as water treatment) could also be taken into account for reducing the overall costs.184,185Experimental studies and computational simulations of catalytic conversion for microalgae to transportation fuels are at the early stages of development compared to processes that use other biomass-derived feedstocks. This work has summarized recent progress on the catalytic conversion of microalgae-based biomass to transportation fuels including hydrothermal liquefaction, transesterification and catalytic hydroprocessing methods. Also included is a fundamental understanding of the process using computational modelling. Both catalyst development and mechanism modelling strongly impact technological development for microalgae to transportation fuels.
For conversion processes for microalgae, some pioneering work is being performed on the heterogeneous catalytic HTL for algae conversion, but homogeneous catalytic HTL is the current widely used method for processing different microalgae feedstocks. Hydrothermal liquefaction of microalgae to a petroleum-equivalent crude oil is considered to be a promising conversion technology. The technology can convert wet and whole microalgae to bio-fuels without the requirement of initially-dry microalgae. Laboratory-scale tests show that hydrothermal liquefaction could convert microalgae with high and low lipid contents and its residues into bio-oil. Catalysts, process conditions and the microalgal species strongly affect the bio-oil yields and properties. However, hydrothermal liquefaction of microalgae faces the challenges of achieving high bio-oil yield, improving the oil quality (e.g. removing nitrogen) and treating/recycling waste water from the process. Furthermore, during scale up of the process, technical difficulties arise when pumping algae slurry into high pressure reactors and when designing a large reactor because the process involves complex reactions for which mechanisms are not yet clearly understood.
To make the HTL a viable process, extensive research is needed to develop catalysts and new reaction media to increase conversion efficiency and reduce the temperature and pressure of the process while maintaining high reaction rates. Characterization of the products, including primary crude-oil, gas, aqueous phases and algae residues, is necessary for optimizing the catalytic upgrading of crude-oil, generation of high-value co-products and minimization of waste and overall costs. In addition, research on development of heterogeneous catalysts for upgrading bio-crude oil and on the design of advanced processes involving combining bio-fuel production, co-products and waste recycling are also needed.
Transesterification has a long history in industry and plays an important role in biofuel production as well. Transesterification of algal oil is normally done with alcohol solvents and bases or acids serving as the catalyst. Compared to other catalytic reactions, the transesterification process for biofuel is relatively intolerant to water and FFAs in the feed, especially for homogeneous catalysts. In addition, the complicated post-treatment process also limits the homogeneous catalytic transesterification process. Instead, heterogeneous transesterification catalysts, such as solid acid catalysts, can greatly improve the economics of biodiesel production, without multiple reaction steps and post-treatment steps. Therefore additional attention should be paid to the development of a heterogeneous transesterification process for biofuel production from a microalgae feedstock.
Most of the current research on the catalytic conversion of microalgae to fuels remains in the early stages of studying simple model compounds, C16–C18 FFAs, instead of using more complex lipids from microalgae. Precious metal-supported catalysts have been widely reported for the catalytic conversion of FFAs to linear hydrocarbons. The development of active non-precious metal catalysts with high selectivity toward green diesel, which are capable of working at mild operating conditions, is a great challenge for microalgae conversion. Chemical solvents, sometimes with H2 under high temperature and pressure, have been applied to these conversion reactions. Novel processes that require less use of H2 and minimal solvents will be a benefit for minimizing the process costs for fuel production. Co-processing of microalgae with other feedstocks in an optimized catalytic commercial process could also help reduce the overall cost.
To provide a feasible cost for the production of green diesel from microalgal lipids, active non-precious metal catalysts with high selectivity toward green diesel, which are also capable of working at mild operating conditions for real microalgae oil, are required. Identifying and optimizing the commercial catalytic petroleum process for microalgae co-feeding can also help reduce the capital investment. Novel processes with decreased usage of H2 and solvents may also be required to minimize the overall cost of fuel production.
In addition, the conversion of by-products from the processes, i.e., glycerol from transesterification and algae residues (mainly protein and carbonhydrates) to high value chemicals, should also be taken into account.
In this review, we summarized the current progress of computational modelling as applied to the catalytic conversion of bio-oil to high-grade biodiesel. Compared with applications in other fields, the computational modelling work performed so far for biodiesel production and upgrading is still very limited. Due to limited computer resources and lack of accumulated knowledge in this relatively new field, most computational modelling work on the catalytic conversion of microalgae has been done on simplified model systems, although such approaches have been successfully applied to other catalyzed chemical reactions.187,188 Therefore, more simulation research in this field is expected to be conducted in the future. Further modelling work should be focused on the following aspects:
(1) Although modelling of simplified model systems could provide basic and useful information, direct simulation of algal systems is highly desired. To reach this goal, new and efficient methodologies should be developed that are capable of simulating large real systems.
(2) Catalysts play important roles in biofuel conversion. Identification of the proper catalysts for specific reactions to obtain desired products is difficult solely by experimental methods. Based on known catalysts, computational modelling can screen and identify promising candidates from vast numbers of trial compounds and provide useful information about active sites, optimized structures and composition, and possible synthesis routes. Understanding the mechanisms of catalysts in biofuel conversions is the key issue for improving production processes. Theoretical simulations could provide insight into the catalysis mechanism, especially concerning deactivation, and therefore can guide the experimental research and production process.
(3) Biofuel upgrading involves several process stages. For different stages, one or more modelling schemes can be used. To explore the mechanism and structure, atomistic level ab initio and density functional theory approaches are essential to obtain information about molecular reactivity, structures and the catalytically active sites. To explore the reaction kinetics, a combination of ab initio calculations with molecular dynamic or Monte Carlo simulations can be used to describe the kinetic properties, and the corresponding dependence on temperature and pressure. To explore the reactions and mass transfer in reactors, chemical engineering, bioengineering and computational fluid dynamics (CFD) modelling are needed to describe the distributions of products and energy transfer. The parameters used in CFD modelling could also be obtained from atomistic modelling. In order to assess and optimize the entire upgrading process, the multi-scale modelling approach, which combines atomistic level, mesoscale and continuum methods with process modelling is the ultimate approach to simulate biofuel production processes.
Disclaimer
Reference herein to any specific commercial product, process or service by trade name, trademark, manufacturer or otherwise does not necessarily constitute or imply its endorsement, recommendation or favouring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.Acknowledgements
This work was performed partially in support of the National Energy Technology Laboratory's Office of Research and Development under contract number DE-FE-0004000 with activity number 4000.5.660.251.003. The authors thank Dr S. Chen and Dr D. C. Sorescu for reading part of the manuscript, and appreciate proofreading by J. Fink and C. Wamsley (internal technical writers).References
- EERE, Biomass multi-year program plan, 2012 Search PubMed http://www1.eere.energy.gov/biomass/pdfs/mypp_april_2012.pdf .
- R. Serna-Guerrero, Y. Belmabkhout and A. Sayari, Chem. Eng. Sci., 2010, 65, 4166–4172 CrossRef CAS.
- Y. Ghasemi, S. Rasoul-Amini, A. T. Naseri, N. Montazeri-Najafabady, M. A. Mobasher and F. Dabbagh, Appl. Biochem. Microbiol., 2012, 48, 126–144 CrossRef CAS.
- R. Luque, J. C. Lovett, B. Datta, J. Clancy, J. M. Campelo and A. A. Romero, Energy Environ. Sci., 2010, 3, 1706–1721 CAS.
- P. T. Pienkos, L. Laurens and A. Aden, American Scientist, 2011, 99, 474–481 Search PubMed.
- M. S. Wigmosta, A. M. Coleman, R. J. Skaggs, M. H. Huesemann and L. J. Lane, Water Resour. Res., 2011, 47, W00H04, DOI:10.1029/2010WR009966.
- M. Olivares-Marin, M. Castro-Diaz, T. C. Drage and M. M. Maroto-Valer, Sep. Purif. Technol., 2010, 73, 415–420 CrossRef CAS.
- EIA, How dependent are we on foreign oil, 2011 Search PubMed http://205.254.135.7/energy_in_brief/foreign_oil_dependence.cfm .
- DOE-EERE, Algae: 2011 Platform Review Report, 2011 Search PubMed http://www1.eere.energy.gov/biomass/pdfs/2011_algae_review.pdf .
- EPA, Regulation of Fuels and Fuel Additives: Changes to Renewable Fuel Standard Program, 2010 Search PubMed http://www.gpo.gov/fdsys/pkg/FR-2010-03-26/pdf/2010-3851.pdf .
- H. Herzog, E. Drake and E. Adams, CO2 capture, reuse, and storage technologies for mitigating global climate change, DOE Order No. DE-AF22-96PC01257, 1997 Search PubMed.
- R. Pate, G. Klise and B. Wu, Appl. Energy, 2011, 88, 3377–3388 CrossRef CAS.
- A. F. Clarens, H. Nassau, E. P. Resurreccion, M. A. White and L. M. Colosi, Environ. Sci. Technol., 2011, 45, 7554–7560 CrossRef CAS.
- F. Bo and W. Q. Liu, Environ. Sci. Technol., 2010, 44, 3640–3640 CrossRef.
- P. J. L. Williams and L. M. L. Laurens, Energy Environ. Sci., 2010, 3, 554–590 CAS.
- D. H. Lee, Bioresour. Technol., 2011, 102, 43–49 CrossRef CAS.
- P. H. Pfromm, V. Amanor-Boadu and R. Nelson, Bioresour. Technol., 2011, 102, 1185–1193 CrossRef CAS.
- A. Singh, P. S. Nigam and J. D. Murphy, Bioresour. Technol., 2011, 102, 10–16 CrossRef CAS.
- D. J. Stepan, R. E. Shockey, T. A. Moe and R. Dorn, U.S. Department of Energy Report 2002-EERC-02-03, 2002 Search PubMed.
- A. Demirbas and M. F. Demirbas, Energy Convers. Manage., 2011, 52, 163–170 CrossRef.
- T. M. Mata, A. A. Martins and N. S. Caetano, Renewable Sustainable Energy Rev., 2010, 14, 217–232 CrossRef CAS.
- G. Andrich, U. Nesti, F. Venturi, A. Zinnai and R. Fiorentini, Eur. J. Lipid Sci. Technol., 2005, 107, 381–386 CrossRef CAS.
- G. Andrich, A. Zinnai, U. Nesti, F. Venturi and R. Fiorentini, Acta Aliment., 2006, 35, 195–203 CrossRef CAS.
- A. P. Carvalho, L. A. Meireles and F. X. Malcata, Biotechnology Progress, 2006, 22, 1490–1506 CAS.
- Y. Chisti, Biotechnol. Adv., 2007, 25, 294–306 CrossRef CAS.
- N. Kosaric and J. Velikonja, FEMS Microbiol. Rev., 1995, 16, 111–142 CrossRef CAS.
- Q. Hu, M. Sommerfeld, E. Jarvis, M. Ghirardi, M. Posewitz, M. Seibert and A. Darzins, Plant J., 2008, 54, 621–639 CrossRef CAS.
- M. P. Mansour, D. M. F. Frampton, P. D. Nichols, J. K. Volkman and S. I. Blackburn, J. Appl. Phycol., 2005, 17, 287–300 CrossRef CAS.
- I. Kubickova, M. Snare, K. Eranen, P. Maki-Arvela and D. Y. Murzin, Catal. Today, 2005, 106, 197–200 CrossRef CAS.
- S. V. Ghadge and H. Raheman, Biomass Bioenergy, 2005, 28, 601–605 CrossRef CAS.
- N. H. Tran, J. R. Bartlett, G. S. K. Kannangara, A. S. Milev, H. Volk and M. A. Wilson, Fuel, 2010, 89, 265–274 CrossRef CAS.
- L. Brennan and P. Owende, Renewable Sustainable Energy Rev., 2010, 14, 557–577 CrossRef CAS.
- H. H. Khoo, P. N. Sharratt, P. Das, R. K. Balasubramanian, P. K. Naraharisetti and S. Shaik, Bioresour. Technol., 2011, 102, 5800–5807 CrossRef CAS.
- V. Patil, K. Q. Tran and H. R. Giselrod, Int. J. Mol. Sci., 2008, 9, 1188–1195 CrossRef CAS.
- S. Amin, Energy Convers. Manage., 2009, 50, 1834–1840 CrossRef CAS.
- P. Bondioli, Top. Catal., 2004, 27, 77–82 CrossRef CAS.
- A. Demirbas, Energy Convers. Manage., 2009, 50, 2782–2801 CrossRef CAS.
- Z. Y. Du, Y. C. Li, X. Q. Wang, Y. Q. Wan, Q. Chen, C. G. Wang, X. Y. Lin, Y. H. Liu, P. Chen and R. Ruan, Bioresour. Technol., 2011, 102, 4890–4896 CrossRef CAS.
- R. Li, Z. P. Zhong, B. S. Jin and A. J. Zheng, Energy Fuels, 2012, 26, 2996–3002 CrossRef CAS.
- H. Fukuda, A. Kondo and H. Noda, Journal of Bioscience and Bioengineering, 2001, 92, 405–416 CAS.
- F. Ma and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef CAS.
- B. Smith, H. C. Greenwell and A. Whiting, Energy Environ. Sci., 2009, 2, 262–271 CAS.
- S. Sawayama, S. Inoue, Y. Dote and S. Y. Yokoyama, Energy Convers. Manage., 1995, 36, 729–731 CrossRef CAS.
- A. A. Peterson, F. Vogel, R. P. Lachance, M. Froling, M. J. Antal and J. W. Tester, Energy Environ. Sci., 2008, 1, 32–65 CAS.
- B. C. Wu, S. Ramon, C. A. DeLuca, E. K. Payne, US Patent No. 0050502, 2010 Search PubMed.
- M. Koberg and A. Gedanken, Energy Environ. Sci., 2012, 5, 7460–7469 CAS.
- R. O. Idem, S. P. R. Katikaneni and N. N. Bakhshi, Fuel Process. Technol., 1997, 51, 101–125 CrossRef CAS.
- A. Leung, D. G. B. Boocock and S. K. Konar, Energy Fuels, 1995, 9, 913–920 CrossRef CAS.
- T. Kalnes, T. Marker and D. R. Shonnard, Int. J. Chem. React. Eng., 2007, 5, A48 Search PubMed.
- S. Lestari, I. Simakova, A. Tokarev, P. Maki-Arvela, K. Eranen and D. Y. Murzin, Catal. Lett., 2008, 122, 247–251 CrossRef CAS.
- J. Myllyoja, P. Aalto, P. Savolainen, V. Purola, V. Alopaeus, J. Gronqvist, US Patent US2007/010682 A1, 2007 Search PubMed.
- M. F. Demirbas, Appl. Energy, 2009, 86, S151–S161 CrossRef.
- D. E. Resasco and S. Crossley, AIChE J., 2009, 55, 1082–1089 CrossRef CAS.
- S. A. Scott, M. P. Davey, J. S. Dennis, I. Horst, C. J. Howe, D. J. Lea-Smith and A. G. Smith, Curr. Opin. Biotechnol., 2010, 21, 277–286 CrossRef CAS.
- V. A. Yakovlev, S. A. Khromova, O. V. Sherstyuk, V. O. Dundich, D. Y. Ermakov, V. M. Novopashina, M. Y. Lebedev, O. Bulavchenko and V. N. Parmon, Catal. Today, 2009, 144, 362–366 CrossRef CAS.
- S. K. Hoekman, A. Broch, C. Robbins, E. Ceniceros and M. Natarajan, Renewable Sustainable Energy Rev., 2012, 16, 143–169 CrossRef CAS.
- G. Pokoo-Aikins, A. Nadim, M. M. El-Halwagi and V. Mahalec, Clean Technol. Environ. Policy, 2010, 12, 239–254 CrossRef CAS.
- H. M. Amaro, A. C. Guedes and F. X. Malcata, Appl. Energy, 2011, 88, 3402–3410 CrossRef CAS.
- J. Lu, C. Sheahan and P. C. Fu, Energy Environ. Sci., 2011, 4, 2451–2466 CAS.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 CAS.
- A. Melis, Energy Environ. Sci., 2012, 5, 5531–5539 CAS.
- S. P. Zou, Y. L. Wu, M. D. Yang, C. Li and J. M. Tong, Energy Environ. Sci., 2010, 3, 1073–1078 CAS.
- P. G. Duan and P. E. Savage, Bioresour. Technol., 2011, 102, 1899–1906 CrossRef CAS.
- H. R. Appell, Y. C. Fu, E. G. Illig, F. W. Steffgen, R. D. Miller, Report of Investigations 8013, Washington, DC: Bureau of Mines. 1975 Search PubMed.
- H. R. Appell, J. C. Tosh, US Patent No. 3,733, 255, 1973 Search PubMed.
- S. M. Kohan, Biomass conversion processes for energy and fuels, Plenum Press, New York, 1981 Search PubMed.
- K. S. Ocfemia, Y. Zhang and T. Funk, Transactions of the ASABE, 2006, 49, 1897–1904 Search PubMed.
- Y. Zhang, in Biofuels from Agricultural Wastes and Byproducts, ed. H. P. Blaschek, T. Ezeji and J. Scheffran, Wiley-Blackwell, 2010 Search PubMed.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- B. J. He, Y. Zhang, T. L. Funk, G. L. Riskowski and Y. Yin, Transactions of the ASAE, 2000, 43, 1827–1833 CAS.
- S. Karagoz, T. Bhaskar, A. Muto and Y. Sakata, J. Chem. Technol. Biotechnol., 2005, 80, 1097–1102 CrossRef CAS.
- S. Karagoz, T. Bhaskar, A. Muto and Y. Sakata, Bioresour. Technol., 2006, 97, 90–98 CrossRef.
- D. Zhou, L. A. Zhang, S. C. Zhang, H. B. Fu and J. M. Chen, Energy Fuels, 2010, 24, 4054–4061 CrossRef CAS.
- Y. Dote, S. Sawayama, S. Inoue, T. Minowa and S. Yokoyama, Fuel, 1994, 73, 1855–1857 CrossRef CAS.
- L. G. Alba, C. Torri, C. Samori, J. van der Spek, D. Fabbri, S. R. A. Kersten and D. W. F. Brilman, Energy Fuels, 2012, 26, 642–657 CrossRef.
- P. Biller and A. B. Ross, Bioresour. Technol., 2011, 102, 215–225 CrossRef CAS.
- T. M. Brown, P. G. Duan and P. E. Savage, Energy Fuels, 2010, 24, 3639–3646 CrossRef CAS.
- T. Minowa, S. Yokoyama, M. Kishimoto and T. Okakura, Fuel, 1995, 74, 1735–1738 CrossRef CAS.
- A. B. Ross, P. Biller, M. L. Kubacki, H. Li, A. Lea-Langton and J. M. Jones, Fuel, 2010, 89, 2234–2243 CrossRef CAS.
- Y. F. Yang, C. P. Feng, Y. Inamori and T. Maekawa, Resour., Conserv. Recycl., 2004, 43, 21–33 CrossRef.
- G. Yu, Y. H. Zhang, L. Schideman, T. Funk and Z. C. Wang, Energy Environ. Sci., 2011, 4, 4587–4595 CAS.
- D. R. Vardon, B. K. Sharma, G. V. Blazina, K. Rajagopalan and T. J. Strathmann, Bioresour. Technol., 2012, 109, 178–187 CrossRef CAS.
- S. P. Zou, Y. L. Wu, M. D. Yang, K. Imdad, C. Li and J. M. Tong, Energy, 2010, 35, 5406–5411 CrossRef CAS.
- K. Anastasakis and A. B. Ross, Bioresour. Technol., 2011, 102, 4876–4883 CrossRef CAS.
- D. M. Li, L. M. Chen, D. Xu, X. W. Zhang, N. H. Ye, F. J. Chen and S. L. Chen, Bioresour. Technol., 2012, 104, 737–742 CrossRef CAS.
- S. Sawayama, T. Minowa and S. Y. Yokoyama, Biomass Bioenergy, 1999, 17, 33–39 CrossRef CAS.
- S. S. Toor, L. Rosendahl and A. Rudolf, Energy, 2011, 36, 2328–2342 CrossRef CAS.
- C. Torri, L. G. Alba, C. Samori, D. Fabbri and D. W. F. Brilman, Energy Fuels, 2012, 26, 658–671 CrossRef CAS.
- U. Jena and K. C. Das, Production of Biocrude Oil from Microalgae via Thermochemical Liquefaction Process, Amer. Soc. Agricultural & Biological Engineers, St Joseph, 2009 Search PubMed.
- T. O. Matsui, A. Nishihara, C. Ueda, M. Ohtsuki, N. O. Ikenaga and T. Suzuki, Fuel, 1997, 76, 1043–1048 CrossRef CAS.
- G. Yu, Y. Zhang, L. Schideman, T. L. Funk and Z. Wang, Transactions of the ASABE, 2011, 54, 239–246 Search PubMed.
- D. R. Vardon, B. K. Sharma, J. Scott, G. Yu, Z. C. Wang, L. Schideman, Y. H. Zhang and T. J. Strathmann, Bioresour. Technol., 2011, 102, 8295–8303 CrossRef CAS.
- P. G. Duan and P. E. Savage, Ind. Eng. Chem. Res., 2011, 50, 52–61 CrossRef CAS.
- P. J. Valdez, J. G. Dickinson and P. E. Savage, Energy Fuels, 2011, 25, 3235–3243 CrossRef CAS.
- T. Minowa and S. Sawayama, Fuel, 1999, 78, 1213–1215 CrossRef CAS.
- C. M. Beal, C. H. Smith, M. E. Webber, R. S. Ruoff and R. E. Hebner, BioEnergy Res., 2011, 4, 36–60 CrossRef.
- B. Cheirsilp, A. H-Kittikun and S. Limkatanyu, Biochem. Eng. J., 2008, 42, 261–269 CrossRef CAS.
- B. Freedman, R. O. Butterfield and E. H. Pryde, J. Am. Oil Chem. Soc., 1986, 63, 1375–1380 CrossRef CAS.
- K. Komers, F. Skopal, R. Stloukal and J. Machek, Eur. J. Lipid Sci. Technol., 2002, 104, 728–737 CrossRef CAS.
- R. Tesser, L. Casale, D. Verde, M. Di Serio and E. Santacesaria, Chem. Eng. J., 2010, 157, 539–550 CrossRef CAS.
- R. Tesser, L. Casale, D. Verde, M. Di Serio and E. Santacesaria, Chem. Eng. J., 2009, 154, 25–33 CrossRef CAS.
- A. F. Chang and Y. A. Liu, Ind. Eng. Chem. Res., 2010, 49, 1197–1213 CrossRef CAS.
- P. G. Cao, A. Y. Tremblay and M. A. Dube, Ind. Eng. Chem. Res., 2009, 48, 2533–2541 CrossRef CAS.
- M. Canakci and J. Van Gerpen, Transactions of the ASAE, 1999, 42, 1203–1210 CAS.
- B. Liu and Z. Zhao, J. Chem. Technol. Biotechnol., 2007, 82, 775–780 CrossRef CAS.
- B. Freedman, E. H. Pryde and T. L. Mounts, J. Am. Oil Chem. Soc., 1984, 61, 1638–1643 CrossRef CAS.
- K. Ngaosuwan, X. H. Mo, J. G. Goodwin and P. Praserthdam, Top. Catal., 2010, 53, 783–794 CrossRef CAS.
- M. G. M. D'Oca, P. L. Haertel, D. C. de Moraes, F. J. P. Callegaro, M. H. S. Kurz, E. G. Primel, R. M. Clementin and J. A. Moron-Villarreyes, Fuel, 2011, 90, 912–916 CrossRef.
- M. G. M. D'Oca, C. V. Viegas, J. S. Lemoes, E. K. Miyasaki, J. A. Moron-Villarreyes, E. G. Primel and P. C. Abreu, Biomass Bioenergy, 2011, 35, 1533–1538 CrossRef.
- X. L. Miao and Q. Y. Wu, Bioresour. Technol., 2006, 97, 841–846 CrossRef CAS.
- P. D. Patil, V. G. Gude, A. Mannarswamy, P. Cooke, S. Munson-McGee, N. Nirmalakhandan, P. Lammers and S. G. Deng, Bioresour. Technol., 2011, 102, 1399–1405 CrossRef CAS.
- A. A. Refaat, S. T. El Sheltawy and K. U. Sadek, IJEST, 2008, 5, 315–322 CAS.
- M. B. Johnson and Z. Y. Wen, Energy Fuels, 2009, 23, 5179–5183 CrossRef CAS.
- R. Alenezi, G. A. Leeke, J. M. Winterbottom, R. C. D. Santos and A. R. Khan, Energy Convers. Manage., 2010, 51, 1055–1059 CrossRef CAS.
- P. D. Patil, V. G. Gude, A. Mannarswamy, P. Cooke, N. Nirmalakhandan, P. Lammers and S. G. Deng, Fuel, 2012, 97, 822–831 CrossRef CAS.
- P. D. Patil, V. G. Gude, A. Mannarswamy, S. G. Deng, P. Cooke, S. Munson-McGee, I. Rhodes, P. Lammers and N. Nirmalakhandan, Bioresour. Technol., 2011, 102, 118–122 CrossRef CAS.
- A. P. S. Chouhan and A. K. Sarma, Renewable Sustainable Energy Rev., 2011, 15, 4378–4399 CrossRef CAS.
- S. L. Yan, S. Mohan, C. DiMaggio, M. Kim, K. Y. S. Ng and S. O. Salley, Fuel, 2010, 89, 2844–2852 CrossRef CAS.
- E. S. Umdu, M. Tuncer and E. Seker, Bioresour. Technol., 2009, 100, 2828–2831 CrossRef CAS.
- Y. S. Li, S. A. Lian, D. M. Tong, R. L. Song, W. Y. Yang, Y. Fan, R. W. Qing and C. W. Hu, Appl. Energy, 2011, 88, 3313–3317 CrossRef CAS.
- C. V. McNeff, L. C. McNeff, B. Yan, D. T. Nowlan, M. Rasmussen, A. E. Gyberg, B. J. Krohn, R. L. Fedie and T. R. Hoye, Appl. Catal., A, 2008, 343, 39–48 CrossRef CAS.
- B. J. Krohn, C. V. McNeff, B. W. Yan and D. Nowlan, Bioresour. Technol., 2011, 102, 94–100 CrossRef CAS.
- M. Kim, S. O. Salley and K. Y. S. Ng, Energy Fuels, 2008, 22, 3594–3599 CrossRef CAS.
- Z. Q. Zhang, Y. X. Qu, S. I. Wang and J. D. Wang, J. Mol. Catal. A: Chem., 2010, 323, 91–100 CrossRef CAS.
- G. M. Turner, E. C. Sherer and G. C. Shields, Int. J. Quantum Chem., 1995, 56, 103–112 CrossRef.
- E. C. Sherer, G. M. Turner and G. C. Shields, Int. J. Quantum Chem., 1995, 56, 83–93 CrossRef.
- A. Kubatova, J. St'avova, W. S. Seames, Y. Luo, S. M. Sadrameli, M. J. Linnen, G. V. Baglayeva, I. P. Smoliakova and E. I. Kozliak, Energy Fuels, 2012, 26, 672–685 CrossRef CAS.
- S. Adhikari, S. D. Fernando and A. Haryanto, Chem. Eng. Technol., 2009, 32, 541–547 CrossRef CAS.
- A. Wawrzetz, B. Peng, A. Hrabar, A. Jentys, A. A. Lemonidou and J. A. Lercher, J. Catal., 2010, 269, 411–420 CrossRef CAS.
- N. Worz, A. Brandner and P. Claus, J. Phys. Chem. C, 2010, 114, 1164–1172 Search PubMed.
- L. He, J. M. S. Parra, E. A. Blekkan and D. Chen, Energy Environ. Sci., 2010, 3, 1046–1056 CAS.
- P. Gallezot, Catal. Today, 2011, 167, 31–36 CrossRef CAS.
- T. Yoshikawa, T. Tago, A. Nakamura, A. Konaka, M. Mukaida and T. Masuda, Res. Chem. Intermed., 2011, 37, 1247–1256 CrossRef CAS.
- D. Ghosh, I. F. Sobro and P. C. Hallenbeck, Bioresour. Technol., 2012, 106, 154–160 CrossRef CAS.
- D. Ghosh, A. Tourigny and P. C. Hallenbeck, Int. J. Hydrogen Energy, 2012, 37, 2273–2277 CrossRef CAS.
- L. Zhou, E. Al-Zaini and A. A. Adesina, Fuel, 2012 DOI:10.1016/j.fuel.2012.05.042.
- S. Nitayavardhana and S. K. Khanal, Bioresour. Technol., 2011, 102, 5808–5814 CrossRef CAS.
- G. Anitescu and T. J. Bruno, Energy Fuels, 2012, 26, 324–348 CrossRef CAS.
- F. D. Mercader, M. J. Groeneveld, S. R. A. Kersten, N. W. J. Way, C. J. Schaverien and J. A. Hogendoorn, Appl. Catal., B, 2010, 96, 57–66 CrossRef.
- L. W. Hillen, G. Pollard, L. V. Wake and N. White, Biotechnol. Bioeng., 1982, 24, 193–205 CrossRef CAS.
- L. W. Hillen, L. V. Wake and D. R. Warren, Fuel, 1980, 59, 446–447 CrossRef CAS.
- P. Biller, R. Riley and A. B. Ross, Bioresour. Technol., 2011, 102, 4841–4848 CrossRef CAS.
- N. Akiya and P. E. Savage, Chem. Rev., 2002, 102, 2725–2750 CrossRef CAS.
- D. Kusdiana and S. Saka, Bioresour. Technol., 2004, 91, 289–295 CrossRef CAS.
- R. B. Levine, T. Pinnarat and P. E. Savage, Energy Fuels, 2010, 24, 5235–5243 CrossRef CAS.
- J. Fu, X. Y. Lu and P. E. Savage, Energy Environ. Sci., 2010, 3, 311–317 CAS.
- J. Fu, F. Shi, L. T. Thompson, X. Y. Lu and P. E. Savage, ACS Catal., 2011, 1, 227–231 CrossRef CAS.
- M. Watanabe, T. Iida and H. Inomata, Energy Convers. Manage., 2006, 47, 3344–3350 CrossRef CAS.
- H. Kitazato, S. Asaoka and H. Iwamoto, J. Jpn. Pet. Inst., 1989, 32, 28–34 CrossRef CAS.
- R. K. Sharma and N. N. Bakhshi, Bioresour. Technol., 1991, 35, 57–66 CrossRef CAS.
- J. Ancheyta, S. Sanchez and M. A. Rodriguez, Catal. Today, 2005, 109, 76–92 CrossRef CAS.
- J. Martinez, J. L. Sanchez, J. Ancheyta and R. S. Ruiz, Catal. Rev. Sci. Eng., 2010, 52, 60–105 CAS.
- G. F. Froment, Catal. Today, 1999, 52, 153–163 CrossRef CAS.
- P. C. Krishna and P. Balasubramanian, Ind. Eng. Chem. Res., 2009, 48, 6608–6617 CrossRef CAS.
- J. W. Thybaut and G. B. Marin, Chem. Eng. Technol., 2003, 26, 509–514 CrossRef CAS.
- M. Mitsios, D. Guillaume, P. Galtier and D. Schweich, Ind. Eng. Chem. Res., 2009, 48, 3284–3292 CrossRef CAS.
- G. G. Martens, G. B. Marin, J. A. Martens, P. A. Jacobs and G. V. Baroni, J. Catal., 2000, 195, 253–267 CrossRef CAS.
- J. W. Thybaut, G. B. Marin, G. V. Baron, P. A. Jacobs and J. A. Martens, J. Catal., 2001, 202, 324–339 CrossRef CAS.
- M. V. Frash, V. B. Kazansky, A. M. Rigby and R. A. van Santen, J. Phys. Chem. B, 1998, 102, 2232–2238 CrossRef CAS.
- T. Isoda, S. Maemoto, K. Kusakabe and S. Morooka, Energy Fuels, 1999, 13, 617–623 CrossRef CAS.
- V. B. Kazansky, M. V. Frash and R. A. vanSanten, Appl. Catal., A, 1996, 146, 225–247 CrossRef CAS.
- A. M. Rigby, G. J. Kramer and R. A. vanSanten, J. Catal., 1997, 170, 1–10 CrossRef CAS.
- N. Bhutani, G. P. Rangaiah and A. K. Ray, Ind. Eng. Chem. Res., 2006, 45, 7807–7816 CrossRef CAS.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- A. Brennfuhrer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef.
- S. Lestari, P. Maki-Arvela, I. Simakova, J. Beltramini, G. Q. M. Lu and D. Y. Murzin, Catal. Lett., 2009, 130, 48–51 CrossRef CAS.
- W. F. Maier, W. Roth, I. Thies and P. V. Schleyer, Chem. Ber., 1982, 115, 808–812 CrossRef CAS.
- P. Maki-Arvela, I. Kubickova, M. Snare, K. Eranen and D. Y. Murzin, Energy Fuels, 2007, 21, 30–41 CrossRef.
- M. Snare, I. Kubickova, P. Maki-Arvela, D. Chichova, K. Eranen and D. Y. Murzin, Fuel, 2008, 87, 933–945 CrossRef CAS.
- M. Snare, I. Kubickova, P. Maki-Arvela, K. Eranen, J. Warna and D. Y. Murzin, Chem. Eng. J., 2007, 134, 29–34 CrossRef CAS.
- M. Snare and D. Y. Murzin, Ind. Eng. Chem. Res., 2006, 45, 6875–6875 CrossRef.
- M. Snare, I. Kubickova, P. Maki-Arvela, K. Eranen and D. Y. Murzin, Ind. Eng. Chem. Res., 2006, 45, 5708–5715 CrossRef.
- I. Simakova, O. Simakova, P. Maki-Arvela, A. Simakov, M. Estrada and D. Y. Murzin, Appl. Catal., A, 2009, 355, 100–108 CrossRef CAS.
- J. G. Immer, M. J. Kelly and H. H. Lamb, Appl. Catal., A, 2010, 375, 134–139 CrossRef CAS.
- R. L. Augustine, Heterogeneous Catalysis for the Synthetic Chemist, Marcel Dekker, New York, 1996 Search PubMed.
- T. Morgan, D. Grubb, E. Santillan-Jimenez and M. Crocker, Top. Catal., 2010, 53, 820–829 CrossRef CAS.
- D. Kubicka and L. Kaluza, Appl. Catal., A, 2010, 372, 199–208 CrossRef CAS.
- X. D. Lei, W. Lu, Q. Peng, H. Y. Li, T. Chen, S. L. Xu and F. Z. Zhang, Appl. Catal., A, 2011, 399, 87–92 CrossRef CAS.
- D. L. Geatches, S. J. Clark and H. C. Greenwell, J. Phys. Chem. A, 2010, 114, 3569–3575 CrossRef CAS.
- D. L. Geatches, H. C. Greenwell and S. J. Clark, J. Phys. Chem. A, 2011, 115, 2658–2667 CrossRef CAS.
- W. K. Metcalfe, J. M. Simmie and H. J. Curran, J. Phys. Chem. A, 2010, 114, 5478–5484 CrossRef CAS.
- Y. Chisti, Trends Biotechnol., 2008, 26, 126–131 CrossRef CAS.
- Y. Chisti and J. Y. Yan, Appl. Energy, 2011, 88, 3277–3279 CrossRef.
- T. Shirvani, X. Y. Yan, O. R. Inderwildi, P. P. Edwards and D. A. King, Energy Environ. Sci., 2011, 4, 3773–3778 CAS.
- R. Davis, A. Aden and P. T. Pienkos, Appl. Energy, 2011, 88, 3524–3531 CrossRef.
- L. Amer, B. Adhikari and J. Pellegrino, Bioresour. Technol., 2011, 102, 9350–9359 CrossRef CAS.
- M. Andersson, J. L. Yuan and B. Sunden, Appl. Energy, 2010, 87, 1461–1476 CrossRef CAS.
- M. Salciccioli, M. Stamatakis, S. Caratzoulas and D. G. Vlachos, Chem. Eng. Sci., 2011, 66, 4319–4355 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |